
Abstract
Selexipag is an orally available selective IP prostacyclin-receptor agonist licensed since 2016 for the therapy of pulmonary arterial hypertension (PAH). We aimed to describe real-life data of patients with pulmonary hypertension (PH) treated with selexipag. We analyzed all patients initiated with selexipag from July 2016 to April 2018 at the Department of Internal Medicine V, University of Munich. Non-invasive and invasive parameters corresponding to the risk assessment were collected at baseline and follow-up (FU). Furthermore, we recorded tolerability. Twenty-six patients were treated with selexipag, of whom 23 had PAH and three had chronic thromboembolic PH. At baseline, most patients were in function class (FC) II or III (42% and 54%, respectively). All patients were under medical treatment for PH, mostly dual therapy (92%). One or more side effects were noted in 19 patients, while seven reported no side-effects. FU assessment was available in 20 patients after 149 ± 80 days of treatment. Nt-proBNP (median, baseline 1641 pg/mL, FU 1185 pg/mL, P = 0.05) and PVR (mean ± SD, baseline 8.5 ± 4.3 WU, FU 5.6 ± 1.1 WU; P < 0.05) improved significantly. At FU, at least one risk assessment parameter improved in nine patients (45%), all parameters remained in the same risk group in seven patients (35%), and at least one parameter deteriorated in four patients (20%). Interestingly, patients with any side effect throughout the dose titration had a better treatment response than those without any side effects. In our real-life cohort, the majority of patients with PH treated with selexipag showed a stable or improved risk assessment at FU.
Introduction
Pulmonary arterial hypertension (PAH) is caused by remodeling of small pulmonary vessels leading to a progressive increase in pulmonary vascular resistance (PVR) and, ultimately, to right ventricular (RV) failure and death. 1 The mortality risk of patients with PAH can by assessed by invasive and non-invasive parameters including World Health Organization functional class (WHO FC), brain natriuretic peptide (BNP), 6-min walk distance (6MWD), cardiac index (CI), and mean right atrial pressure (mRAP). Current treatments for PAH target prostacyclin, endothelin-1, and nitric oxide pathways; drugs targeting each of these pathways may be combined to increase treatment effects. Guidelines recommend combination therapy if initial risk is not low and escalation of therapy if risk is not low at reassessment. 2
Selexipag is the first orally available, highly selective prostacyclin (IP) receptor agonist, approved in the therapy of PAH in the European Union since May 2016 for patients in WHO FC II or higher. The phase III trial (GRIPHON) showed that, among patients with PAH, the risk of the primary composite end point of death or a complication related to PAH was significantly lower with selexipag than with placebo. 3
Secondary endpoint analysis showed a small but significant increase in the 6MWD; however, WHO FC did not change in most of the patients. Exploratory endpoint analysis of BNP showed a significant decrease. Hemodynamic parameters were not assessed as part of the study.
Several other drugs that target the prostacyclin pathway are licensed in Europe for pulmonary hypertension (PH) in more advanced disease (WHO FC III and higher), but all of these are prostacyclin analogues using other routes of administration such as inhalation and parenteral route. Intravenous prostanoid therapy is considered one of the most effective treatment options in PAH, as it was shown to improve survival even in the short term. 4 However, prostanoids have not been consistently used, even in the most seriously ill patients, 5 due to the complex and time-consuming delivery and dose-limiting side effects.6,7
With respect to the GRIPHON trial results, but also the other available therapies, the role of selexipag in clinical practice needs to be defined. Moreover, patients in real-life cohorts do not always correspond to the study population as they can have more complicated disease, multiple co-morbidities, and more variable individual treatment regimens.
Hence, the aim of our study was to describe real-life data on treatment with selexipag by assessing, first, tolerability and, second, efficacy as measured by current risk assessment parameters including hemodynamics.
Materials and methods
Selection of patients
All patients with PH, in whom treatment with selexipag was initiated from July 2016 to April 2018 at the Department of Internal Medicine V, University of Munich, were included and analyzed retrospectively. The study was approved by the local ethics committee (No 18-611). Diagnosis of PAH was confirmed by right heart catheterization (RHC) in all patients. At baseline, all patients already received a stable treatment for PH, with mono or dual therapy that did not include prostanoids. One patient had been treated with prostanoids previously, but not at the time of selexipag initiation. Selexipag was added to the baseline treatment. The University of Munich Institutional Review Board approved this study (no. 18-611).
Procedures
Non-invasive and invasive parameters were collected at baseline and follow-up (FU) including the determination of the WHO-FC, 6MWD, nt-proBNP, tricuspid annular plane systolic excursion (TAPSE), right atrial area (RAA), mean pulmonary arterial pressure (mPAP), mean right atrial pressure (mRAP), CI, and pulmonary vascular resistance (PVR). In addition, using six of these parameters (WHO-FC, 6MWD, nt-proBNP, RAA, mRAP, CI), the risk assessment was defined at baseline and at FU as suggested in current guidelines. 2
Baseline was defined as the time of stable medication before starting selexipag. FU data were collected after 4–6 months of selexipag treatment. If RHC was performed, non-invasive parameters were obtained at the same visit. RHC was performed 2–4 h after the morning dose of PAH treatments.
Dosage titration
Dosage titration was performed as recommended starting with 200 µg twice daily and increasing weekly in 200 -µg twice-daily increments until unmanageable side effects occurred. The dose was then decreased by 200 µg in both daily doses. Further uptitration was suggested to patients as soon as all side effects subsided, with slower increases, and 200 µg increments daily also allowed. If unmanageable side effects occurred again, dosage was decreased by 200 µg once or twice daily and reached dosage was considered the maximal maintenance dosage.
We treated side effects with supportive therapy using antiemetic, antidiarrheal, and/or analgetic drugs. As first-line therapy we used metoclopramide 10 mg (up to three times per day) to treat nausea, loperamid 2 mg (up to six times per day) to treat diarrhea, and ibuprofen 400 mg (up to three times per day) to treat pain. Dosage titration and supply of selexipag was supported by a PH nurse from an external patient service who kept in touch with patients and physicians from the first day of therapy with selexipag onwards. Patients were called at least weekly, questioned about occurrence of side effects (i.e. headache, flush, jaw pain, diarrhea, nausea/emesis, and musculosceletal pain) and this was documented by the patient service on a standardized questionnaire. Patients were advised about supportive treatment and dosage titration after consulting the treating physician if necessary.
Medical examinations
WHO FC was determined by the treating physician at each visit. The 6-min walking test (6MWT) was carried out according to ATS guidelines. 8 Transthoracic echocardiography was performed in the left lateral decubitus position and two-dimensional and M-mode echocardiograms as well as CW and Tissue Doppler measurements were obtained. RHC was performed by using a Swan-Ganz catheter. Cardiac output was measured by thermodilution.
Statistical analysis
We tested data for normal distribution using D’Agostina and Pearson test. Continuous data are presented as mean with standard deviation when distributed normally or as the median with interquartile range (IQR) otherwise. Discrete data are given as counts or as percentages. Differences between baseline and follow-up were analyzed by paired t-test when distributed normally or Wilcoxon matched-pairs signed rank test otherwise. Values of P < 0.05 were considered significant. All statistical analyses were performed with Prism 7 (La Jolla, CA, USA).
Results
Patient cohort
From July 2016 to April 2018, 26 patients were treated with selexipag at the Department of Internal Medicine V, University of Munich.
Baseline characteristics of all patients treated with selexipag (n = 26).
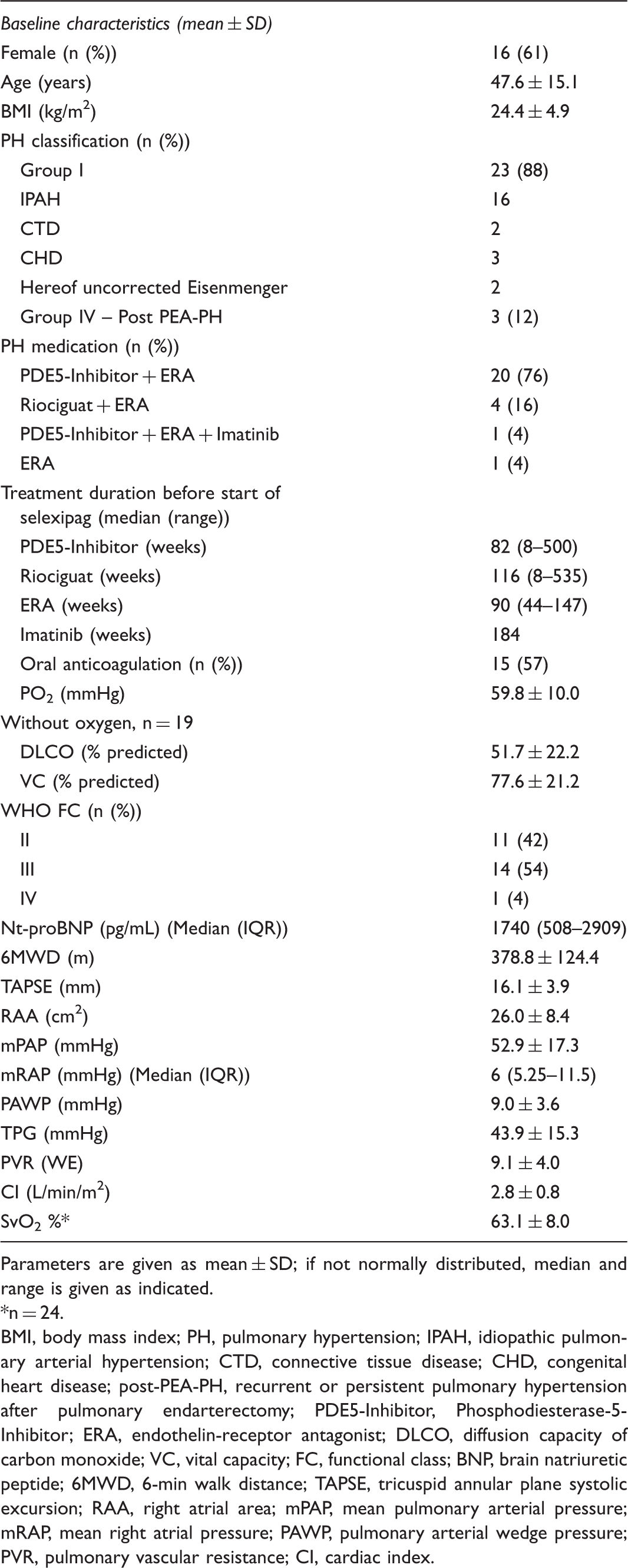
Parameters are given as mean ± SD; if not normally distributed, median and range is given as indicated.
n = 24.
BMI, body mass index; PH, pulmonary hypertension; IPAH, idiopathic pulmonary arterial hypertension; CTD, connective tissue disease; CHD, congenital heart disease; post-PEA-PH, recurrent or persistent pulmonary hypertension after pulmonary endarterectomy; PDE5-Inhibitor, Phosphodiesterase-5-Inhibitor; ERA, endothelin-receptor antagonist; DLCO, diffusion capacity of carbon monoxide; VC, vital capacity; FC, functional class; BNP, brain natriuretic peptide; 6MWD, 6-min walk distance; TAPSE, tricuspid annular plane systolic excursion; RAA, right atrial area; mPAP, mean pulmonary arterial pressure; mRAP, mean right atrial pressure; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance; CI, cardiac index.
Invasive FU data are available in 16 patients, non-invasive parameters in 20 patients (Fig. 1). Two patients stopped selexipag because of intolerable side-effects, two died before FU, one was switched to intravenous treprostinil because of cardiac decompensation before FU, and one was lost to FU.
Description of the study cohort. FU, follow-up.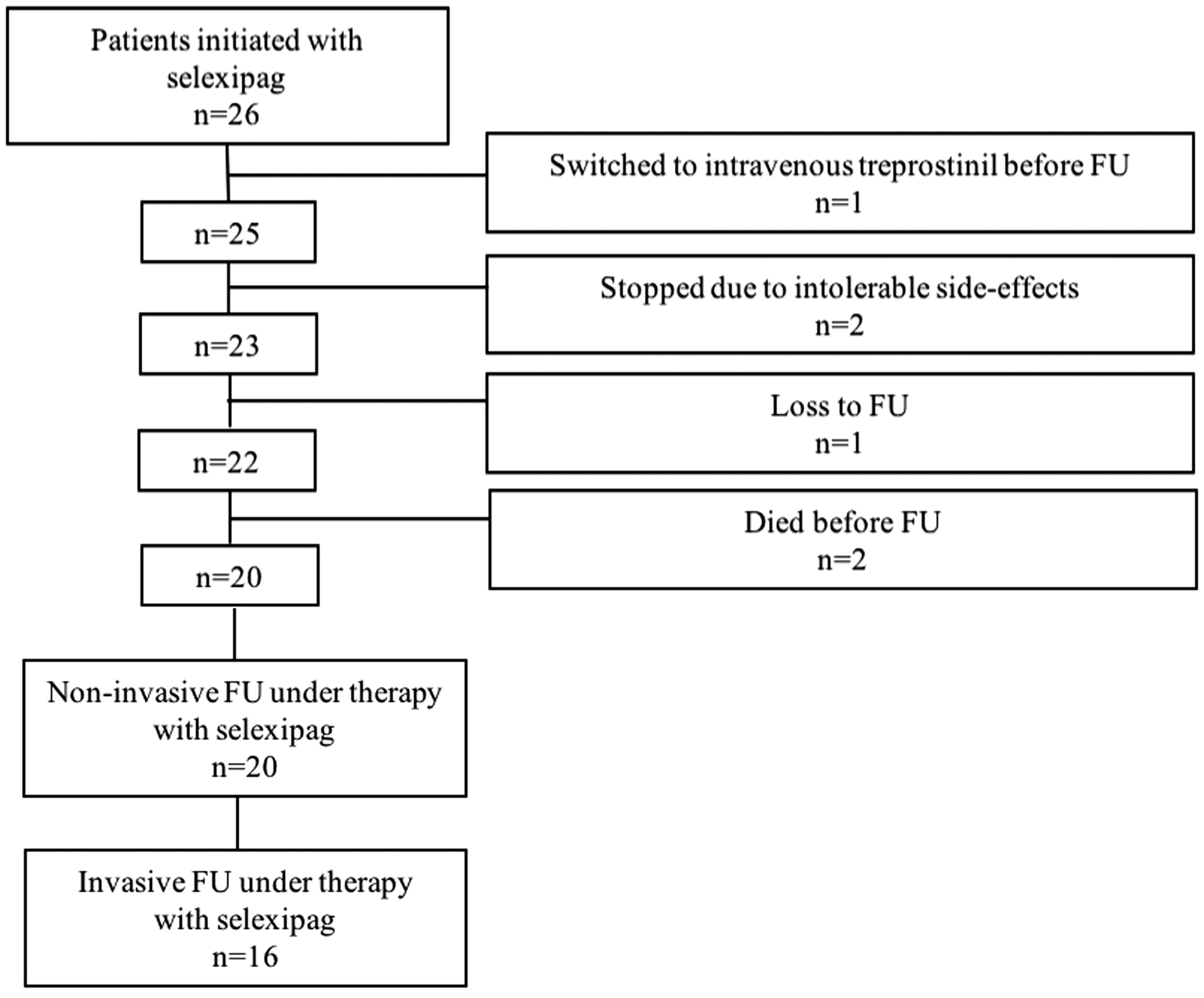
Maintenance dosage and tolerability
Twenty-two patients continued treatment to reach a maximal maintenance dosage, which was 2900 µg/day at median (IQR = 1550–3200 µg/day). The median time to reach the maximal maintenance dosage was 90 days (range = 56–272 days). The maximal maintenance dosage was low (<1000 µg/day) in 9% of patients, medium (1000–2000 µg/day) in 27%, and high (>2000 µg/day) in 64% of patients (Fig. 2a). The highest recommended dose of 3200 µg/day was achieved by nearly half of the patients. Weight adapted dosage calculation shows that the majority of patients reach a dosage of 20–60 µg/kg/day (Fig. 2b).
Individual maintenance dosage of selexipag; (a) dosage in ug/day; (b) dosage in ug/kg/day.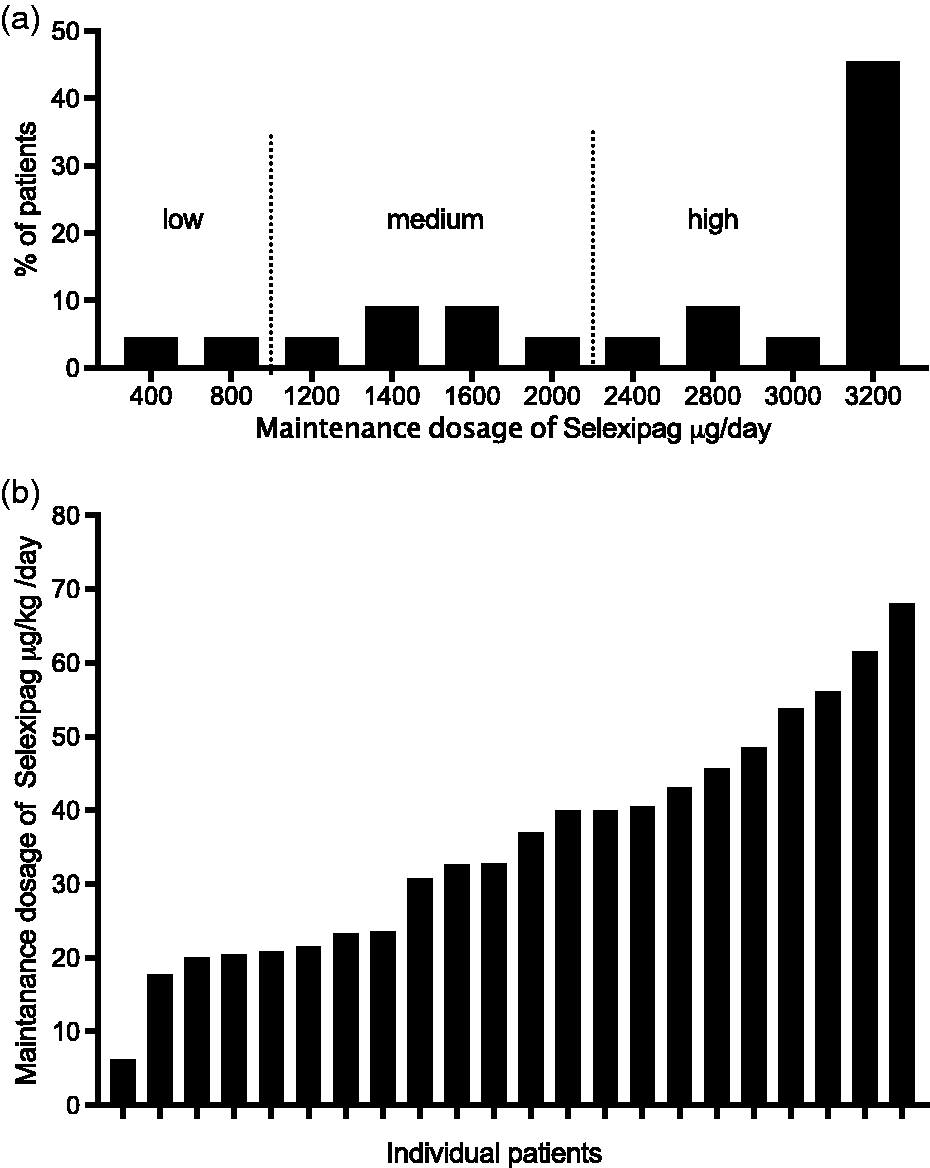
In our cohort, 7/26 patients reported none of the questioned side effects throughout the entire duration of dosage titration and titrated up to 3200 µg/day within 56 days, whereas two patients stopped treatment because of side effects (Fig. 3). The most commonly reported side effects in our patients were musculoskeletal pain (extremity pain and myalgia), diarrhea, and nausea/emesis (38% each). No unknown side effects were reported.
Side effects reported under selexipag therapy (n = 26).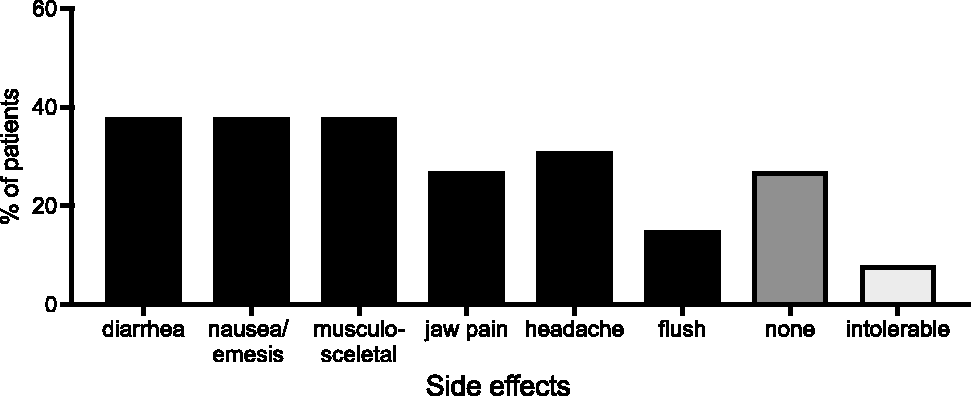
Efficacy
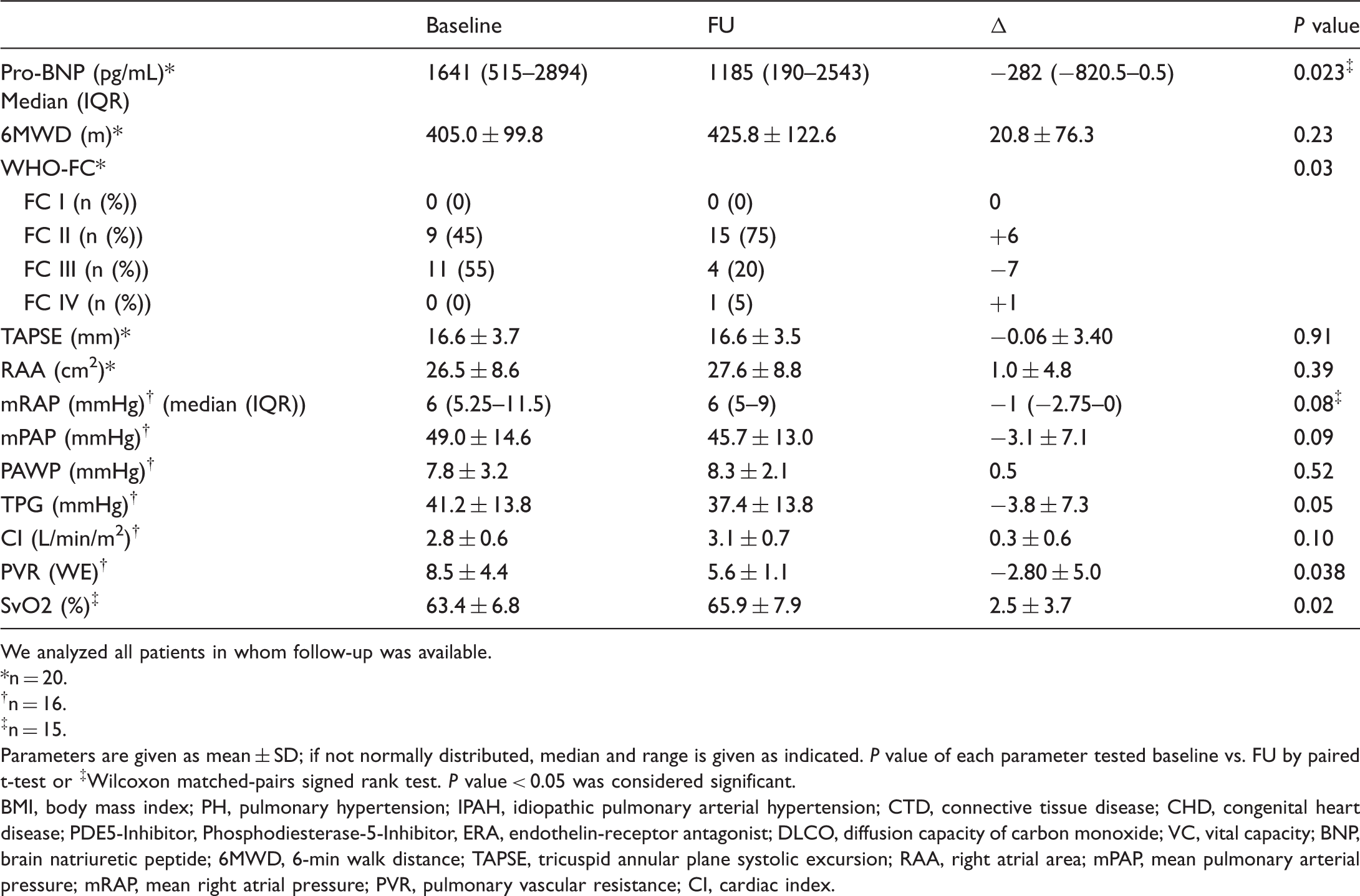
FU assessment was recorded at 149 ± 80 days after starting therapy with selexipag. Nt-proBNP (n = 20; median, baseline 1641 pg/mL; FU 1185 pg/mL, P < 0.05), mixed venous blood oxygen saturation SvO2 (n = 15; mean ± SD, baseline 63.4 ± 6.8%; FU 65.9 ± 7.1%; P < 0.05), and PVR (n = 16; mean ± SD, baseline 8.5 ± 4.3 WU; FU 5.6 ± 1.1 WU; P < 0.05) improved significantly (Table 2). mRAP, mPAP, and CI improved numerically, without reaching statistical significance (ΔmRAP = −1 mmHg, p(mRAP) = 0.08; (ΔmPAP = −3.1 ± 7.1 mmHg p(mPAP) = 0.09; p(CI) = 0.10); 6MWD, TAPSE, and RAA showed no relevant changes. Moreover, WHO FC was improved in seven patients, stable in 11 patients, and deteriorated in two patients (Fig. 4).
WHO FC at baseline and FU (n = 20): nine patients were in FC II at baseline. Of these, eight remained stable and one had FC III at follow-up. Eleven patients were in FC III at baseline. Of these, seven improved to FC II, three remained stable, and one deteriorated. Risk assessment parameters at baseline and follow-up. We analyzed all patients in whom follow-up was available. n = 20. n = 16. n = 15. Parameters are given as mean ± SD; if not normally distributed, median and range is given as indicated. Pvalue of each parameter tested baseline vs. FU by paired t-test or ‡Wilcoxon matched-pairs signed rank test. Pvalue < 0.05 was considered significant. BMI, body mass index; PH, pulmonary hypertension; IPAH, idiopathic pulmonary arterial hypertension; CTD, connective tissue disease; CHD, congenital heart disease; PDE5-Inhibitor, Phosphodiesterase-5-Inhibitor, ERA, endothelin-receptor antagonist; DLCO, diffusion capacity of carbon monoxide; VC, vital capacity; BNP, brain natriuretic peptide; 6MWD, 6-min walk distance; TAPSE, tricuspid annular plane systolic excursion; RAA, right atrial area; mPAP, mean pulmonary arterial pressure; mRAP, mean right atrial pressure; PVR, pulmonary vascular resistance; CI, cardiac index.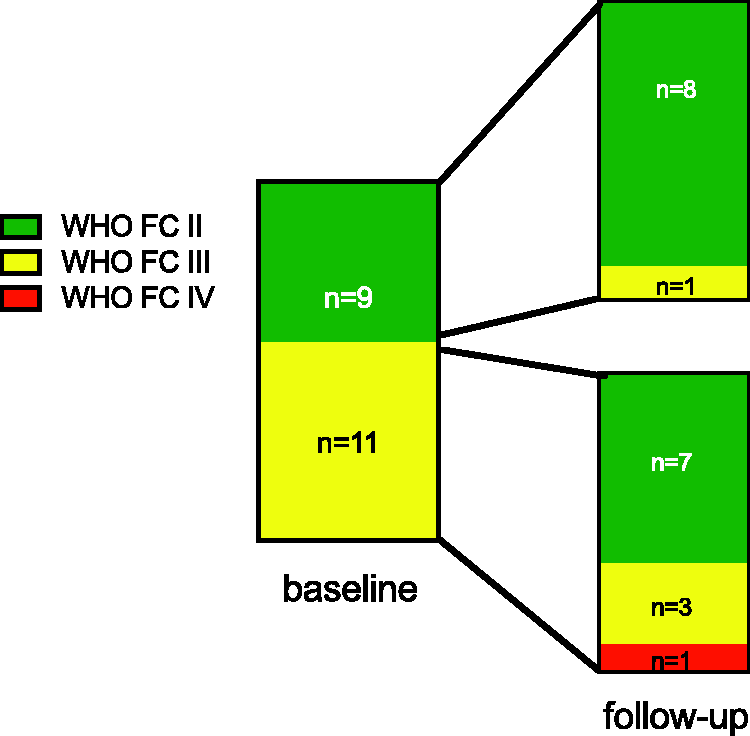
Regarding non-invasive risk assessment (n = 20) using the three parameters WHO FC, BNP, and 6MWD, as recently suggested,
9
we found that at baseline eight patients (40%) had no low-risk criteria, five patients (25%) had one low-risk criterion, five patients (25%) had two low-risk criteria, and two patients (10%) had three low-risk criteria. At follow-up, the distribution was four (20%), six (30%), four (20%), and six (30%) patients with no, one, two, and three low-risk criteria, respectively (Fig. 5a).
Change of risk assessment under treatment with selexipag. The number of parameters in the low-risk group was counted for each patient at baseline and FU. (a) Only three non-invasive parameters were counted (WHO FC, nt-proBNP, 6MWD). (b) Six parameters were counted (WHO FC, nt-proBNP, 6MWD, right atrial area, CI, RAP).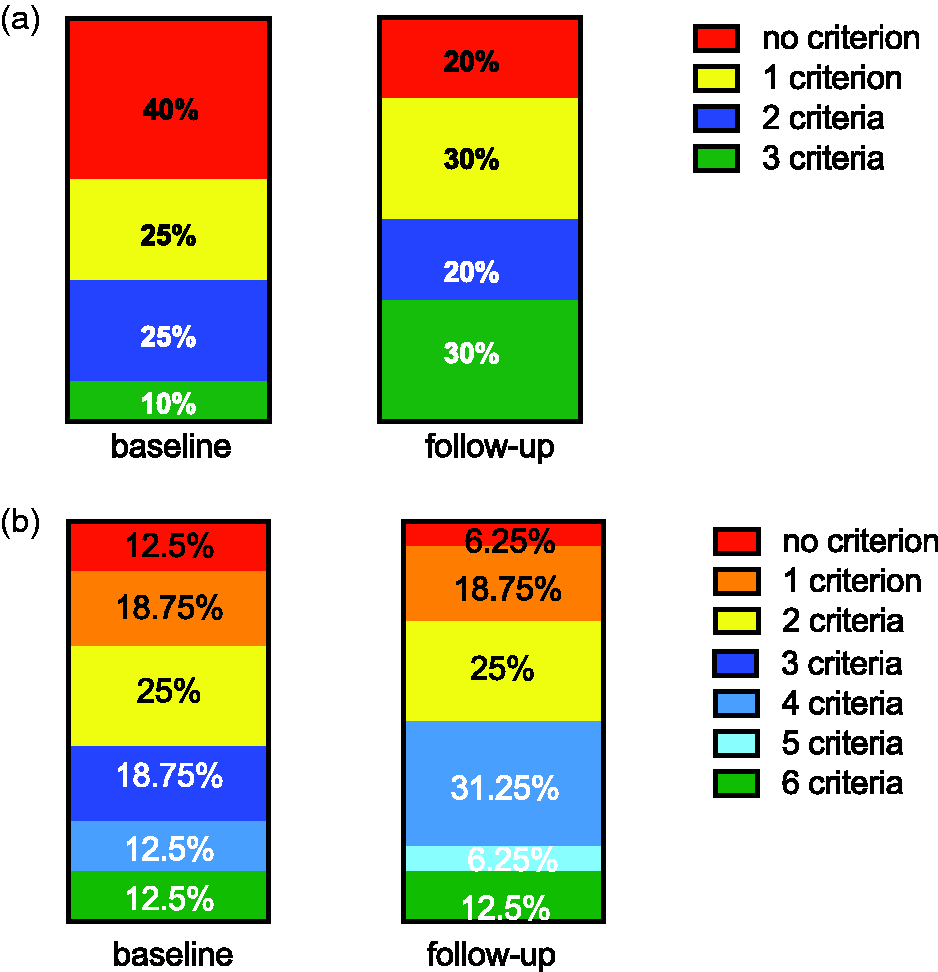
When adding echocardiographic (RAA) and hemodynamic parameters (CI and mRAP, n = 16), we found that at baseline, two patients had no low-risk criteria, three patients had one low-risk criterion, four patients had two low-risk criteria, three patients had three low-risk criteria, two patients had four low-risk criteria, no patients had five low-risk criteria, and two patients had six low-risk criteria. At FU, this improved to one, three, four, no, five, one, and two patients, respectively (Fig. 5b).
When looking at individual patients, we found that risk assessment improved in nine patients (45%), remained stable in seven patients (35%), and deteriorated in four patients (20%) (Suppl. Fig. 1).
We further analyzed possible correlations between the maximal maintenance dosage and the treatment response in terms of risk assessment. Here, we found no correlation (Pearson r = −0.13, P = 0.56). Interestingly, both patients who had a low maximal maintenance dosage improved their risk assessment from two to four low-risk parameters. Thus, we investigated a possible correlation between occurrence of side effects and treatment response. We found that patient with any side effect (n ≥ 1) throughout the dose titration had a better treatment response than those without any side effects assessed by change of number of low-risk parameters (median difference = 2, P = 0.038 by Mann–Whitney U test) and PVR (mean difference ± SD = −2.99 WU ± 1.21, P = 0.027 by t-test).
Patients of special interest
A 50-year-old female patient with IPAH diagnosed in 2007 was on combination therapy, including imatinib, when selexipag was initiated. After diagnosis, she first received PDE-5-inhibitor, then an additional ERA. In 2013, she had severe deterioration and i.v. Ilomedin was added. She remained highly symptomatic in a high-risk constellation. Thus, imatinib was added three months later and the patient was listed for lung transplantation at this point. Hereafter, within months, she improved markedly and she was taken off the transplant list. Because of recurrent catheter infections and patient refusal of further i.v. therapy, Ilomedin was ended in 2016. When selexipag became available three months later, selexipag was initiated and uptitrated to 3200 µg/day. The patient is currently stable and all values are in the low-risk category.
Next, we separately looked at patients with diagnostic subgroups outside the approved indication for selexipag. Here, we found that of three patients with chronic thromboembolic pulmonary hypertension (CTEPH) persisting after pulmonary endarterectomy, two patients had an improved risk assessment under selexipag, whereas one patient remained in the high-risk category under oral triple-therapy with riociguat, ERA, and selexipag 3200 µg/day (Suppl. Fig. 1, middle, red line). After 10 months of selexipag treatment, he was switched to parenteral prostanoid, initially s.c. treprostinil, subsequently i.v. treprostinil with a current dosage of 44 ng/kg body weight/min. As of now, 14 months after the start of treprostinil, the patient is clinically stable but has not improved his risk category compared to the treatment with selexipag, remaining at high risk.
Of the two patients with uncorrected VSD and Eisenmenger reaction one had improved risk assessment while the other one was unchanged (Suppl. Fig. 1).
Patients not included in the efficacy analysis
Six patients were not included in the efficacy analysis because no FU was available (Fig. 1). A 39-year-old female patient with severe CTD-PAH (systemic sclerosis) was in WHO FC IV when selexipag was initiated. After six weeks, she was switched to i.v. treprostinil before FU due to further clinical worsening. She also had other organ manifestations of systemic sclerosis and was not a candidate for lung transplantation. She further deteriorated and died five months after initiation of i.v. treprostinil due to right heart failure.
Two female patients stopped the treatment due to intolerable side effects. One was 62 years old and had CTD-PAH; the other was 50 years old and had IPAH. At baseline, both had a dual oral therapy (PDE-5-inhibitor ERA and riociguat + ERA) and an intermediate-risk classification, which remained unchanged throughout the observation period of the study.
One patient moved to another PH center and FU data could not be retrieved.
Two patients died before FU. Both died at home and no autopsy was performed. The clinical cause of death retained by the family doctor was heart failure in both cases.
A 62-year-old woman with severe IPAH diagnosed four years prior was on therapy with PDE-5-inhibior and ERA. Due to a high-risk constellation, an escalation of therapy with i.v. prostanoid and evaluation for lung transplantation was recommended, but both were declined by the patient. Instead, selexipag was initiated. The patient died three months later when the dosage of selexipag was 1000 µg/day.
A 58-year-old man with severe IPAH diagnosed three years prior was on therapy with PDE-5-inhibior and ERA when selexipag was initiated. Due to a high-risk constellation, he was evaluated for lung transplantation, but this option was rejected due to multiple co-morbidities including previous lung operation because of pleural empyema, neurologic disease, and ulcus cruris with recurrent infections with drug-resistant bacteria. He titrated selexipag up to a maintenance dosage of 1600 µg/day but did not keep further appointments in the PH clinic. However, he continued treatment under direction of his generalist. He died 13 months after initiation of selexipag.
Both patients who died had severe, progressive disease but were not eligible for or declined further treatment options. The clinical cause of death recorded was progression of PAH with right heart failure not linked to the treatment in both cases, and this was in line with the judgement of our center.
Discussion
This is the first study describing real-life data on treatment with selexipag in patients with PH.
In the licensing phase III trial (GRIPHON), 3 the primary composite endpoint of death or a complication related to PAH was reduced under therapy with selexipag. The effect of selexipag with respect to the primary endpoint was similar in the subgroup of patients who were not receiving treatment for the disease at baseline and in the subgroup of patients who were already receiving treatment at baseline, including those who were receiving a combination of two therapies. 3 In contrast to the GRIPHON trial, all of our patients received PH medication at baseline, most of them a dual combination therapy. Compared to our study, the mean age was similar, but the female percentage was higher in the GRIPHON trial. 3 Regarding PAH etiology, most of our patients also had IPAH. However, we also included few patients with CTEPH persisting after PEA and uncorrected CHD patients with Eisenmenger syndrome.
While hemodynamic measurements were not included in the GRIPHON trial, these were assessed at baseline and FU in the majority of our patients. Here we found a significant decrease in PVR at FU. A similar improvement in PVR was found in two phase II trials after 16–17 weeks of treatment,10,11including patients who were already on a dual therapy with PDE5-I and ERA. 10
The number of patients in the high dosage group was higher in our study than in the GRIPHON trial (63% vs. 43%). This may be explained by a more flexible uptitration regimen that was allowed from the beginning in our real-life study compared to a stricter titration scheme up to the maintenance phase in the study. In addition, larger experience of the physicians with the drug, as well as patient education and proactive use of supportive treatment, might have facilitated higher dosages. Interestingly, similar to the GRIPHON trial, we found no association of the dosage with the treatment response.
As expected, side effects typical for the prostacyclin pathway occurred frequently during the dosage titration of the selexipag (Fig. 2). However, the absolute frequency of side effects reported by the patients was lower than expected from the GRIPHON trial. For example, headache was reported by only 27% of our patients compared to 65%. This difference might be explained by lower threshold for reporting during the randomized controlled trial (RCT) study. Here, headache was also reported by 33% of placebo patients, so that the number accountable to the drug (32%) is comparable to our real-life findings. Remarkably, 27% of patients in our study did not report any side effects whereas in the GRIPHON trial only 13% of patients treated with selexipag and 50% of placebo patients reported no side effects.
Selexipag and prostanoids are usually uptitrated until intolerable side effects occur. The rationale behind this is the assumption that side effects may indicate a higher circulating dose of the drug and therefore be related to a beneficial treatment response. However, this assumption is not evidence-based. Interestingly, our study favors this assumption as we found that patients without any side effects showed less pronounced treatment response than those with side effects as measured by change of low-risk parameters and change of PVR under therapy. This is in contrast to a recently published study that analyzed 908 patients from four RCTs of subcutaneous treprostinil and found that the occurrence of gastrointestinal side effects was related to a higher mortality while other side effects showed no associations to outcome. 12
This association of side effects and treatment response under selexipag therapy warrants further investigation in larger cohorts. Additionally, it raises the question if dosage titration beyond the recommended maximal dosage of 2 × 1600 µg/day might be beneficial in some patients, as this has already been suggested in some case reports. 13
The present study should be interpreted with careful consideration of its limitations. We report observational data on a limited number of individuals from a single center. Moreover, there is no placebo group and thus all analyses are paired comparison of baseline to FU.
In the presented real-life cohort, several patients belonged to diagnostic subgroups that were not studied in the GRIPHON study: three patients had CTEPH persisting after PEA and two patients had non-corrected VSD and Eisenmenger reaction. Two of the patients with CTEPH showed a clear benefit while one patient deteriorated further. Of the patients with Eisenmenger syndrome, one improved and the other was stable. Prostanoid treatments have been used successfully as an add-on therapy in such patients who are not sufficiently treated with other drugs, even though none of the prostanoids is licensed for these diagnoses.14–16Further studies are necessary to evaluate if selexipag is beneficial in these indications.
Conclusion
In sum, our real-life data show significant improvement of functional, serological, and hemodynamic parameters under treatment with selexipag. This is also reflected by an improved risk assessment in nearly half of the treated patients, which might translate into longer event-free survival. Further investigations are warranted regarding a possible association of occurrence of side effects and treatment response.
Supplemental Material
Supplemental material for Real-life data on Selexipag for the treatment of pulmonary hypertension
Supplemental Material for Real-life data on Selexipag for the treatment of pulmonary hypertension by Michaela Barnikel, Nikolaus Kneidinger, Friederike Klenner, Andrea Waelde, Paola Arnold, Torben Sonneck, Jürgen Behr, Claus Neurohr and Katrin Milger in Pulmonary Circulation
Footnotes
Conflict of interest
The author(s) declare the following conflicts of interest: KM has received honoraria for lectures from Actelion, MSD, Novartis; JB has received honoraria for lectures and/or consultancy from Actelion, Bayer, Boehringer, Gilead, GSK, InterMune, Lilly, MSD, Novartis, and Pfizer; CN received honoraria for lectures and/or consultancy from Actelion, Bayer, GSK, and United Therapeutics; NK received honoraria for lectures and/or consultancy from Actelion, Astra Zeneca, Boehringer Ingelheim, Novartis, Pfizer, and Roche; MB, TS, PA, and AW declare no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
*Equal contributors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.