
Abstract

Effect of cigarette smoking and honey on lung inflammation by lipopolysaccharides (LPS) in mice
Department of Animal Medical Science, Kyoto Sangyo University, Japan; University of California Davis, USA
Cigarette smoke (CS) is a risk factor of pulmonary diseases. Honey is used as a traditional medicine for colds and skin inflammation. It is known that lipopolysaccharides (LPS) induce lung inflammation. We previously reported that CS inhibited immune functions. However, the effect of CS and honey on LPS-induced lung inflammation is unclear. In this study, we investigated whether CS and honey affect LPS-induced lung inflammation. Mice were exposed to CS for ten days. After exposure to CS, mice inhaled 600 µg of Japanese honey and one day later mice inhaled 60 µg LPS by intranasal administration. After one day, bronchoalveolar lavage cells were obtained. Expressions of TLR4, CD14 surface antigen, and reactive oxygen species production were measured by FACS. Cytokines and NF-κB mRNA expressions were assayed by RT-PCR. Neutrophils were significantly increased with LPS inhalation. Expression of TLR4 in neutrophils was significantly decreased by CS. Hydrogen peroxide production from neutrophils was significantly increased by CS. IL-1β, TNF-α, CXCL1, and NF-κB mRNA expression of neutrophils were not different from CS. Honey inhibited infiltration of neutrophils to the lung, IL-1β, and CXCL1 mRNA expression. These results suggest that the recognition of neutrophil is inhibited by CS. This inhibition may result in increased pulmonary infection and exacerbated infection. Honey indicated anti-inflammation activity via the suppression of infiltration of neutrophils to the lung. Honey may be a candidate as an anti-inflammatory drug in pulmonary disease.
The importance of ECHO findings, RV/LV ratio, and cardiac biomarkers for prognosis and follow-up in patients with pulmonary embolism
Departments of Pulmonary Diseases, Cardiology, Radiology, Gazi University School of Medicine, Ankara, Turkey
Early and late adverse outcomes can be seen in patients with pulmonary embolism (PE). This study was designed to find out the importance of blood proBNP and troponin T values, tricuspid annular plane systolic excursion (TAPSE), and pulmonary artery pressure (PAP) received from echocardiography and right ventricle/left ventricle (RV/LV) ratio obtained from pulmonary CT angiography for early prognosis. Moreover, we also aimed to find out the role of these parameters to predict permanent high PAP, known as chronic thromboembolic pulmonary hypertension (CTEPH). Eighty patients with PE were enrolled in the study. All patients’ proBNP, troponin T values, and RV/LV ratio were noted. PAP and TAPSE values were also recorded at admission and after three months of anticoagulant therapy. In the whole group, there were positive correlations between serum biomarkers, PAP value, and RV/LV ratio, where there were negative correlations with TAPSE value at admission. Eleven patients had early adverse clinical outcome. With a threshold of 148 pg/mL, proBNP had a sensitivity of 81.8% and specificity of 52.3% for predicting early adverse outcomes. PAP > 35 mmHg at admission was a significant predictor of early adverse outcomes. Eleven patients had elevated PAP (≥35 mmHg) after three months of anticoagulation. These patients had higher PAP, RV/LV ratio, and proBNP levels at admission than the others. Furthermore, a PAP value of ≥41 mmHg at admission was found to be an independent risk factor for predicting CTEPH (OR = 7.37, P = 0.038). PE patients with high cardiac biomarkers and RV/LV ratio should be monitored closely in early period of the disease whereas patients who have PAP ≥ 41 mmHg should be followed up carefully for the risk of CTEPH.
Increased interleukin-1 levels are an early hallmark of pulmonary remodeling in Fra-2 mouse model of scleroderma
Medical University of Graz, Austria; Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria; Institute of Cancer Research of the Medical University of Vienna, Austria
Pulmonary hypertension (PH) and pulmonary fibrosis (PF) worsen the clinical outcome of scleroderma (SSc) patients. However, the factors responsible for vascular and parenchymal remodeling are still unclear. Here, we have investigated the sequence of events leading to pulmonary remodeling in SSc. The longitudinal development and progression of SSc was studied in the Fra-2 TG mouse model. Signaling pathways and extracellular matrix protein expression were investigated in primary human pulmonary arterial smooth muscle cells (PASMCs) and parenchymal fibroblasts (PFs) after IL-1α or IL-1β stimulation. Human lung tissue and plasma samples were applied to translate our findings to SSc patients. In Fra-2 TG mice, vascular remodeling was already evident at eight weeks, before the development of pulmonary fibrosis, and accompanied by elevated IL-1α levels. IL-1 activated the JNK and p38 kinases in PASMCs and PFs, whereas NF-κB and ERK were activated in PFs and PASMCs, respectively. IL-1 stimulation downregulated collagen 1 and α-Sma, but increased the expression of IL-6 and tenascin C (TNC) in both cell types, without affecting their proliferation. Similarly, in vivo elevated IL-1α in Fra-2 TG mice was followed by increased IL-6 and TNC at 16 and 24 weeks on mRNA and protein levels concomitant with enhanced phosphorylation of STAT3. Circulating TNC levels were increased in patients with manifested lung fibrosis (mean ± SD = 61.3 ± 38.8 ng/mL) compared to healthy controls (19.2 ± 6.9 ng/mL), patients with PH (27.6 ± 8.2 ng/mL), and SSc without PF (29.5 ± 17.9 ng/mL). In conclusion, increased Fra-2/IL-1 levels exert indirect pro-fibrotic effects on resident pulmonary cells through IL-6 and TNC. Furthermore, TNC serves as a potential marker to discriminate patients with and without PF.
p22phox-dependent NADPH oxidase in chronic obstructive pulmonary disease and pulmonary hypertension
Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria; Charité - Universitätsmedizin Berlin, Germany; Medical University Vienna, Austria; University of Minnesota, Minneapolis, MN, USA; Medical University of Graz, Austria
The presence of pulmonary hypertension (PH) has been associated with reduced survival among chronic obstructive pulmonary disease (COPD) patients. Alveolar hypoxia is considered to be a major contributor to the development of pulmonary arterial remodeling. The Nox family of NADPH oxidases is emerging as a key disease-related factor in various vascular diseases, but currently its role in hypoxia-induced pulmonary remodeling in COPD remains unclear. p22phox expression was evaluated in patients with severe COPD who underwent lung transplantation and was correlated with clinical parameters. p22phox-/- knockout mice were used to investigate HPV and the development of hypoxia-induced PH. In COPD patients, p22phox expression significantly correlates with mean pulmonary arterial pressure and oxygenation index and inversely with the diffusing capacity of the lung for carbon monoxide. Phase 2 of acute HPV is significantly impaired in p22phox-/- mice, whereas the pulmonary vasopressor response to other agents is unchanged. The pulmonary vascular resistance is unaffected by p22phox deficiency under normoxia, but significantly reduced under hypoxia. In the chronic hypoxic setting, lack of p22phox is associated with improved right ventricular function and decreased pulmonary vascular remodeling. These data delineate the pivotal role of NADPH oxidase in the pulmonary circulation under hypoxia, showing that a functional NADPH oxidase system is important under physiological and pathological conditions, such as COPD in the lung, and thus might have diagnostic and therapeutic value in the human disease.
A young patient with pulmonary arterial hypertension
Istanbul University, Turkey
Pulmonary hypertension (PH) is a serious disease that could lead to death. Due to its non-specific symptoms, it might be diagnosed after a delay of 2.5 years. Here, we report an early diagnosed young patient with PH. A 30-year-old woman admitted with progressive dyspnea for five years. TTE revealed high (70 mmHg) pulmonary artery systolic pressure (PASP) and no left heart pathology. After the evaluation, she had the diagnosis of idiopathic pulmonary arterial hypertension. In her physical examination, attenuated second heart sound was auscultated. ProBNP was high (260 ng/mL). Right heart catheterization (RHC) revealed mean pulmonary arterial pressure (mPAP) = 54 mmHg, pulmonary capillary wedge pressure = 10 mmHg. Six-minute walk distance (6MWD) was 320 m and she desaturated during the test. Bosentan was started. She improved with initial treatment (6MWD = 520 m) but she had progression at the 30th month (6MWD = 310 m, proBNP = 324 ng/mL, PASP = 110 mmHg). Inhaled ilioprost was added. At the 60th month, sildenafil was added due to progression of dyspnea. At the 72nd month, progression with bilateral pretibial edema in lower extremities was detected (functional class [FC] III, 6MWD = 280 m, proBNP = 382 ng/mL, mPAP = 75 mmHg). Intravenous epoprostenol infusion was considered due to clinical detoration. At the 90th month her FC was I and 6WMD increased to 500 m with i.v. epoprostenol 25 ng/kg/min. In conclusion, early diagnosis of PH improves the survival of patients. If there is no satisfying answer gained during monotherapy, it should be switched to combination therapy. Patients with progressive dyspnea under the treatment of combination therapy should start i.v. epoprostenol treatment without delay.
Intravenous epoprostenol in pulmonary hypertension: experience of a single center
Istanbul University, Turkey
Epoprostenol infusion in pulmonary hypertension (PH) is the only treatment which is shown to improve survival in advanced disease. The study was performed in Istanbul University Istanbul Medical Faculty Pulmonary Hypertension Center. Patients with PH who were treated with i.v. epoprostenol between June 2014 and October 2016 were evaluated. In total, 15 patients were evaluated (11 women, 4 men). Thirteen were in Group I (ten with idiopathic pulmonary hypertension [IPAH], one with Eisenmenger syndrome, two with scleroderma). Two patients had inoperable chronic thromboembolism pulmonary hypertension (CTEPH). Mean age was 55 years (age range = 31–79 years). At baseline, four of the patients were in functional class (FC) III and 11 were in FC IV.
Average systolic pulmonary arterial pressure (PAP) was 112 mmHg (range = 80–160 mmHg) (by TTE) and two patients had moderate to severe pericardial effusion. Average mean PAP was 51 mmHg, pulmonary capillary wedge pressure was 10 mmHg, pulmonary vascular resistance was 11.9 mmHg (by right heart catheterization). Seven patients could perform 6-min walk test and walk distance was 258 m. ProBNP was 8275 pg/mL. Mean treatment duration of i.v. epoprotenol was 6.5 month (range = 1–27 months). Seven patients died during the follow-up period. Three of them discontinued treatment due to catheter infection, thrombocytopenia, jaw pain). FC improved in most patients who continued treatment. Walk distance increased 62 m. FC improved from IV to II. During the two-year follow-up, FC and walk distance of patients with PH was improved with i.v. epoprestenol.
Pulmonary hypertension in interstitial lung disease: Ege University experience
Ege University Hospital, Izmir, Turkey; Dokuz Eylul University Hospital, Izmir, Turkey
The aim of this study was to assess the frequency and prognostic significance of pulmonary hypertension (PH) in our patients with interstitial lung disease (ILD). Patients with a diagnosis of ILD and who had been assessed for PH at Ege University Hospital between 2009 and 2016 were reviewed. The study population is 672 patients referred with ILD who were investigated for PH (260 men, 412 women). The mean age was 56 years (age range = 13–86 years). The diagnoses included a wide range of interstitial lung diseases. The mean follow-up from presentation was 50 months (range = 1–206), during which time 145 patients (22%) died. All patients had echocardiography. The mean systolic pulmonary arterial pressure (SPAP) value was 42 mmHg (range = 10–130 mmHg). By Cox regression analysis, the SPAP value is a strong predictor of survival (P < 0.001). On Kaplan–Meier survival analysis, mean survival from first presentation for those (n = 488) with an SPAP ≤ 35 mmHg on echo was 161 months (95% CI = 145–176), compared to 78 months (95% CI = 62–95) for those (n = 184) with an SPAP > 35 mmHg (P < 0.001). Eighty-three patients went on to right heart catheterization (RHC), of whom 52 were found to have PH. Receiver operating curve (ROC) analyses were performed to investigate possible predictors of PH on RHC. The areas under the curve were: pulmonary artery diameter = 0.728; pulmonary artery/aorta ratio = 0.676; pulmonary artery /vertebral body ratio = 0.680; SPAP > 35 mmHg = 0.702; TVR on ECHO = 0.687; brain naturetic peptide = 0.729; FVC%/DLCO% ratio = 0.663. In conclusion, assessment of pulmonary pressure is of prognostic value in the broad spectrum of patients with ILD. SPAP is a non-invasive method applicable to all patients and yields a statistically significant stratification of patients. Pulmonary artery diameter, BNP, and SPAP predict PH by RHC.
Clinicians need to be aware of risk of PH in ILD and to appropriately investigate. The development of PH has a significant impact on survival.
Iron deficiency in hypoxic pulmonary hypertension results in increased circulating erythropoietin which is associated with attenuated pulmonary hypertension
University of Colorado, Denver, CO, USA
Iron deficiency in pulmonary arterial hypertension (PAH) is prevalent and associated with worsened morbidity and mortality. However, in models of pulmonary hypertension (PH), there are conflicting data regarding whether iron deficiency is protective or pathogenic in PH. We have demonstrated a protective effect of iron deficiency in a murine model of PH and hypothesize that the protective effect is driven by increased renal expression of erythropoietin (EPO), which acts in a paracrine manner to attenuate PH. C57BL6 mice were exposed to three weeks of normoxia (Nx) or hypoxia (Hx) (10% FiO2, at Denver barometric pressure), in addition to either normal diet (ND) or iron deficient diet/environment (FeD). The animals then underwent terminal right ventricular catheterization. Whole lung and renal tissue, as well as serum, were utilized for analysis of hypoxia-inducible factor (HIF) transcripts/protein. Finally, intraperitoneally dosed soluble EPO- receptor (sEPO-R) and vehicle were given to animals exposed to hypoxia and iron-deficient conditions. Under hypoxic conditions, iron deficiency led to significant attenuation of RVSP (NxND = 19.11 mmHg, HxND = 32.6 mmHg, HxFeD = 25.82 mmHg, P < 0.05). In lung tissue, we saw the expected rise in HIF target gene transcript levels in hypoxia, but this was not altered by iron deficiency. However, we saw a significant rise in circulating EPO in HxFeD (serum, Nx = 211.3 ng, Hx = 536.0 ng, Hx + FeD = 2340 ng). With blockade of circulating erythropoietin utilizing sEPO-R, we saw restoration of the PH phenotype in HxFeD (HxFeD + vehicle = 27.03 mmHg, HxFeD + sEPO-R = 37.84 mmHg). We have demonstrated that iron deficiency exerts a protective effect in experimental PH. Iron deficiency with hypoxia increases renal HIF activity resulting in significant increases in EPO expression, which may exert protective pleiotropic effects on the pulmonary vasculature, resulting in attenuation of hypoxic rise in RVSP. Future work will delineate the mechanisms by which EPO-signaling modulates the pulmonary vasculature.
In experimental hypoxic PH, iron deficiency results in attenuated RVSP, which is restored with blockade of endogenous erythropoietin.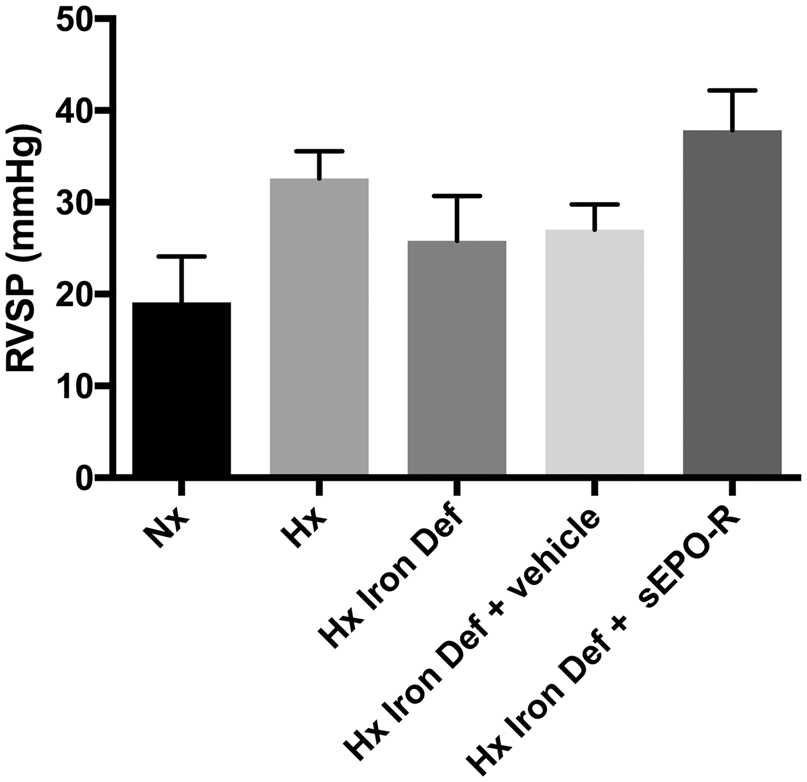
Comparing survival in pulmonary hypertension due to chronic lung disease with pulmonary arterial hypertension
University of Minnesota Medical School, Minneapolis, MN, USA
Pulmonary hypertension (PH) due to chronic lung disease (WHO Group 3) increases cost and is associated with worse survival when compared to patients with chronic lung disease without PH. However, the clinical characteristics of patients with WHO Group 3 PH are not well described. The aim of this study is to determine the clinical characteristics, hemodynamics, right ventricular (RV) function, and survival in patients with WHO Group 3 PH and to compare it to patients with pulmonary arterial hypertension (WHO Group 1). We studied all patients with WHO Group 3 PH (n = 98) and WHO Group 1 PAH (n = 162) referred to our center. We used the Kaplan–Meier method and Cox proportional hazard analysis to determine survival and independent predictors of mortality. WHO Group 3 patients were older and had a higher proportion of men than Group 1 patients. When assessing invasively measured hemodynamics, Group 3 patients had lower mean pulmonary arterial pressure (40 ± 10 vs. 45 ± 14, P = 0.002), lower pulmonary vascular resistance (7.5 ± 3.4 vs. 10.5 ± 6.4, P < 0.001), and higher pulmonary arterial compliance (1.7 ± 0.9 vs. 1.3 ± 0.7, P < 0.001) than Group 1 patients. However, Group 3 patients walked less on the 6-min walk test (237 ± 112 vs. 332 ± 140 m, P < 0.001) and had lower RV fractional area change (29 ± 10% vs. 33 ± 11%, P = 0.024) when compared to Group 1 patients. Finally, group 3 PH had a worse survival when compared to Group 1 PAH. Group 3 PH patients walked shorter distances, had more RV dysfunction, and worse survival despite having less severe pulmonary vascular disease when compared to Group 1 patients.
Right ventricle on fire: interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension
University of Minnesota Medical School, Minneapolis, MN, USA; Department of Medicine, Queen’s University, Kingston, ON, Canada; Scripps Hospital, San Diego, CA, USA
The objective was to determine if interleukin-6 (IL6) and right ventricular (RV) function are independently associated in pulmonary arterial hypertension (PAH). Elevated serum levels of IL6 identify PAH patients at increased risk of mortality, but the mechanism underlying this observation remains incompletely defined. Interestingly, pre-clinical and clinical data have revealed an association between IL6 and cardiac dysfunction, but the effect of IL6 on RV function, the major determinant of long-term outcomes in PAH, has not been examined. We performed a single-center study of 40 patients with PAH from an institutional registry and analyzed how RV function was related to IL6. IL6 had a negative logarithmic relationship with RV function as determined by echocardiography. There were no significant relationships between IL6 and mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), and only a weak association with pulmonary arterial compliance (PAC). When the cohort was divided by median IL6 level, patients with higher IL6 had significantly worse RV function on echocardiography, higher right atrial pressures, and reduced cardiac index and stroke volume despite having similar mPAP, PVR, and PAC. Finally, on multivariate analysis, IL6 was associated with RV function even after adjusting for PVR and PAC. IL6 is independently associated with RV function in PAH, and patients with higher IL6 levels had more severe RV dysfunction. These findings suggest IL6 may contribute to RV dysfunction and thus could explain the poor survival in PAH patients with elevated IL6.
Colchicine-induced microtubule depolymerization increases junctophilin-2 and improves right ventricular function in experimental pulmonary arterial hypertension
University of Minnesota Medical School, Minneapolis, MN, USA; Department of Medicine, Queen’s University, Kingston, ON, Canada; Scripps Hospital, San Diego, CA, USA
Pulmonary arterial hypertension (PAH) is a rare but lethal cardiovascular disorder characterized by pulmonary vascular remodeling leading to right ventricular (RV) dysfunction. Currently, PAH therapies primarily target the pulmonary vasculature despite the fact that RV function is the major determinant of clinical outcomes. Here, we investigated the hypothesis that colchicine, a microtubule depolymerizing agent, improves RV function by increasing junctophilin-2 by targeting a pathologically remodeled microtubule cytoskeleton.
Western blot, confocal microscopy, tissue analysis, echocardiography, cardiac catheterization, and treadmill exercise were used to examine how colchicine altered phenotypic severity of the monocrotaline (MCT) PAH rat model. One week after MCT injection, rats were treated with phosphate buffered saline or colchicine for three weeks. In the RV of MCT rats, there was increased microtubule density, reduced junctophilin-2 protein levels with loss of t-tubule localization pattern, and t-tubule disarray. Colchicine increased junctophilin-2 levels and improved localization patterns and partially corrected t-tubule morphology. Also, colchicine reduced RV hypertrophy and improved RV function. Colchicine also regressed pulmonary vascular disease (PVD), evident by a lower mean pulmonary arterial pressure, lower total pulmonary vascular resistance, and increased pulmonary artery (PA) acceleration time. Colchicine enhanced RV–PA coupling suggesting that the observed improvement in RV function was not solely due to regression of PVD. Finally, colchicine-treated rats had increased exercise capacity and improved survival. Colchicine increases junctophilin-2 expression and normalizes t-tubule architecture of RV cardiomyocytes in MCT rats leading to improved RV function and RV–PA coupling.
Improved platelet aggregation following administration of phosphodiesterase-5 inhibitors in patients with Eisenmenger syndrome
Pro-Sangue Foundation, São Paulo, Brazil; University of São Paulo School of Medicine, Brazil
Platelet aggregation (PA) has been shown to be decreased in pulmonary arterial hypertension (PAH) associated with Eisenmenger syndrome (PAH-ES). Platelets probably become “exhausted” following chronic endogenous activation, with decreased response when aggregation is analyzed in vitro.
We analyzed platelet aggregation before and during administration of phosphodiesterase-5 inhibitors (PDE-5is, sildenafil and tadalafil) in PAH-ES adults. The mean age of 23 naïve patients was age 25 years (age range = 18–40 years). Sildenafil (20 mg t.i.d., n = 11) or tadalafil (single daily dose of 40 mg, n = 12) were administered orally and laboratory data were obtained at baseline, three and six months of treatment. Whole blood ADP-induced PA was analyzed by the impedance method. At baseline, patients had decreased oxygen saturation (O2 sat. 87% [80–92%]), increased hematocrit (Htc 54% [49–61%]), and a platelet count of 210 (180–261) × 109 pl/L. Compared to normal, PA was 47% decreased and directly influenced by platelet count (R2 = 0.54, P < 0.001). Treatment with PDE-5is was followed by decrease in Htc (P < 0.001), although resting O2 sat., did not change. There was a mild decrease in platelet count (P = 0.093), particularly in the sildenafil group. Compared to baseline, PA improved (P = 0.048) with further increase when it was normalized by platelet count (P = 0.014). There were no differences between treatment groups. In PAH-ES patients, in vitro PA was improved by treatment with PDE-5is. We speculate that this might be due to improvement of chronic endogenous platelet activation.
Sirt1 activity is necessary for the protective effects of the female sex in the response to chronic hypoxia: a role for Sirt1 in estrogen signaling
Ottawa Hospital Research Institute, Ottawa, ON, Canada; University of Ottawa, Ottawa, ON, Canada
Sirtuin-1 (Sirt1) is an NAD+-dependent deacetylase that is strongly implicated in maintenance of endothelium homeostasis. We have shown that pulmonary hypertension (PH) in response to chronic hypoxia (CH) is exaggerated in Sirt1-deficient mice. Since the estrogen receptor has been identified as a target of Sirt1 deacetylation, we hypothesized that the protective effect of female sex in CH is dependent on Sirt1 activity. Male and female mice lacking Sirt1 catalytic activity (sirt1Y/Y) and their wild-type (WT) littermates were exposed to chronic hypoxia (CH; 10% O2) for 21 days. Right ventricular hypertrophy at three weeks of CH was significantly lower in female (RV/LV + S = 0.41 ± 0.01) compared to male WT mice (0.47 ± 0.03; n > 8/group, P < 0.05), whereas right ventricle systolic pressures (RVSP) were similar (29 ± 1 and 31 ± 2 mmHg, respectively). The sex difference in RV remodeling was lost in mutant mice deficient in Sirt1 activity, and both male and female sirt1Y/Y mice exhibited similar exaggerated increases in RVSP (39 ± 3 vs. 40 ± 2 mmHg, respectively) and RVH (0.54 ± 0.02 vs. 0.55 ± 0.02; n > 16/group) at three weeks. Interestingly, WT female mice also exhibited more modest increases in hematocrit levels in response to CH compared to males (59 ± 1 vs. 65 ± 1%, respectively; n > 13/group, P < 0.001), which was lost in sirt1Y/Y mice (71 ± 2 vs. 71 ± 1%, respectively; n >12/group). Lack of Sirt1 activity abolished the protective effect of female sex on RV remodeling and erythrocytosis in the CH model of PH, consistent with a role of Sirt1 in moderating the hypoxic response, most likely through estrogen signaling.
Role of female sex hormones in modulating the severe pulmonary arterial hypertension phenotype induced by VEGFR2 inhibition in a “hyper-responsive” colony of Sprague Dawley rats
Ottawa Hospital Research Institute, Ottawa, ON, Canada; University of Ottawa, Ottawa, ON, Canada
Our group has reported that a specific colony of Sprague Dawley (SD) rats exhibit hyper-responsive (HR) phenotype to VEGFR2 inhibitor (SU5416, SU) and develop severe pulmonary arterial hypertension (PAH) in response to single injection of SU, even in the absence of chronic hypoxia (CH). Interestingly, the HR phenotype showed strong sex dependence. Therefore, in this project we explored the role of female sex hormones in modifying the HR phenotype in the SD HR rats. Male and female rats were injected with SU (20 mg/kg, s.c.) or vehicle and right ventricular systolic pressure (RVSP) was measured after seven weeks. In response to SU alone, 72% of male rats exhibited HR phenotype, with mean RVSP of 97 ± 18 mmHg, compared to only 27% of the female rats. Oophorectomy (OVX) resulted in a marked increase in the HR in female rats to 71% compared to 33% in non-operated female rats. Moreover, estradiol replacement completely abrogated the HR phenotype in both male and OVX female rats. Conversely, progesterone treatment inhibited HR phenotype only in OVX female rats but not in the male SD rats. These data support a role of female sex hormones in modulating the development of severe PAH in response to SU-alone in a unique colony of SD rats that exhibit increased sensitivity to VEGFR2 blockade. SD HR rats exhibit exquisite sensitivity to estrogen and thus provide a unique opportunity to further define the complex interactions between hormonal and genetic modifiers in the development of PAH.
Identification of a novel BMP signaling regulator to attenuate pulmonary arterial hypertension
Peking Union Medical College, Beijing, China; University of Cambridge School of Clinical Medicine, Cambridge, UK; The Anne McLaren Laboratory for Regenerative Medicine, University of Cambridge, Cambridge, UK
Deficiency of bone morphogenetic protein type II receptor (BMPRII) signaling is causally implicated in the pathobiology of pulmonary arterial hypertension (PAH). Targeting of BMPRII signaling to improve the function of vascular endothelium could be desirable therapeutic intervention for PAH. Heterozygous germline mutations of the BMP type II receptor are responsible for the majority of cases of heritable PAH and 15–40% idiopathic PAH. In this study, we aim to clarify the key target of BMPRII in pulmonary vasculature and apply into the drug screening. We identified that the Id (Inhibitor of DNA binding) family of basic helix–loop–helix proteins is a major transcriptional target of BMP signaling in human pulmonary artery smooth muscle cells (PASMCs) and is directly affected by mutations or deficiency of BMPR-II (QPCR). We are now employing genomic recombineering to generate a human Id1/3 promoter derived dual reporter in human embryonic stem cells derived vascular cells, and set up a platform with it to screen small molecules library to identify novel compounds for enhancing Id gene transcription in relevant cell types as potential therapies for PAH. Also, we generated hESCs-R899X with CRISPR/Cas9 to demonstrate the effect of heritable PAH (HPAH) bearing mutation on endothelial differentiation and tube formation. We demonstrated that a BMP signaling upregulator effectively rescued the expression of Apelin in heterozygous autologous cells by fluorescent immunostaining. The combination of stem cell technology and cellular molecular mechanism of PAH enable the development of PAH targeted therapy.
Endothelial exosomes induced by aldosterone increase fibrillar collagen levels in pulmonary artery smooth muscle cells
Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Elevated levels of the hormone aldosterone (ALDO) are implicated in the pathobiology of vascular fibrosis in pulmonary arterial hypertension (PAH). We have demonstrated previously that human pulmonary artery endothelial cell (HPAEC) treatment with ALDO upregulates fibrosis proteins in co-cultured human pulmonary artery smooth muscle cells (HPASMCs) in vitro; however, the mechanism by which to account for this effect is not known. We used systems biology to predict the protein NEDD9 as a critical node regulated by ALDO that controls fibrosis signaling in silico. To validate this finding, cells were treated with vehicle (V) control or ALDO (10-7 mol/L) for 24 h. Compared to V-treated cells, ALDO increased NEDD9 significantly in HPAECs by 95% (P < 0.05), but not in HPASMCs (P = NS) by immunoblot. However, NEDD9 was increased by 62% (P < 0.05) in HPASMCs co-cultured with ALDO-treated HPAECs compared to ALDO-treated HPASMC monolayers. We hypothesized that ALDO induced cross-talk between cell types through a mechanism involving exosomes. To test this hypothesis, HPASMCs were incubated for 24 h with exosomes from ALDO-treated HPAECs (ALDO-Exosome). Compared to V-treated cells, ALDO-Exosome increased NEDD9-positivity by 46% (P < 0.001) and collagen III by 31% (P < 0.001), by immunofluorescence and immunoblotting, respectively. Compared to control HPAECs, NEDD9 positivity was also increased in HPAECS from PAH patients by 170% (P < 0.05), which correlated with collagen III levels (r = 0.77, P < 0.02). These data demonstrate that ALDO activates pro-fibrotic transcellular signaling between HPAECs-HPASMCs by a mechanism involving endothelial exosomes. Identifying NEDD9-collagen III signaling in the pathobiology of vascular fibrosis has important therapeutic implications for PAH patients.
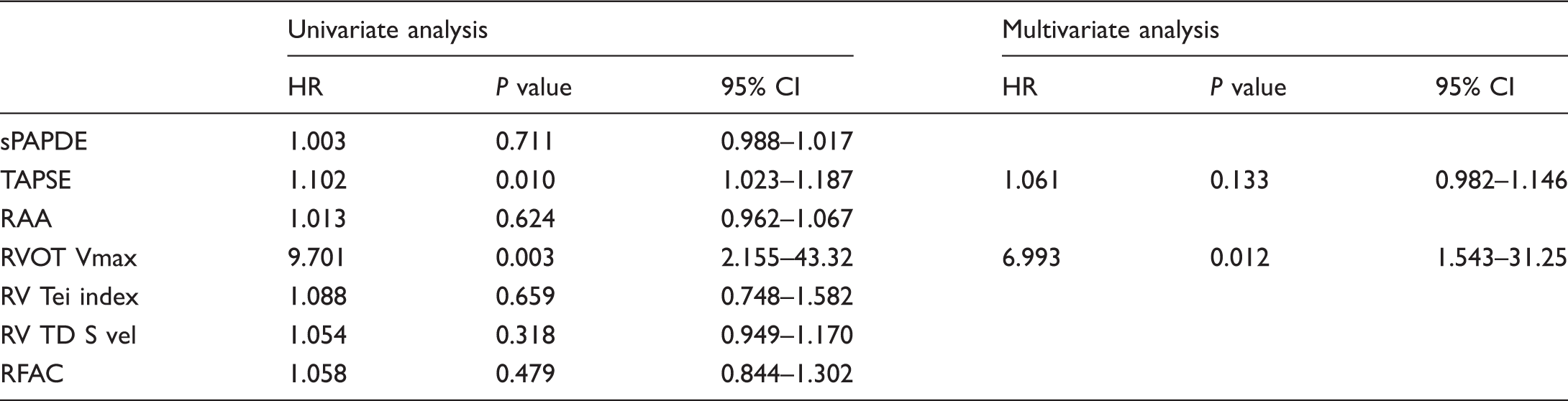
A simple and valuable echocardiographic parameter for predicting prognosis in pulmonary hypertension; RVOT maximal systolic velocity
School of Medicine and Faculty of Nursing, Dokuz Eylül University, Izmir, Turkey
Univariate and multivariate Cox regression analyses of echocardiographic parameters in predicting mortality in PAH patients.
Increased mitochondrial motility in pulmonary arterial smooth muscle cells in pulmonary arterial hypertension
Department of Medicine, Queen’s University, Kingston, ON, Canada
Pulmonary arterial hypertension (PAH) is a pathological state characterized by elevated pulmonary arterial pressure (PAP) and increased pulmonary vascular resistance (PVR). A key feature of PAH is hyperproliferation and apoptosis-resistance in pulmonary arterial smooth muscle cell (PASMC). This phenotype is related in part to mitochondrial abnormalities including increased fragmentation and a shift to aerobic glycolysis. However, we have noted altered mitochondrial motility in PAH as well. Higher mitochondrial motility is associated with increased cell proliferation in a previous study. Mitochondria from PAH and normal PASMC were imaged by loading with tetramethylrhodamine methyl ester perchlorate (TMRM), a mitochondrial-targeted, potentiometric dye. High-resolution Z-stack and time-lapse imaging were performed using a Leica 3D STED super resolution confocal microscope. Size (µm3), shape (sphericity), and position of every mitochondrion were tracked every 5 s for 5 min and measured with LAS-X software (Leica). Mean velocity of each mitochondrion was measured as total distance (nm)/total time (s) tracked by Imaris software (Bitplane). Expression levels of mitochondrial motility-related proteins (Miro, Milton, kinesin and dynein) in whole cell lysate were quantified by western blot analyses using ß-actin as internal control. Mitochondrial velocity is significantly increased in PAH PASMCs compared to normal PASMCs (61.1 ± 1.8 nm/s vs. 52.9 ± 1.7 nm/s, P = 0.0009). A higher percentage of mitochondria are moving at a high speed (>100 nm/s) in PAH PASMCs (28.5% in PAH vs. 20.4% in normal). There is no significant change in the expression levels of Miro, Milton, kinesin and dynein in PAH PASMCs. Mitochondrial are fragmented and their motility is significantly increased in PAH PASMCs. Mitochondrial motility-related proteins are largely unchanged in PAH PASMCs. We next intend to investigate functional changes in the calcium sensor in Miro and potential alteration in GTPase activity as a basis for dysregulated motility.
Role of altered mitochondrial metabolism in right ventricular fibroblasts in determining fibrosis in the right ventricle in pulmonary arterial hypertension
Department of Medicine, Queen’s University, Kingston, ON, Canada
In pulmonary arterial hypertension (PAH), fibrosis of the right ventricle (RV) is associated with reduced RV compliance. The role of RV fibrosis in PAH is under-appreciated. Fibroblasts from the RV of rats with monocrotaline (MCT)-induced PAH have increased mitochondrial fission and a metabolic shift to glycolysis that promote fibroblast proliferation and RV fibrosis. Sprague Dawley rats (n = 54) received a single injection of MCT or saline. Drinking water containing either no supplement (n = 34) or the pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (DCA; 0.75 g/L, n = 20) was started from day 7 post injection. At week 4, PAH was confirmed by echocardiography. Postmortem, the RV was stained for collagen using picrosirius red. Vimentin (+) RV fibroblasts were enzymatically isolated. Proliferation, metabolism, mitochondrial fragmentation, pyruvate dehydrogenase (PDH) activity, and the expression of Glut1, PDK, and procollagen were measured in cells cultured with or without DCA. Compared to control, MCT rats displayed more RV fibrosis. mRNA expression of Glut1 and PDK-1&3, and procollagen-III protein increased in MCT vs. control fibroblasts. MCT-derived fibroblasts displayed mitochondrial fragmentation and had significantly higher proliferation rates, lower oxygen consumption rate (OCR), and decreased PDH activity than control fibroblasts. DCA partially restored OCR and reduced collagen production in MCT-derived fibroblasts, and reduced RV fibrosis and improved RV function of MCT rats. In experimental PAH, RV fibroblasts manifest increased mitochondrial fragmentation and a shift to glycolysis that drives collagen production and fibrosis. These mitochondrial-metabolic changes are similar to those that occur in pulmonary artery smooth muscle cells and can be therapeutically targeted with DCA to decrease fibrosis and improve RV function.
The prognostic value of Qp/Qs ratio in pulmonary hypertension patients without left-to-right shunts
School of Medicine and Faculty of Nursing, Dokuz Eylül University, Izmir, Turkey
Cox regression analysis of hemodynamic parameters in prediction of death.
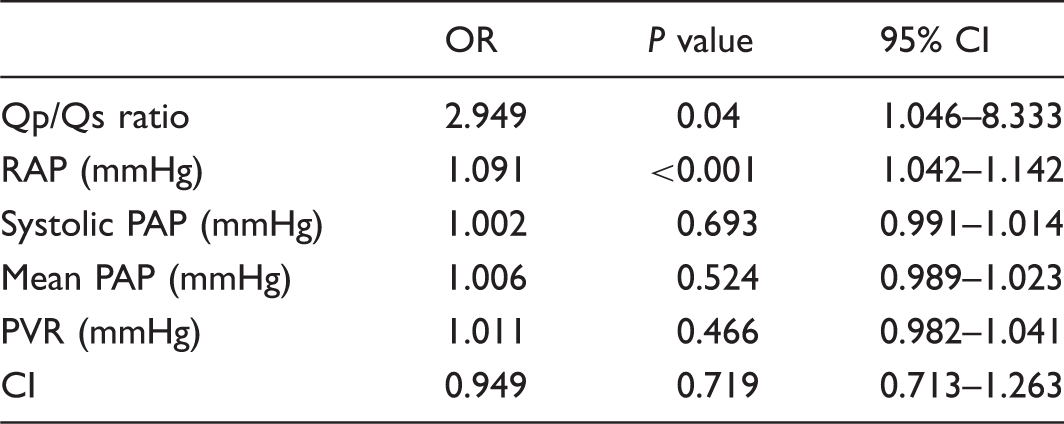
RAP, right atrial pressure; PAP, pulmonary artery pressure; PVR, pulmonary vascular resistance; CI, cardiac index.
Discriminant validity of the 6-min walk distance for muscle weakness in patients with pulmonary arterial hypertension
Dokuz Eylül University, Izmir, Turkey
The association between muscle strength and functional exercise capacity is well-known in pulmonary arterial hypertension (PAH) patients. Assessment of muscle strength and functional capacity are important for quality of life. However, in clinical practice, objective measurements of respiratory and peripheral muscle strength take time and require special equipment, unlike the 6-min walk test (6MWT). A distance more than 380 m on therapy is associated with improved survival in PAH. The aim was to determine the discriminant validity utilization of the 6-min walk distance (6MWD) for comparison of peripheral and respiratory muscle weakness in PAH patients. In total, 24 PAH patients, including 11 patients with >380 m on 6MWT and 13 with <380 m, were included. Mean pulmonary arterial pressure (mPAP) was measured by heart catheterization. Exercise capacity was assessed by the 6MWT. Respiratory muscle strength was assessed by maximal inspiratory and expiratory pressures (MIP-MEP). Peripheral muscle strength (knee extension, shoulder flexion, shoulder abduction, elbow flexion, and grip strength) was measured with hand-held and handgrip dynamometer. There were no significant differences in mPAP between the two groups (P > 0.05). The patients with <380 m on the 6MWD had significantly less MIP (P = 0.002), shoulder flexion strength (P = 0.016), shoulder abduction strength (P = 0.019), elbow flexion strength (P = 0.004), grip strength (P < 0.001), and knee strength (P = 0.037) than the other group. The results of this study suggest that the 6MWD can discriminate the patients with PAH with impaired peripheral and respiratory muscle strength.
Role of metabolic syndrome in pulmonary hypertension associated with left heart disease
Department of Medicine, Laval University, Quebec City, QC, Canada
Heart failure with preserved ejection fraction (HFpEF) affects over 50% patients with heart failure. Over 50% of HFpEF patients develop pulmonary hypertension (WHO2-PH) with pulmonary vascular remodeling which leads to worse prognosis. Interestingly, 94% of patients with PH due to heart diseases and 34.3% of patients with idiopathic pulmonary arterial hypertension (PAH) display at least two characteristics of metabolic syndrome (MS) suggesting its implication in pathogenesis. Our hypothesis is: MS and associated adipose tissue inflammation may act as a trigger in clinical complication of HFpEF into PH by exacerbating aberrant pulmonary vascular remodeling and right and left ventricles dysfunction. We developed a new model of WHO2-PH due to HFpEF and MS using Wistar rats. HFpEF is induced by supra-coronary aortic banding (SAB). Chronic MS is induced by high fat diet (HF) and/or olanzapine (4 mg/kg/2 days) for nine weeks. Only SAB rats with HF + olanzapine develop PH (≤25 mmHg). Increased visceral and mediastinal fat accumulation (CT scan) is associated with increased plasmatic level of leptin and increased pulmonary levels of leptin, leptin receptor, p-STAT3, IL-6 and CD68, and down-expression of PPARdelta which strongly correlates with PH severity. Olanzapine exposure on preadipocytes in vitro leads to obese adipocyte differentiation and leptin over-expression. Leptin exposure on macrophages leads to PPARdelta downregulation and M1 inflammatory response. Leptin-exposed macrophages supernatants increased pulmonary smooth muscle cells proliferation which is implicated aberrant pulmonary vascular remodeling. HFpEF is not sufficient to induce PH. Chronic MS lead to adipose tissue inflammation which worsen PH development in case of HFpEF.
Is hypoxemia at high altitudes in young infants related to neurologic respiratory control changes or secondary to an increase in the vascular pulmonary reactivity?
Universidad Nacional de Colombia, Clínica De La Mujer, Fundacion HOMI, Fundación CardioInfantil, Universidad Del Bosque, Bogotá, Colombia
Our research has shown that at 2640 m as well as at 3200 m of altitude above sea level, there is a significant gap in the oxygen saturation (SpO2) between 25% of infants (aged 1–4 months) in comparison with the remaining 75%. To explain this fact, two hypotheses can be raised: (1) 25% of infants have a higher Apnea/Hipopnea/Periodic Breathing index associated with high altitude, which induces hypoxemia; and (2) 25% of infants have higher pulmonary vascular reactivity (VPR) induced by the hypobaric hypoxia in comparison with the remaining 75%. Our research found that the central apnea index does not change as altitude increases, and that lower saturations are not associated with the periodic breathing characteristic of high altitude. In conclusion, lower SpO2 is not secondary to neurological respiratory control alterations related with high altitude. This means that the right hypothesis is probably the one related to an increase in VPR; in consequence, pulmonary vasoconstriction would come first and hypoxemia afterwards. Our main conclusions are: at given conditions – young infants at high altitudes – up to 25% of healthy individuals have significant lower SpO2 values in comparison with the remaining 75%. This fact is not explained by respiratory control alterations related with high altitude. The explanation is probably related to an increase in VPR when exposed to hypobaric hypoxia.
Inhibiting PINK1-and PKA-mediated phosphorylation of mitofusin 2 prevents proteasomal degradation: a new mitofusin 2 construct displays enhanced efficacy in hyperproliferative diseases
Department of Medicine, Queen’s University, Kingston, ON, Canada
Downregulation of the mitochondrial fusion mediator mitofusin 2 (Mfn2) contributes to unrestricted cell proliferation and apoptosis resistance in pulmonary arterial hypertension (PAH) and lung cancer. Our hypothesis was that the mechanism of depressed Mfn2 levels in PAH and lung cancer involves phosphorylation of Mfn2 by PTEN-induced putative kinase 1 (PINK1) and protein kinase A (PKA). We compared wild-type Mfn2 (Mfn2-WT) to mutant Mfn2 constructs that were rendered phosphorylation-deficient (Mfn2-PD) by S-442-A substitution, or constitutively phosphorylated, by a S-442-D substitution (Mfn2-CP). Constructs were delivered in PAH pulmonary artery smooth muscle cells (PASMC) and A549 lung cancer cells by adenoviral vector and their effects on cell proliferation, apoptosis, mitochondrial fusion, and tumor regression were studied. Mitochondria in PAH and lung cancer cells were fragmented and Mfn2 levels were depressed. Mfn2-WT increased Mfn2 expression, decreased proliferation, and increased apoptosis and mitochondrial fusion. Compared to Mfn2-WT, Mfn2-PD infection caused greater Mfn2 expression, suppression of proliferation, and apoptosis induction. Conversely, Mfn2-CP failed to increase expression of Mfn2 or restore mitochondrial fusion and failed to inhibit cell proliferation or induce apoptosis. Pharmacological inhibition of PKA or proteasome blockade increased Mfn2 expression and enhanced mitochondrial fusion. Silencing PINK1 and PKA C-α increased Mfn2 expression, inhibited cell proliferation, induced apoptosis, and increased mitochondrial fusion. Mfn2-PD gene therapy caused greater regression of tumor growth than Mfn2-WT in a xenotransplantation model of lung cancer. PINK1 and PKA induced phosphorylation of Mfn2 triggers proteasomal degradation of this fusion mediator. Rendering Mfn2 resistant to phosphorylation at serine-442 enhances its antiproliferative, proapoptotic effects. This strategy may have therapeutic implications against PAH and lung cancer.
Development of new pre-clinical model of pulmonary hypertension associated with left heart disease
Department of Medicine, Laval University, Quebec City, QC, Canada
Heart failure (HF) with preserved ejection fraction (HFpEF) affects more than 50% of all patients with HF. Fifty percent of HFpEF culminates to pulmonary hypertension (PH), which confers poor prognosis. Furthermore, metabolic syndrome (MS), which affects more than 34% of adults worldwide, is strongly associated with PH (34.3%) and HFpEF (prevalence of 35%). Taken together, these results suggest that MS and HFpEF, which are two major healthcare challenges in western society, may predispose to PH. Unfortunately studies supporting this hypothesis remain limited by the lack of good pre-clinical models that preclude extensive investigation. We developed a new animal model for WHO2 PH associating HFpEF and MS. Using supra-coronary-aortic banding, we induced HFpEF in wistar rats characterized by left ventricle (LV) hypertrophy, rise in LVSP, LVEDP, E/E’ ratio, and no significant modification of LV ejection fraction, ten weeks post surgery. We then induced MS with high fat diet and/or daily injection of olanzapine (4 mg/kg/2 days) for nine weeks. Establishment of MS was confirmed by rise of hepatic triglyceride, increased visceral fat, and high blood pressure. Compared to healthy and sham surgery, only HFpEF-MS rats displayed PH characterized by significant increase of PAP, RVSP, PAAT, and vascular remodeling of distal pulmonary arteries. Interestingly, both mediastinal and visceral fat displayed strong correlation with PH severity (RVSP) suggesting that fat accumulation exacerbates PH associated with HFpEF. In the present study, we: (1) developed a new model for WHO2 PH associating HFpEF and SM; and (2) demonstrated that MS worsens PH associated with HFpEF.
Paradoxical impact of cholesterol on lipid packing and cell stiffness
Department of Medicine, University of Illinois at Chicago, Chicago, IL, USA
Cell stiffness or deformability is a fundamental property that is expected to play a major role in multiple cellular functions. It is well-known that cell stiffness is dominated by the intracellular cytoskeleton that together with the plasma membrane forms a membrane–cytoskeleton envelope. However, our understanding of how lipid composition of plasma membrane regulates physical properties of the underlying cytoskeleton is only starting to emerge. Cholesterol is one of the major lipid components of the plasma membrane in all mammalian cells and its impact on the physical properties of the membrane lipid bilayer is well studied, both in pure lipid environments, such as liposomes or lipid monolayers, and in cellular membranes. Our studies demonstrate, however, that an increase in the rigidity and lipid packing of the bilayer does not translate into an increase in the overall stiffness of the cellular envelope, a bi-component system where the sub-membrane cytoskeleton underlies the membrane lipid bilayer. In contrast, it appears that there is an inverse relationship between the lipid order of the membrane bilayer and the stiffness of the cellular envelope that is dominated by the cortical cytoskeleton. First, we showed that cholesterol depletion results in significant stiffening and loss of deformability of vascular endothelial cells by measuring progressive membrane deformation in response to negative pressure applied by a glass micropipette. These data were verified by measuring endothelial elastic modulus using Atomic Force Microscopy. Furthermore, using a combination of experimental and computational techniques, we show that incorporation of oxysterols and other oxidized lipids into endothelial membranes that disrupt lipid packing result significant increase in endothelial elastic modulus and increase in endothelial force generation. In contrast, loading the cells with cholesterol rescue all the effects described above.
Characterizing the progression of PAH in the left and right pulmonary arteries of a PAH rat animal model
Bioengineering Department at the University of Illinois at Chicago, Chicago, IL, USA
Pulmonary arterial hypertension (PAH) is an aggressive disease with a 48% three-year average survival. To study PAH, Sprague Dawley rats were followed longitudinally for up to four weeks post monocrotaline injections. During open chest surgeries, blood pressure and flow from the pulmonary artery were measured simultaneously. Post hemodynamic studies, the right and left pulmonary arteries (RPA and LPA, respectively) were harvested for mechanical testing and structural investigation. Mean pressure rose from 24.1 ± 5.0 to 44.4 ± 6.3 mmHg while flow overall remained constant during the disease progression. Using a three-element Windkessel model, distal resistance was found to increase exponentially while compliance decreased linearly as a function of the disease, indicating a different remodeling process according to location. From mechanical testing stiffness increase in the circumferential direction was found in advanced PAH, but it was only statistically significant in the RPA (Young’s modulus went from 3.4 ± 3.1 to 22.9 ± 12.7 kilo mmHg). Dampening properties of the LPA vessels decreased continuously as the disease progressed, while in the RPA the relaxation time initially increased and then decreased showing a different adaptive response. Structure of the LPA and RPA changed in the advanced stage of PAH. In both vessels, there was over a 10% increase in collagen content assessed by Masson’s Trichrome stained histology slides. Through multiphoton microscopy imaging analysis, collagen fibers were found to go from being crimped and randomly organized in the normotensive state to uncrimped and highly aligned towards the axial (0°) direction in hypertension. While the distribution of collagen fibers of both vessel types were aligned towards 14° (90° and –90° correspond to the circumferential direction), they had a second distribution aligned at –28° for the RPA and –8° for the LPA. This indicates that there is a second direction of preference in the RPA, while in the LPA it is mainly just one family of fibers.
Real World Analysis of Riociguat Data (REWARD) for CTEPH from a single center
Division of Pulmonary Disease and Critical Care Medicine, University of California, San Francisco Fresno, CA, USA
Baseline characteristics, demographics, hemodynamics, and outcomes.
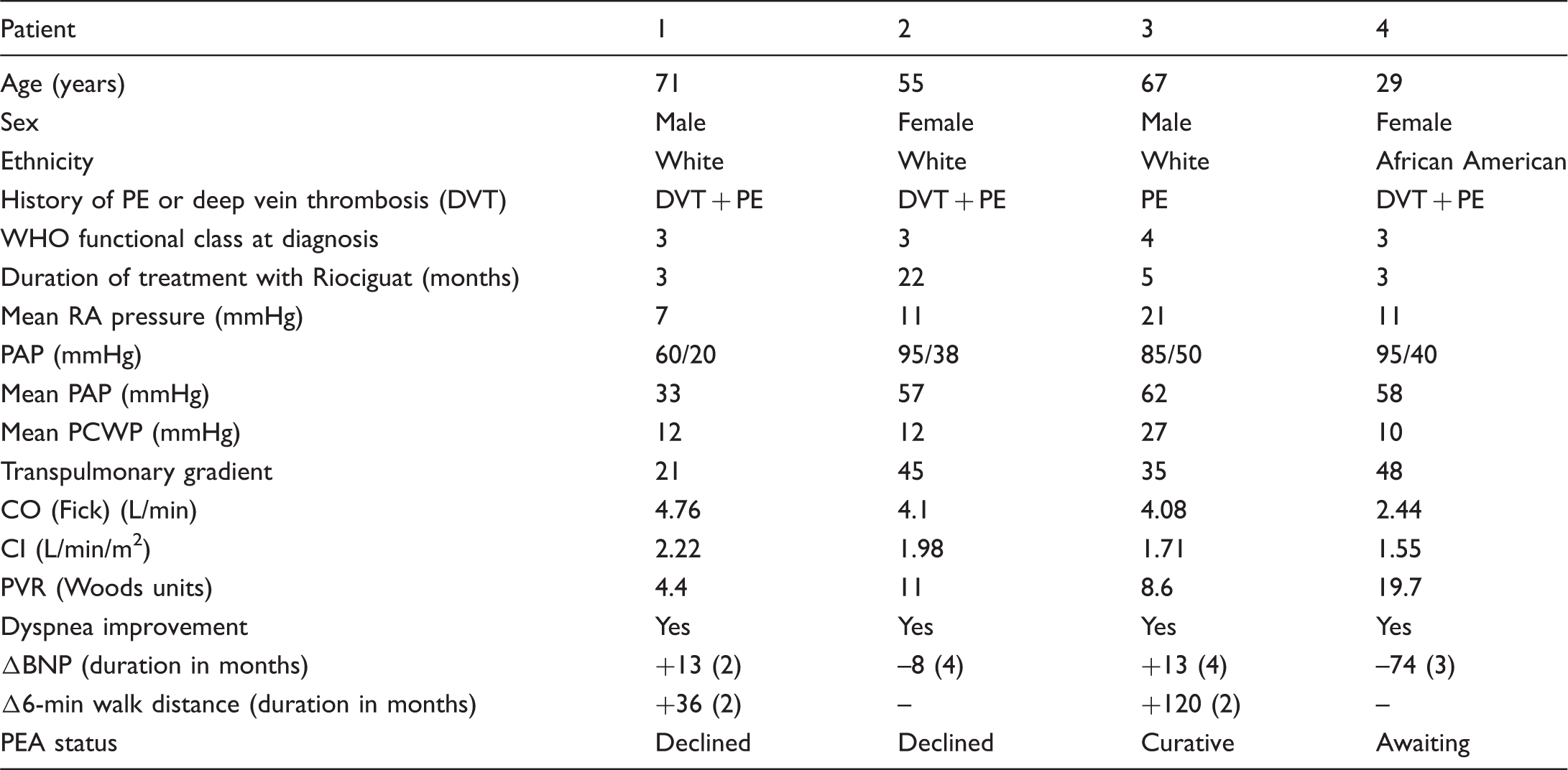
Riociguat appeared to be effective in improving symptoms (SOB) in patients with CTEPH after as early as two months and was also well tolerated. Therefore, it may be considered as bridging therapy as well while a patient is awaiting surgery/surgical assessment.
HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension
Department of Medicine, Laval University, Quebec City, QC, Canada
Pulmonary arterial hypertension (PAH) is a vascular remodeling disease of complex etiology. Despite environmental stressful conditions, pulmonary artery smooth muscle cells (PASMCs) and endothelial cells (PAECs) exhibit a pro-proliferative and anti-apoptotic phenotype. HDAC6 is a cytoplasmic histone deacetylase overexpressed in response to stress and implicated in the regulation of multiple pro-survival mechanisms in cancer cells. Therefore, we hypothesized that HDAC6 expression is increased in PAH-PASMCs and PAH-PAECs allowing them to survive and proliferate, thus contributing to vascular remodeling in PAH. Using a multidisciplinary and translational approach, we aimed to demonstrate that HDAC6 inhibition is a promising strategy to improve PAH. HDAC6 is significantly upregulated (immunoblot) in lungs, distal PAs, and isolated PASMCs and PAECs from ten PAH patients and five experimental PAH animals (five Sugen/hypoxia rats, five monocrotaline rats, and five chronic hypoxic mice) compared to controls. Molecular (siRNA) and pharmacological inhibition (Tubastatin and ACY-775) of HDAC6 reduces dose-dependent PAH-PASMC/PAEC proliferation (Ki67 assay) and resistance to apoptosis (Annexin V assay) in vitro sparing control cells. Mechanistically, we demonstrated that HDAC6 deacetylates Ku70, blocking the translocation of Bax to mitochondria and preventing apoptosis. In vivo inhibition of HDAC6 (Tubastatin A 25 mg/kg/day for 2 weeks) significantly improved (n = 5 to 10 P < 0.05) established PAH by decreasing mean PA pressure and total pulmonary resistance, and increasing cardiac output in two experimental models (Sugen/hypoxia and monocrotaline). In addition, we showed that HDAC6 inhibitor can be safely given in combination with currently approved PAH therapies (macitentan and tadalafil). Finally, Hdac6 knockout mice have significantly lower right ventricle systolic pressure in response to three weeks of chronic-hypoxia compared to wild-type mice. We showed for the first time that HDAC6 is implicated in PAH development and represents a new promising therapeutic target to improve PAH.
Selective accumulation of Hsp90 in mitochondria as an adaptive strategy to face stress in pulmonary arterial hypertension
Department of Medicine, Laval University, Quebec City, QC, Canada
Pulmonary arterial hypertension (PAH) is a vascular remodeling disease with a poor prognosis and no therapeutic option. Although the causal pathomechanisms contributing to remodeling of the pulmonary vascular bed in PAH are still unclear, several features, including hyperproliferation and resistance to apoptosis of pulmonary smooth muscle cells (PASMCs) sustained by oncogenic pathways activation and metabolic alterations, have led to the emergence of the cancer-like concept. The molecular chaperone heat shock protein 90 (Hsp90), by interacting with its client proteins, is directly associated with malignant growth and proliferation. In addition to being highly expressed in the cytosol, Hsp90 exists in a subcellular pool compartmentalized in the mitochondria (mtHsp90) of tumor cells, but not in normal cells, where it promotes cell survival. We hypothesized that Hsp90 upregulation in PAH triggers PASMC proliferation and resistance to apoptosis. We showed that Hsp90 is upregulated in lungs and PASMCs isolated from distal pulmonary arteries from PAH patients compared to control donors. Using pharmacological inhibitors, we demonstrated that cytosolic Hsp90 stabilizes the expression of numerous clients’ proteins overexpressed in PAH that promote cell growth and survival. More importantly, we demonstrated that Hsp90 is specifically expressed in PAH-PASMC mitochondria (immunoblot, dual immunofluorescence, and immunogold electronic microscopy) and not in healthy cells. Whereas cytosolic Hsp90 inhibition displays a lack of absolute specificity for PAH-PASMCs, selective inhibition of mtHsp90 activity using Gamitrinib decreased PAH-PASMC proliferation (Ki67 labeling) and resistance to apoptosis (Annexin V assay) without affecting control cells. In PAH-PASMCs, mtHSP90 accumulation prevents the accumulation of mitochondrial DNA damage and maintains bioenergetics functions (Seahorse). In the fawn-hooded rat and monocrotaline- induced models of PAH, mtHsp90 inhibition reduces PA remodeling thus improving pulmonary hemodynamic parameters. We demonstrated that accumulation of mtHsp90 is a cardinal feature of PAH-PASMCs, contributing to the development of vascular lesions.
Upregulation of FOXM1 in PAH promotes DNA repair and cell survival
Department of Medicine, Laval University, Quebec City, QC, Canada
Pulmonary arterial hypertension (PAH) is a disease of the pulmonary vasculature, defined by an elevated pulmonary vascular resistance (PVR), leading to right heart failure and premature death. PAH is characterized by enhanced pulmonary artery smooth muscle (PASMC) proliferation and suppressed apoptosis within the pulmonary artery (PA) wall. Targeting the cancer-like of PASMCs represents an attractive therapeutic avenue to tackle PAH. Overexpression of the transcription factor FOXM1 has been shown to be a key driver of cancer progression through the stimulation of DNA repair, cell proliferation, and cell survival. Using a multidisciplinary and translational approach, we aimed to demonstrate that upregulation of FOXM1 in PAH-PASMCs triggers proliferation and resistance to apoptosis. FOXM1 is significantly increased (immunoblot and immunofluorescence) in lungs, distal PAs, and isolated PASMCs from PAH patients as well as in the monocrotaline (MCT)-induced PAH model compared to controls. We demonstrated that, in response to growth factors like PDGF and IGF1, Akt is activated (increased P-Akt), phosphorylates FOXO3a and inhibits it, allowing the upregulation of FOXM1. Moreover, we found that inhibition of FOXM1 in PAH-PASMCs increases DNA damage (γH2AX) and reduces expression of NBS1 and PLK1 (immunoblot), two factors upregulated in PAH-PASMCs (immunoblot and immunofluorescence) and involved in DNA repair and cell cycle progression. Consistently, pharmacological inhibition of FOXM1 using Thiostrepton dose-dependently reduces PAH-PASMC proliferation (Ki67 labeling) and resistance to apoptosis (Annexin V assay) in vitro. Inhibition of FOXM1 with Thiostrepton improves established PAH (right heart catheterization) in the MCT model. We showed for the first time that FOXM1 is overexpressed in human PAH and implicated in the pro-proliferative and anti-apoptotic phenotype of PAH-PASMCs.
NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension
Universities of Giessen and Marburg Lung Center, Giessen, Germany; Institute for Cardiovascular Physiology, Goethe University Frankfurt, Germany
Chronic exposure to hypoxia induces a pronounced remodeling process of the pulmonary vasculature, leading to pulmonary hypertension (PH). In this regard, reactive oxygen species (ROS), produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox), are discussed as triggers for pulmonary vascular remodeling. Recently, we identified Nox4 to be a source of ROS involved in proliferation of pulmonary arterial smooth muscle cells (PASMC). In addition, we observed elevated Nox4 expression in mice suffering from chronic hypoxia-induced PH, in isolated human and rat PASMC in hypoxia, and in lungs of patients suffering from idiopathic pulmonary arterial hypertension (PAH). Here, we aim to investigate the role of Nox4 in PH in vivo, using constitutive and inducible Nox4 knockout mice. Hypoxia-induced pathology was analyzed in isolated perfused lungs, by measuring hemodynamic values and in histological sections vascular muscularization was quantified. The degree of PH was assessed by right ventricular systolic pressure measurements and analysis of right heart. Neither constitutive nor inducible Nox4 knockout mice exhibit any differences in responses to acute or chronic hypoxia when compared to wild-type mice. Elevated Nox4 levels in PH and its role in PASMC proliferation suggest Nox4 to be causal for the development of PH. Therefore, Nox4 appeared to be an interesting target to treat PH. Our current study proves this assumption wrong. Absence and probably also inhibition is not sufficient to prevent hypoxia-induced PH pathogenesis in vivo.
Caveolin-1 loss/dysfunction may in part facilitate metabolic shift in pulmonary hypertension
New York Medical College, Valhalla, NY, USA
We have previously reported progressive loss of endothelial caveolin-1 (cav-1) and enhanced cav-1 expression in smooth muscle cells (SMC) in pulmonary hypertension (PH). Accelerating the PH progression by exposing monocrotaline (M)-treated rats to hypoxia, extensive endothelial damage, enhanced cav-1 expression in SMC, and neointima formation are observed for four weeks. Neointimal cells have normal eNOS and scant cav-1 expression, a set-up for oxidative and nitrosative stress. Cav-1 has been shown to regulate cell proliferation, oxidative stress, and metabolic shift. Recent studies indicate that the metabolic shift is an important aspect of PH. We hypothesized that cav-1 loss/dysfunction may have a role in the metabolic shift observed in PH. To test our hypothesis, we examined rats treated with M, and M + hypobaric hypoxia (M + H), and compared these with the controls (C). At four weeks, M + H gr. revealed significant PH compared with M gr. Cav-1 expression was significantly reduced in M gr; but closer to normal in the M + H gr. The expression of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), MnSOD, and Glut 1 was significantly increased in the M + H gr. Interestingly, in cancer, loss of cav-1 function leads to oxidative stress resulting in the activation of Nrf2 and translocation to nucleus where it activates MnSOD, leading to p53 inhibition and aerobic glycolysis, thus facilitating the survival of proliferating cells. In contrast, cav-1 links free radical pathways to the activation of p53, thus maintains anti-proliferative and apoptotic state. Furthermore, loss of cav-1 function results in increased Glut1 expression which can increase glucose uptake and facilitate aerobic glycolysis. Our preliminary data suggest that cav-1 loss/dysfunction may facilitate metabolic shift in PH.
Molecular imaging of the human pulmonary vascular endothelium in pulmonary hypertension: a phase II safety and proof of principle trial
Research Center of the Montreal Heart Institute; Départment de Médicine et de Radiologie de l’Université de Montréal, Montreal, QC, Canada; Lady Davis Institute and Jewish General Hospital, McGill University, Montreal, QC, Canada; Institut Universitaire de Cardiologie et Pneumologie de Québec, Quebec, QC, Canada; INRS-Institut Armand-Frappier, Laval, QC, Quebec; Montreal Health Innovation Coordination Center, Quebec, QC, Canada
The adrenomedullin receptor is densely expressed in the pulmonary vascular endothelium. PulmoBind, an adrenomedullin receptor ligand, was developed for molecular diagnosis of pulmonary vascular disease (PVD). We evaluated the safety of PulmoBind SPECT imaging and its capacity to detect PVD associated with pulmonary hypertension (PH) in a human phase II study. Thirty patients with pulmonary arterial hypertension (PAH; n = 23) or chronic thromboembolic PH (CTEPH; n = 7) in WHO functional class II (n = 26) or III (n = 4) were compared to 15 healthy controls. Lung SPECT was performed after injection of 15 mCi 99mTc-PulmoBind in supine position. Qualitative and semi-quantitative analyses of lung uptake were performed. Reproducibility of repeated testing was evaluated in controls after one month. PulmoBind injection was well tolerated without any serious adverse event. Imaging was markedly abnormal in PH with ∼50% of participants showing moderate to severe heterogeneity of moderate to severe extent. The abnormalities were unevenly distributed between the right and left lungs as well as within each lung. Segmental defects compatible with pulmonary embolism were present in 7/7 patients with CTEPH and in 2/23 patients with PAH. There were no segmental defects in controls. The PulmoBind activity distribution index, a parameter indicative of heterogeneity, was elevated in PH (65% ± 28%) vs. controls (41% ± 13%; P = 0.0003). In the only participant with vasodilator-responsive idiopathic PAH, PulmoBind lung SPECT was completely normal. Repeat testing one month later in healthy controls was well tolerated and showed no significant variability of PulmoBind distribution. In this phase II study, molecular SPECT imaging of the pulmonary vascular endothelium using 99mTc-PulmoBind was safe and reproducible. PulmoBind showed potential to detect both pulmonary embolism and abnormalities indicative of PVD in PAH. Phase III studies with this novel agent are justified.
Evaluation of pulmonary perfusion using an endothelial cell tracer in supine humans and dogs
Research Center, Montreal Heart Institute; Departments of Medicine Nuclear Medicine, Université de Montréal, Montréal, Québec, QC, Canada; INRS-Institut Armand Frappier, Laval, Québec, QC, Canada
The objective is to evaluate human pulmonary perfusion using a novel endothelial cell tracer. Pulmonary perfusion is not homogeneously distributed and its variations could be of diagnostic value. PulmoBind is a ligand of the adrenomedullin receptor expressed in endothelial cells of lung capillaries. The spatial distribution of human lung perfusion has never been evaluated using a molecular tracer. Normal humans (n = 19) enrolled into the PulmoBind phase I trial were studied (Clinicaltrials.gov NCT01539889). They were injected with 99mTc-PulmoBind for SPECT imaging. Results were compared with 99mTc-PulmoBind in quadruped mammals (dogs, n = 5). Imaging was performed in the supine position and activity was determined as a function of cumulative voxels along different planes. PulmoBind uptake in humans was 58% ± 1% (mean ± SEM) of the injected dose. Dorsal activity was 18.1% ± 2.1% greater than ventral and caudal activity was 25.7% ± 1.6% greater than cranial. Lateral activity was only mildly higher than medial by 7.0% ± 1.0%. In supine dogs, similar but higher PulmoBind gradients were present: dorsal 28.6% ± 2.5%, caudal 34.1% ± 5.0%, and lateral 18.1% ± 2.0%. The perfused pulmonary circulation of supine humans, assessed by an adrenomedullin receptor ligand, is not homogeneously distributed with more prominent distribution in dorsal and caudal regions. It is qualitatively similar to a supine quadruped mammal confirming the presence of a microcirculatory gravitational perfusion gradient detectable with this tracer. Future studies are needed to determine if this novel endothelial cell tracer could be used to detect physiologic and pathologic variations of lung perfusion.
The transpulmonary ratio of endothelin 1 is increased in patients with combined pre- and post-capillary pulmonary hypertension
Divisions of Cardiology and Allergy, Pulmonary, and Critical Care Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
Pulmonary hypertension complicating left heart disease (PH-LHD) is associated with increased morbidity and mortality, especially in patients who develop combined pre- and post-capillary PH (Cpc-PH). Molecular mechanisms underlying PH-LHD are incompletely understood, particularly for individuals with preserved left ventricular ejection fraction (LVEF). We hypothesized that transpulmonary concentrations of biomarkers representing signaling pathways with known effects in the pulmonary circulation could provide insight into the molecular etiology of PH-LHD. Blood samples were collected from the pulmonary artery (PA) and wedge positions of outpatients with normal LVEF referred for right heart catheterization. Hemodynamic tracings were manually reviewed to classify patients as no PH (mean PA pressure [mPAP] <25 mmHg, n = 22) or PH-LHD (mPAP ≥ 25 mmHg and wedge pressure >15 mmHg, n = 20); patients with PH-LHD were further divided into Cpc-PH (diastolic pressure gradient ≥7 mmHg or pulmonary vascular resistance >3 Wood units) and isolated pre-capillary PH (Ipc-PH). A biomarker’s transpulmonary ratio (TPR) was calculated as the quotient of wedge and PA concentrations. The TPR of endothelin 1 (ET-1) was elevated in Cpc-PH (n = 7) compared to Ipc- PH (n = 13) or no PH (Fig. 1). The TPRs of cAMP and cGMP were not different among groups. The difference in ET-1 TPR was due to higher wedge ET-1 in Cpc-PH (3.3 ± 0.8 pg/mL) compared to No PH (1.6 ± 0.9 pg/mL, P < 0.001) or Ipc-PH (1.9 ± 0.8 pg/mL, P < 0.05). Pulmonary vascular resistance (PVR) strongly correlated with wedge ET-1 exclusively in Cpc-PH patients (r = 0.79, P < 0.05).
The TPR of endothelin 1 (ET-1) was elevated in Cpc-PH (n = 7) compared to Ipc- PH (n = 13) or no PH.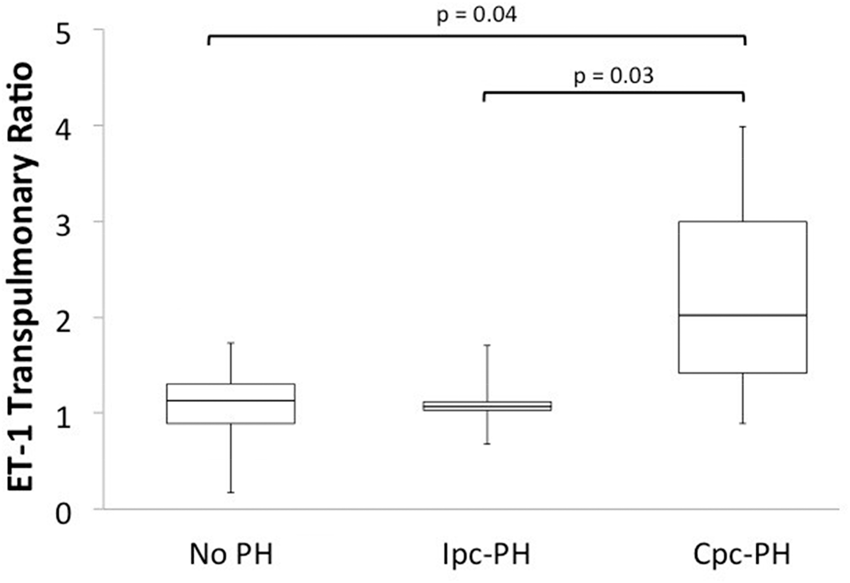
In patients with preserved LVEF, ET-1 TPR is higher in those with Cpc-PH compared to those without PH or with Ipc-PH. The correlation between PVR and wedge ET-1, observed only in the Cpc-PH group, also suggests increased pulmonary vascular responsiveness to ET-1 in these patients. These findings implicate elevated pulmonary ET-1 as a marker of, and potential contributor to, development of Cpc-PH in this population.
Lipopolysaccharides induce survival in lung cell lines: the role of protein-kinases activation
Department of Internal Medicine, University of Giessen, Giessen, Germany; Clinic for Radiotherapy and Radio-oncology, Philipps-University Marburg, Marburg, Germany
Pulmonary infections are common complications in patients with lung diseases and worsen prognosis. These patients often show a worse approach to treatments. Common pathogens found in patients are gram-negative bacteria. Their virulence is caused by lipopolysaccharides (LPS). LPS is known to activate multiple pathways in pulmonary epithelial cells. This could induce an increased survival and proliferation in human lung cell lines. Radiation was used to kill cells. Colony formation assays were performed to show an increased proliferation and survival after LPS treatment and radiation. Cells were treated with different doses of LPS (0, 0,1, 1, and 10 µg/mL) and exposed to ionizing radiation (0, 1, 2, 4, 6, and 8 Gy). A defined number of treated cells were platted on dishes and after ten days the colonies were counted. The plating efficiency and the survival rate were calculated. In parallel, proteins were isolated and proteome arrays were performed. With this proteome array potential target proteins were found. Upregulated target proteins were inhibited in LPS-treated cells before irradiation.
Ionizing radiation induced a reduction in survival. LPS promotes an increased survival in the lung cell line. This effect was dose dependent and most pronounced when 10 µg/mL LPS was used. The Proteome Array shows an upregulation of the cAMP response element-binding protein (CREB) and epidermal growth factor receptor (EGFR) after LPS treatment and radiation. Also, the Src family members Fyn, Lyn, and Fgr were upregulated after LPS treatment and radiation. After CREB binding protein (CBP) or EGFR inhibition the LPS-induced survival was decreased.
The LPS treatment leads to an increased survival and proliferation after radiation. An inhibition of possible target proteins like CREB or EGFR may serve as a potential treatment to overcome this increased survival and proliferation after pulmonary infections.
Parallels between endothelin-1 receptor antagonists in pulmonary arterial hypertension and fetal development
VU University Medical Center/Institute for Cardiovascular Research, Amsterdam, The Netherlands; Erasmus MC – Sophia Children Hospital, Rotterdam, The Netherlands; Charles River, ‘s-Hertogenbosch, The Netherlands; INSERM UMR S 999, LabEx LERMIT, Centre Chirurgical Marie Lannelongue, Le Plessis-Robinson, France; University of Paris-Sud, Kremlin-Bicêtre, France
To prevent drug-induced developmental malformations, drugs with that feature adverse teratogenic effects are regulated via the Pregnancy Prevention Program (PPP). Remarkably, among the ten PPP-enlisted drugs are three endothelin-1 (ET-1) receptor antagonists (ERAs). ERAs are approved for the treatment of pulmonary arterial hypertension (PAH). While ERAs hamper pathological remodeling of the pulmonary vasculature and as such exert beneficial effects in PAH, they disturb fetal development of cardiopulmonary tissues. However, the effects of ERAs in PAH pathobiology and cardiopulmonary fetal development also show remarkable parallels in non-pregnant PAH patients. An important physiologic response of the right ventricle is to induce compensative hypertrophy as adaptation to the increased pulmonary vascular resistance (PVR). To facilitate this process, the right ventricle re-activates a fetal gene program: specific developmental genes. Therefore, inhibition the of ET-1-mediated positive inotropic effects and myocardial fetal gene induction by ERAs may affect right ventricular adaptation to the increased PVR in both the fetus, but also in the adult – and non-pregnant – PAH patient. – a part of the submitted abstract has been published in Reproductive Toxicology, PMID: 26111581.
Evaluation of health-related quality life in patients with pulmonary arterial hypertension
Istanbul University Institute of Cardiology, Cardiology Department, Istanbul, Turkey
Various health-related quality of life (HRQoL) instruments have been employed to measure the effects of pulmonary arterial hypertension (PAH). The aim of this study was to evaluate the HRQoL with the generic and the disease specific HRQoL instrument in patients with PAH. Thirty patients (mean age = 39.3 ± 10.7 years) were enrolled for the study. All patients completed the generic Medical Outcomes Study Short-Form 36 (SF-36) and the disease-specific Minnesota Living With Heart Failure Questionnaire (MLWHFQ) to measure HRQoL. The SF-36 contains eight subscales: physical functioning (PF), role physical (RP), bodily pain (BP), general health (GH), vitality (VT), social functioning (SF), role emotional (RE), and mental health (MH); and two summary scores: physical component summary (PCS) and mental component summary (MCS). The MLWHFQ is divided into three separate scores: physical; emotional; and total scores. MLWHFQ physical, emotional, and total scores were negatively correlated with all subscale and all summary scores of SF-36. Total scores were correlated with PF: r = –0.60, P = 0.0005; RP: r = –0.66, P < 0.0001; BP: r = –0.72, P < 0.0001; GH: r = –0.67, P < 0.0001; VT: r = –0.67, P < 0.0001; SF: r = –0.61, P = 0.003; RE: r = –046, P = 0.01; MH: r = –0.59, P = 0.0006; PCS: r = –0.66, P < 0.0001; and MCS: r = –0.46, P = 0.01. In conclusion, both SF-36 and MLWHFQ can be used to assess HRQL in PAH.
Swinging between Group 1 and Group 2 pulmonary hypertension: a case presentation
Cerrahpasa Medical Faculty, Cardiology and Pulmonary Disease Departments, Istanbul University, Istanbul, Turkey
He was diagnosed as IPAH and since he was not vasoreactive, bosentan therapy was initiated. However, in spite of add on sildenafil, every two to three months he was re-hospitalized with clinical deterioration, without any significant changes in his echocardiographic and hemodynamic findings. In the second year of his follow-up, increase in LA dimensions and IVS thickness were observed. In hemodynamic examination with volume challenge, PAWP increased from 14 mmHg to 22 mmHg. PAH-specific drugs were stopped and he is currently on HFpEF treatment without any hospitalizations for 1.5 years. In conclusion, the need for standard tests to reveal Cpc-PH is obvious.
Evolution of pulmonary pressure in healthy newborns, in the first three months of life, in Bogota, located at 2640 m above sea level
Universidad Nacional de Colombia, Clínica De La Mujer, Medellín, Colombia
The effect of altitude is more significant over 2500 m above sea level (a.s.l.). There is no knowledge about the evolution of pulmonary pressure in healthy newborns at 2500–3000 m a.s.l. The objective was to evaluate by echocardiogram the evolution of pulmonary pressure in healthy newborns in postnatal life at 2640 m a.s.l. This was a prospective, descriptive study. Fifty-two healthy newborns were evaluated by echocardiography, after clinical cardiovascular evaluation, looking to rule out any CV pathology. The first echocardiogram to evaluate the SPP was made around 24 h after delivery, with controls at 72 h of life, one month and three months. The SPP was valuated through the tricuspid regurgitation jet, using the Bernoulli modified equation. It was only accepted when an excellent and repetitive regurgitation spectral curve was registered. The median was 20 h for the first echocardiogram and 96 h for the second one. In the first echocardiogram, in 49 of 52 newborns we were able to get an excellent tricuspid regurgitation curve. The median of the SPP was 30 mm Hg; 25th percentile = 27 mmHg and 75th percentile = 40 mmHg (range = 17–68 mmHg). In five newborns, the SPP was 50 mmHg or higher. In conclusion, 10.2% of healthy neonates that are born at 2640 m a.s.l. without any risk factors in the perinatal period showed delay of the drop of SPP in the first hours of life. The only factor that influenced this finding was the effect of hypobaric hypoxia by altitude. This study brings light on the major frequency of persistent pulmonary hypertension of the newborn at altitude.
Pulmonary hypertension in children at altitude
Universidad Nacional de Colombia, Clínica De La Mujer, Medellín, Colombia; Fundacion HOMI, Fundación CardioInfantil, Universidad Del Bosque, Bogotá, Colombia
The effect of altitude is significant over 2500 m above sea level (a.s.l.). Bogotá, where we carried out the study, is at 2640 m a.s.l. The objective is to show the effect of hypobaric hypoxia on pulmonary hypertension (PH) in children. We include new patients studied in the last 12 months. All had clinical evaluation, BNP, hyperoxia test, and catheterization. One patient did not have catheterization because he died before the procedure. Necropsy showed IPH with severe PVD. All patients had prolonged hyperoxia test and VRT with NO and oxygen. Living at low altitudes was recommended. We had 20 new patients (16 girls [80%]); eight patients were aged three years or less (40%). Twelve patients had idiopathic PH (IPH) (60%), four had coronary heart disease (three developed PH after surgery); two had Chronic Pulmonary Disease (CPD), one had high altitude pulmonary edema, and one group 5 . The majority of patients had severe PH: MPP > 60 mmHg; PR > 20 WU. Altitude gives special characteristics to PH by hypobaric hypoxia, affecting epidemiology, pathogenesis, diagnostic approach, and treatment. PH is frequent, could begin and be severe at an early age, and IPH is the main cause. Hyperreactivity of PV is important and remodeling could be severe very early. Diagnosis and treatment: oxygen plays an important role. In conclusion, at altitude, hypobaric hypoxia gives special characteristics to PH. Because hypobaric hypoxia influences not only other pathologies of group 3 but also pathologies of groups 1 and 5 of the Nice Classification, maybe PH of altitude deserves to be considered as a subgroup of group 3.
Distinctive clinical and hemodynamic characteristics of IPAH patients of Indian descent from Guyana
Mount Sinai Health System, New York, NY, USA
Hemodynamic data.
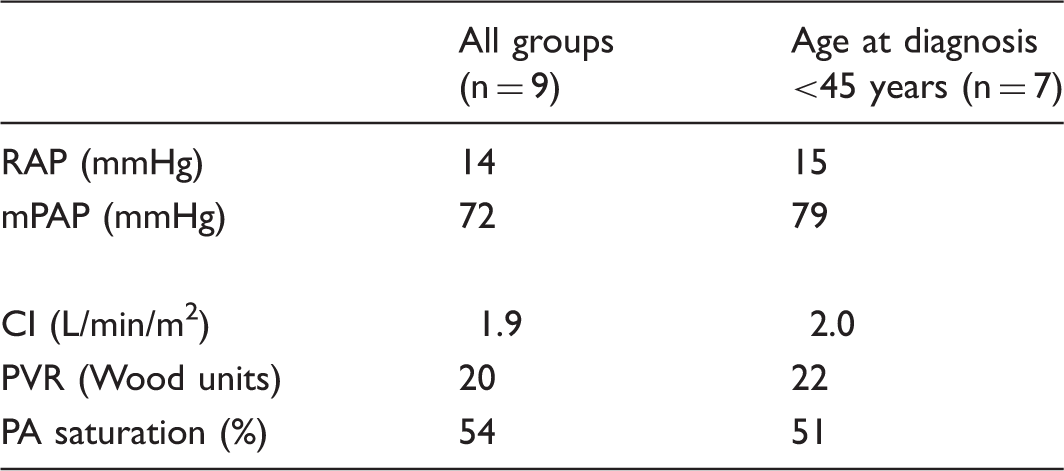
RAP, right atrial pressure; mPAP, mean pulmonary artery pressure; CI, cardiac index; PVR, pulmonary vascular resistance; PA, pulmonary artery.
In this case series of Guyanese patients, we have noted a high prevalence of severe PH with poor response to therapy and poor outcomes. It is unclear if this specific phenotype has either a genetic or environmental explanation, but further studies of this patient population may offer insights in the pathophysiology of PAH.
Effects of riociguat in patients with inoperable CTEPH vs. persistent/recurrent pulmonary hypertension after pulmonary endarterectomy: two-year efficacy results from the CHEST-2 study
Division of Pulmonary, Critical Care, and Sleep Medicine, University of California, San Diego, USA; Clinic for Respiratory Medicine, Hannover Medical School, Hannover, Germany; Department of Internal Medicine, The Ohio State University Medical Center, Columbus, Ohio, USA; Department of Pulmonary Circulation and Thromboembolic Diseases, Medical Center of Postgraduate Education, ECZ-Otwock, Otwock, Poland; Global Clinical Development, Bayer Pharma AG, Wuppertal, Germany; Global Development, Bayer HealthCare Pharmaceuticals Inc., Whippany, New Jersey, USA; University of Giessen and Marburg Lung Center, Giessen, Germany; Department of Medicine, Imperial College London, London, UK
Secondary efficacy endpoints in CHEST-2.
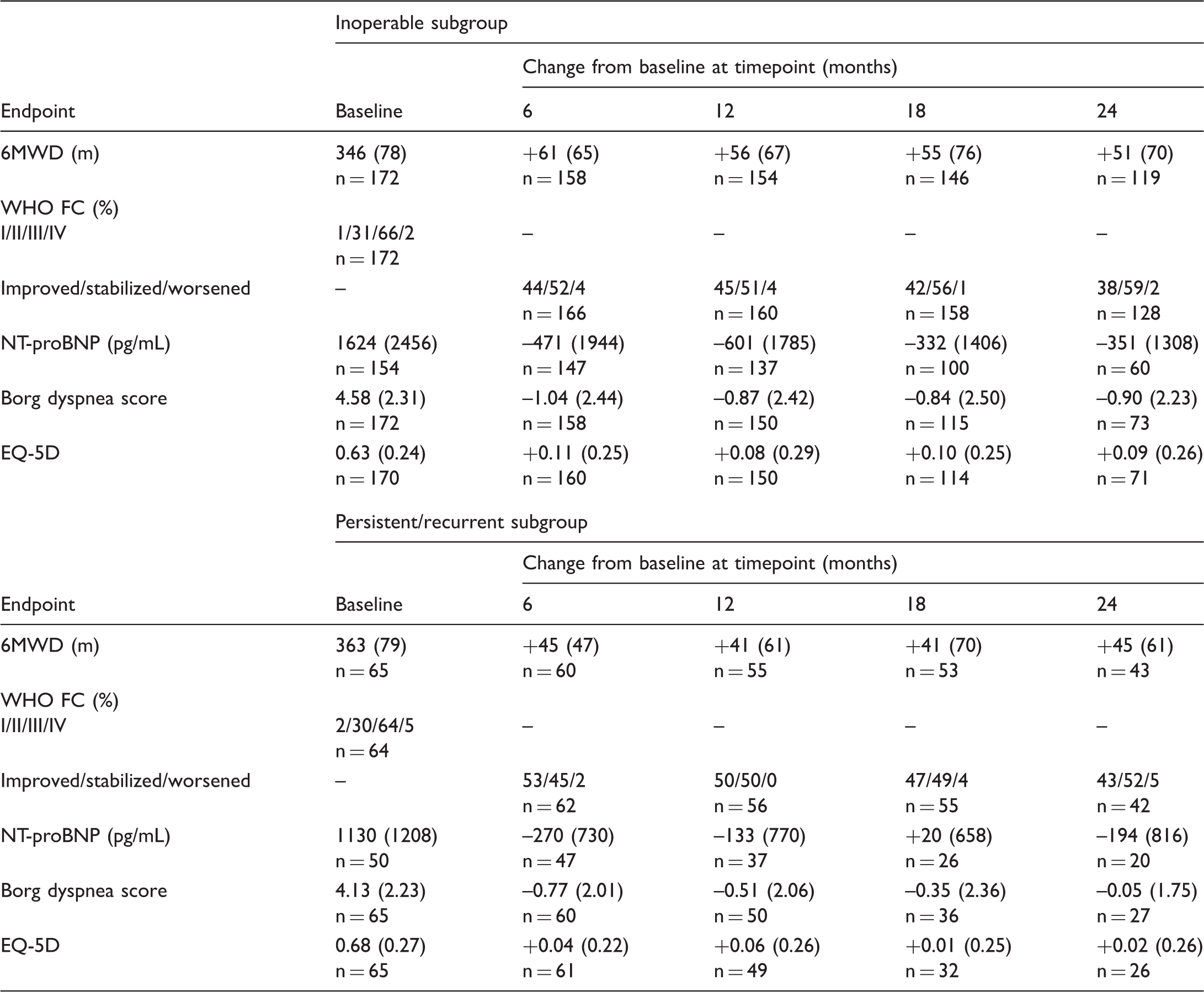
Data are mean (SD) unless otherwise stated. 6MWD, 6-min walk distance; EQ-5D, EuroQol-5 Dimensions questionnaire; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; WHO FC, World Health Organization functional class.
Riociguat for the treatment of patients with chronic thromboembolic pulmonary hypertension (CTEPH): application of criteria for satisfactory clinical response to CHEST-1 and CHEST-2 populations
Cleveland Clinic, Cleveland, OH, USA; Division of Critical Care and Pulmonary Medicine, University of California, Davis Medical Center, Sacramento, CA, USA; Boston University School of Medicine, Boston, MA, USA; Centre for Pulmonary Hypertension, Thoraxclinic, University Hospital Heidelberg, Germany; Clinical Department of Cardiology and Angiology of the First Faculty of Medicine and General Teaching Hospital, Prague, Czech Republic; Chrestos Concept GmbH & Co. KG, Ratingen, Germany; Bayer HealthCare Pharmaceuticals, Whippany, NJ, USA; University of Giessen and Marburg Lung Center, Germany; Department of Medicine, Imperial College London, UK
Riociguat is approved for the treatment of inoperable or persistent/recurrent chronic thromboembolic pulmonary hypertension (CTEPH) following pulmonary endarterectomy (PEA), based on data from the CHEST-1 study and its extension, CHEST-2. The current analysis investigated application of the satisfactory clinical response (SCR) outcome measure (similar to that used in the AIR and AMBITION studies) to the CHEST data. This is the first time SCR has been applied to a database exclusively consisting of CTEPH patients. In CHEST-1, patients received riociguat up to 2.5 mg t.i.d. or placebo for 16 weeks. All patients received riociguat up to 2.5 mg t.i.d. during CHEST-2. SCR status was calculated at CHEST-1 Week 16 and CHEST-2 Week 12. SCR was defined as: ≥10% improvement from baseline in 6-min walk distance; WHO functional class (FC) I/II; and no clinical worsening. SCR status was determined for 250/261 patients in CHEST-1 (163 riociguat, 87 placebo). A greater proportion of riociguat-treated patients achieved SCR compared with placebo patients (39% vs. 13%, respectively). In CHEST-2, SCR status was determined for 224 patients (147 former riociguat, 77 former placebo). Overall, 44% of patients achieved SCR (former riociguat, 46%; former placebo, 40%), with a greater proportion achieved in the inoperable vs. persistent/recurrent CTEPH subgroup (49% vs. 33%, respectively). Patients achieving components of the SCR had greater clinical worsening-free survival in CHEST-2 (Fig. 1). In CHEST-1, riociguat increased the proportion of patients achieving SCR compared with placebo. Further increases were observed during CHEST-2 and the proportion of patients achieving SCR was similar between former placebo and former riociguat patients.
Kaplan–Meier plot showing effect of achieving components of the SCR on clinical worsening-free survival in CHEST-2.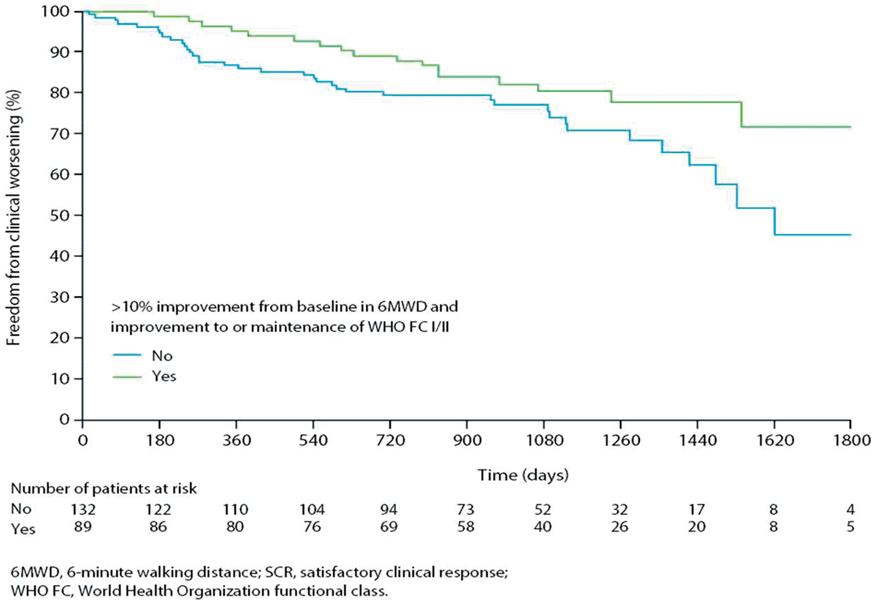
Diastolic pressure gradient predicts operability in children with post-tricuspid shunt and pulmonary arterial hypertension
Children’s Heart Center, Kokilaben Dhirubai Ambani Hospital, Mumbai, India
Persistence or recurrence of pulmonary hypertension (PH) is a major cause of mortality and morbidity after closure of post tricuspid shunt with borderline operability. We sought to identify parameters at cardiac catheterization which could possibly predict persistence of PH two years after surgery. This is a retrospective study. We studied consecutive patients of ventricular septal defect who underwent cardiac catheterization to determine operability and underwent closure of the shunt during 2011–2014. Patients with follow-up of at least two years were included in the study. Mean pulmonary arterial pressure (mPAP) of ≥25 mmHg on cardiac catheterization or peak systolic pressure of ≥40 mmHg on echocardiogram was used to define PH at follow-up. The participants were divided into two groups depending on the presence (group 1) or absence of PH (group 2). Both groups were compared for preoperative cardiac catheterization parameters. DPG was defined as the difference between diastolic PA pressures and mean PA wedge pressures. A total of 17 patients (7 male patients), 11 in group 1, were included in the study. The median age was 5.6 years (age range = 1–20 years) and the median weight was 12.4 kg (range = 6.3–39.4 kg). mPAP in group 1 was 58.1 ± 10.6 mmHg and in group 2 was 37.7 ± 16 mmHg (P = 0.01), The Rp/Rs and Qp/Qs ratio were comparable in both the groups (P = 0.4 and 0.9, respectively). The diastolic PAPs in group 1 were 37.6 ± 8.74 and in group 2 were 22.14 ± 8.27 (P = 0.002). The DPG in group 1 was 27.8 ± 9.22 mmHg and in group 2 was 11.28 ± 6.67 mmHg (P = 0.001). DPG emerged as the most sensitive and specific marker to predict presence of postoperative PH on receiver operator characteristic curve. Higher DPG was a more sensitive and specific marker to predict postoperative PH. If validated further, it may emerge as a very important marker to determine operability.
The relationship between nitric oxide pathway biomarkers and response to riociguat in the RESPITE study of patients with pulmonary arterial hypertension not reaching treatment goals with phosphodiesterase 5 inhibitors (PDE5i)
Division of Pulmonary, Sleep, and Critical Care Medicine, Rhode Island Hospital, Alpert Medical School of Brown University, Providence, RI, USA; Cardiovascular Institute, Allegheny General Hospital, Pittsburgh, PA, USA; Institute of Cellular Medicine, Newcastle University, Newcastle, UK; Center for Pulmonary Vascular Disease and Lady Davis Institute, Jewish General Hospital, McGill University, Montreal, QC, Canada; Department of Cardiology, Erasme University Hospital, Brussels, Belgium; Assistance Publique–Hôpitaux de Paris, Université Paris-Sud, INSERM Unité 999, Le Kremlin–Bicêtre, France; Global Clinical Development, Bayer Pharma AG, Berlin, Germany; Global Clinical Development, Bayer HealthCare Pharmaceuticals, Barcelona, Spain; Chrestos Concept GmbH & Co.KG, Essen, Germany; Clinic for Respiratory Medicine, Hannover Medical School, Hannover, Germany
A proportion of patients with pulmonary arterial hypertension (PAH) treated with phosphodiesterase 5 inhibitors (PDE5i) do not reach/maintain treatment goals. RESPITE investigated whether it is safe, feasible, and beneficial to replace PDE5i with riociguat in patients with an insufficient response to PDE5i. This analysis explored the relationship between nitric oxide signaling biomarkers and response to riociguat. RESPITE (NCT02007629) was a 24-week, open-label, single-arm, Phase IIIb trial in PAH patients in WHO functional class (FC) III, with 6-min walk distance (6MWD) 165–440 m and cardiac index (CI) <3.0 L/min/m2, despite PDE5i treatment for ≥90 days. Patients underwent a PDE5i-free treatment period before receiving riociguat up to 2.5 mg t.i.d.
Change from baseline in biomarkers at Week 24 in RESPITE.
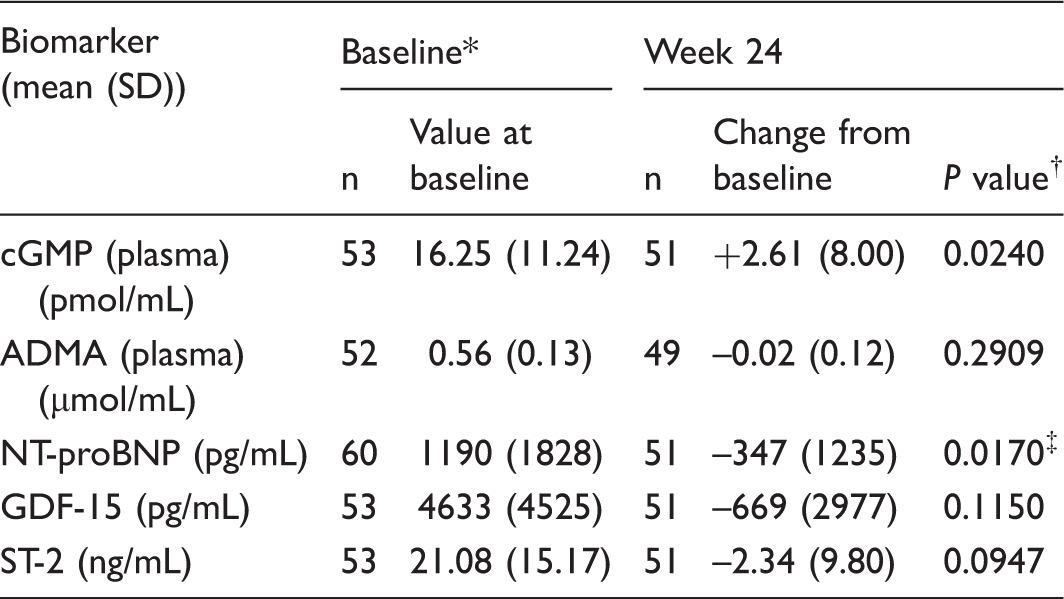
Baseline = the last documented value while still receiving PDE5i.
Pvalues are for change from baseline to Week 24.
P value is for the relative change from baseline in NT-proBNP.
ADMA, asymmetric dimethylarginine; cGMP, cyclic guanosine monophosphate; GDF-15, growth differentiation factor 15; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; PDE5i, phosphodiesterase type 5 inhibitor; ST-2, suppression of tumorigenicity 2.
Application of the criteria for satisfactory clinical response to riociguat treatment of patients with pulmonary arterial hypertension in PATENT-1 and PATENT-2
Columbia University, New York, NY, USA; Division of Critical Care and Pulmonary Medicine, University of California, Davis Medical Center, Sacramento, CA, USA; Department III of Internal Medicine and Cologne Cardiovascular Research Center, Cologne University Heart Center, Cologne, Germany; Chrestos Concept GmbH & Co. KG, Ratingen, Germany; Bayer HealthCare Pharmaceuticals, Whippany, NJ, USA; University of Giessen and Marburg Lung Center, Giessen, Germany; Department of Medicine, Imperial College London, London, UK
Riociguat is approved for treatment of pulmonary arterial hypertension (PAH), based on data from the PATENT-1 study and PATENT-2 extension. This analysis examined PATENT data using a satisfactory clinical response (SCR) outcome measure similar to that used in the AIR and AMBITION studies.
In PATENT-1, patients received riociguat up to 1.5 mg t.i.d., 2.5 mg t.i.d., or placebo for 12 weeks. All patients received riociguat up to 2.5 mg during PATENT-2. The proportion of patients achieving SCR was calculated at PATENT-1 Week 12 and PATENT-2 Week 12. SCR was defined as: a ≥10% improvement in 6-min walk distance (6MWD) compared with baseline, WHO functional class (FC) I/II, and no clinical worsening. In PATENT-1 overall, a higher proportion of riociguat-treated patients achieved SCR vs. placebo (30% vs. 16%). This was also true in treatment-naïve (38% vs. 17%) and pretreated patients (22% vs. 15%). In PATENT-2, 41% of patients achieved SCR overall. A greater proportion of former riociguat patients achieved SCR vs. former placebo (44% vs. 33%), as did treatment-naïve patients vs. pretreated (36% vs. 46%). Patients achieving components of the SCR had improved clinical worsening-free survival in PATENT-2 (Fig. 1). In PATENT-1, riociguat increased the proportion of patients meeting SCR criteria compared with placebo. SCR achievement increased further in PATENT-2, with similar increases observed in the former placebo and former riociguat arms. The proportion of patients achieving SCR in PATENT-2 was similar to that in the AMBITION study, supporting the use of SCR as a clinical measure of efficacy in PAH patients.
Kaplan–Meier plot showing effect of achieving components of the SCR on clinical worsening-free survival in PATENT-2.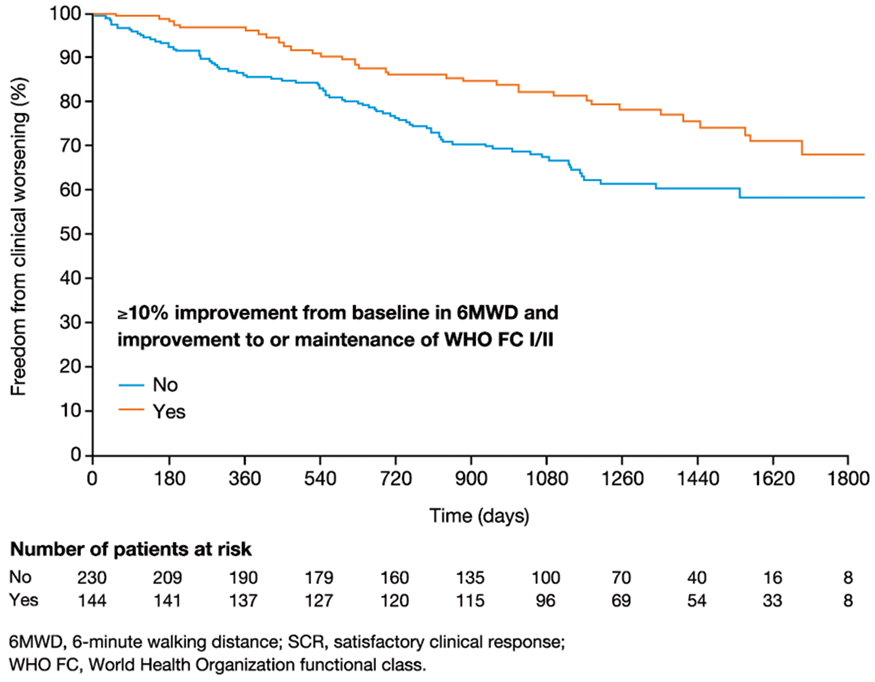
Clinical, laboratory, echocardiographic, and hemodynamic characteristics in relation to survival in pulmonary arterial hypertension associated with congenital heart disease
Kartal Kosuyolu Heart Education and Research Hospital, Istanbul, Turkey
Pulmonary arterial hypertension (PAH) associated with congenital heart disease (APAH- CHD) is classified as a subgroup of PAH that includes four different forms sharing similar clinical and pathological findings. However, attention has been focused on Eisenmenger syndrome (ES) and predictors of the survival in other patients (pts) with APAH-CHD remain to be determined. In this study, we investigated clinical, biochemical, neurohumoral, echocardiographic, and hemodynamic predictors of survival in 134 patients with APAH- CHD (72 women, 62 men; age = 40.7 ± 17.5 years) out of the 341 patients included in our single-center, prospective study. Subgroups of APAH-CHD were as follows: (1) ES (n = 71); (2) PAH associated with prevalent systemic-to-pulmonary shunts (n = 37); and (4) PAH after defect correction (n = 26). Baseline 6-min walk distance (6MWD) was 301.5 ± 127.8 m, pulmonary arterial systolic and mean pressures (PASP, PAMP [mmHg]) were 99.6 ± 32.9 and 64.7 ± 22.85, and transpulmonary and diastolic pressure gradients (TPG, DPG [mmHg]) were 51.8 ± 22.9 and 29.6 ± 20.3, respectively. Pulmonary and systemic vascular resistance (PVR, SVR [WU]) and PVR/SVR ratio were 10.6 ± 9.3, 21.8 ± 9.6, and 0.52 ± 0.37, respectively. Targeted PAH treatments were noted in 100 (75%) patients. Mean follow-up was 30.9 ± 28 months (range = 0.5–122.6). The APAH-CHD compared with idiopathic PAH (IPAH) and PAH associated with connective tissue disease (APAH-CTD) was associated with a better eight-year survival (83 vs. 54.4 and 61%, P < 0.05), and APAH-CHD subgroups showed comparable survival (P > 0.05). The age, sex, functional class, hemoglobin, oxyhemoglobin saturation %, C-reactive protein, uric acid, and brain natriuretic peptide were not related to survival (P > 0.05). Neither tertile definition (<190, 190–330, >331) nor pre-defined cutoff values of 6MWD (m) (<165, 165–440, >440) were related to APAH-CHD survival (P > 0.05). Similarly, tertiles of TPG, DPG, PVR, and PVR/SVR ratio showed comparable survival (P > 0.05). However, pericardial effusion (P = 0.0001) and the highest tertile of right atrial pressure (RAP; mmHg) (>11 vs. 9–11 and <9, P < 0.01) were found to discriminate high-risk patients. In conclusion, APAH-CHD compared with other PAH subgroups is associated with a better survival. However, only pericardial effusion and RAP predict the worst clinical outcome in these patients.
Single-center experience on rheolytic thrombectomy in patients with pulmonary embolism at high risk or intermediate–high risk
Kartal Kosuyolu Heart Education and Research Hospital, Istanbul, Turkey; İH Tanboğa, Ataturk University, Cardiology and Biostatistics, Erzurum, Turkey
Rheolytic thrombectomy (RT) has been used as a percutaneous technology providing a high efficacy with a reduced bleeding risk in patients with pulmonary embolism (PE). In this study, we aimed to present our single-center results on RT in patients with PE at intermediate–high and high risk (IHR, HR). Our study was based on the retrospective analysis of the overall 26 patients (14 women, age = 60 ± 18.2 years) with documented PE who underwent RT using a dedicated system. The systematic work-up including multidetector computed tomography (MDCT), Echo, biomarkers, and PE severity index and its simplified version (PESI, sPESI) were performed in all patients and Qanadli score (QS) was used as the MDCT measure of the thrombotic burden in the pulmonary arteries (PA). The IHR and HR were noted in 22 and four patients, respectively. Clinical, echo, and MDCT measures and median time delay from symptoms to treatment were comparable between the UFT and RT groups (P > 0.05). Failure in the placement of catheters was experienced in one out of 27 RT attempts. Intra-PA infusion of adjuvant tissue-plasminogen activator (t-PA) was needed in only five patients (19.2 %) treated with RT and t-PA dosage was 14.6 ± 5.1 mg. Regardless of the baseline risk status, RT resulted in dramatic improvements in tricuspid annular planary systolic excursion (TAPSE) and tissue systolic velocity (St), PA systolic and mean pressures, QS, right–left ventricle diameter ratio (RV/LV) and right–left atrial diameter ratio (RA/LA), and diameters of main, right, and left PA (P < 0.001 for all). The RT induced short-term bradycardias or conduction disturbances during system activation, and a gross hemoglobinuria for one or two days in all patients. Non-fatal major bleeding was not observed whereas death from treatment failure was documented in two patients (7.6%). Post- discharge PE-related morbidity and mortality was not documented during follow-up period for a median of 310 days (range = 68–983 days). Irrespective of the baseline risk status, RT facilitates the thrombolysis, recovery of pulmonary hemodynamics, and right heart functions with low rates of complications in patients with PE.
Endothelial response to cytokine stimulation is disturbed in pulmonary arterial hypertension: cilium length as a functional marker for dysregulated immunity
Leiden University Medical Center, Leiden, The Netherlands; VU University Medical Center, Amsterdam, The Netherlands
Pulmonary arterial hypertension (PAH) is a lethal syndrome characterized by progressive lung vascular remodeling driven by abnormal endothelial cells (EC) in tandem with excessive inflammation. The primary cilium is a sensory antenna that integrates signaling and fine tunes EC responses to mechanical and chemical stimuli. As such, cilium architecture and function is altered in response to cytokines. Yet, the relation between cilium length and function in context of dysregulated immunity in PAH remains obscure. We hypothesize that primary cilia of EC from patients with PAH are incapable to respond to cytokine stimulations. Primary human pulmonary and mouse embryonic EC were exposed for 24 h to pro- (TNFα, IL1β, and IFNγ) and/or anti-inflammatory (IL-10) cytokines and the length of the primary cilia was quantified. Chronic treatment with all tested inflammatory cytokines leads to a significant elongation of the primary cilia in both control human and mouse EC (by ca. 1 µm, P < 0.001). This structural response is PKA and PKC dependent. Intriguingly, withdrawal of the inflammatory stimulus does not influence cilia length. However, IL-10 prevents and reverses the inflammation-induced cilia elongation in healthy EC but did not influence basal cilia length. Conversely, primary cilia of EC from patients with PAH are significantly longer under baseline conditions compared to controls (2.0 ± 0.7 vs. 2.5 ± 0.4 µm, P < 0.001). These cilia do not elongate further upon inflammatory stimulation and IL-10 treatment does not impact cilia length under any of the tested conditions. PAH EC exert elongated cilia compared to control EC. These cilia do neither respond to pro- nor anti-inflammatory cytokine stimulation suggesting a desensitization of the PAH endothelium. This could represent an adaptation to the chronic state of inflammation present in the PAH vasculature.
The association of adiponectin and pulmonary hypertension in African-Americans: the Jackson Heart Study
Providence VA Medical Center, Providence, RI, USA; Alpert Medical School of Brown University, Providence, RI, USA
Prevalence ratios and 95% confidence intervals (CI) for the association of plasma adiponectin and PH.
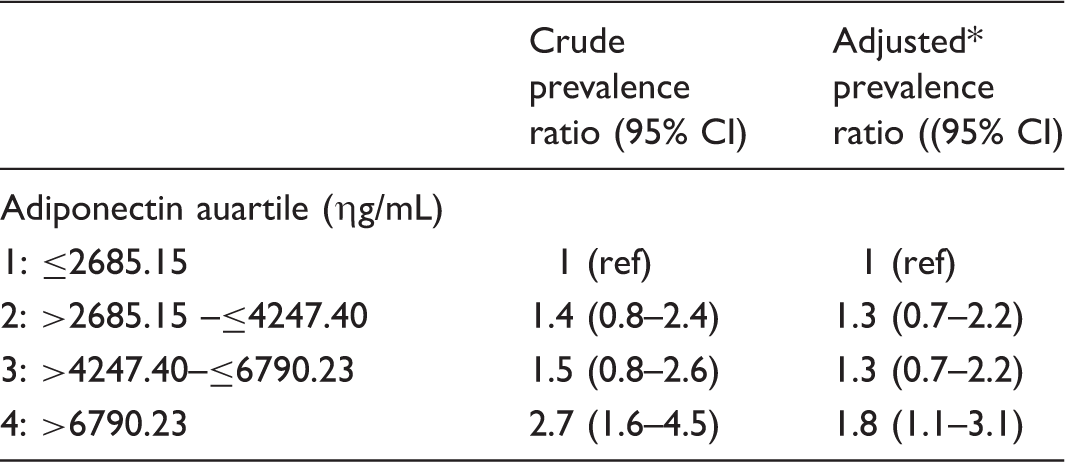
Adjusted for age, sex, BMI category, pulse pressure, hypertension, diabetes, history of coronary heart disease, history of chronic lung disease, spirometry profile, left ventricle ejection fraction, left atrial diameter index, estimated glomerular filtration rate.
Prognostic value of the baseline serum sodium level in 2 independent cohorts of pulmonary arterial hypertension
Bridgeport Hospital, Yale New Haven Health, Bridgeport, CT, USA; Cleveland Clinic, Respiratory Institute, Cleveland, OH, USA; United Therapeutics Inc., Research Triangle Park, NC, USA; Yale University, New Haven, CT, USA
Hyponatremia has prognostic value in heart failure and liver disease, so we studied the potential prognostic value of hyponatremia in pulmonary arterial hypertension (PAH). We performed secondary analyses of two PAH cohorts: (1) United Therapeutics’ randomized clinical trials (including P0:03, P0:04, P0:05, and P0:06) and (2) a long-term PAH registry at the Cleveland Clinic.
We included adult WHO group 1 PH patients. Our hypothesis was that baseline hyponatremia is associated with worse one-year survival. Baseline Na level is negatively correlated with baseline mean right atrial pressure (r = –0.09, P = 0.018; r = –0.089, P = 0.015 in cohorts 1 and 2, respectively). In unadjusted analyses of cohort 1, Na level (as a continuous variable) is associated with one-year mortality (hazard ratio = 0.94, P = 0.035). Hyponatremia (Na ≤ 137) status loses its significance (P = 0.12) in the multivariable regression Cox model when adjusted for functional class (FC) (after applying a stepwise model selection procedure to identify the confounding variable whose presence turns the effect of baseline sodium level into insignificant).
Secondary analyses of two PAH cohorts: (1) United Therapeutics’ randomized clinical trials; and (2) a long-term PAH registry at the Cleveland Clinic.
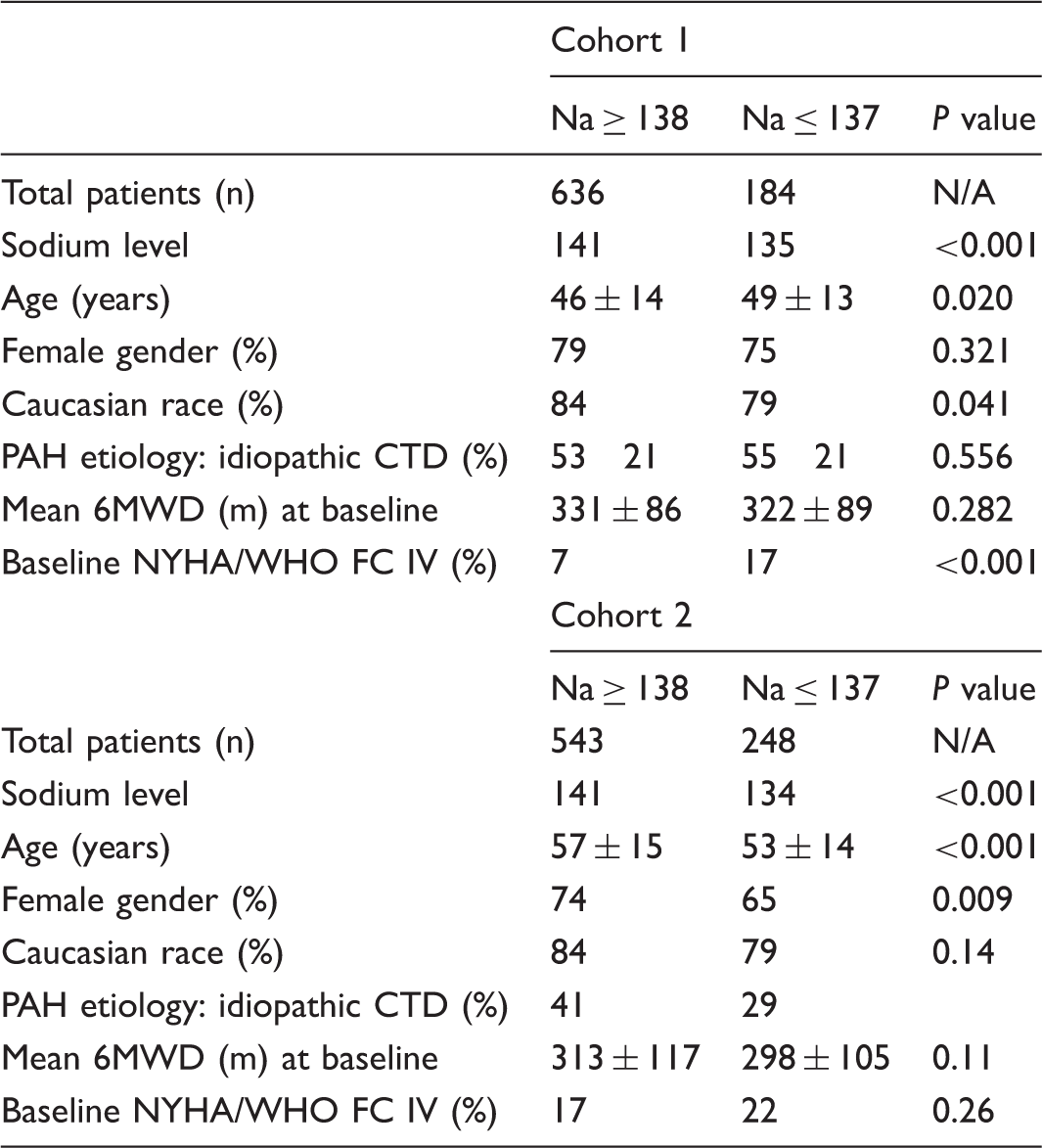
Acknowledgments
We thank United Therapeutics Corporation for making its database available for our secondary analyses.
Evaluation of tumor produced EMAP II as a T lymphocyte apoptosis inducing and potentially immune suppressive mechanism
Faculty of Medicine, Minya University, Minya, Egypt; Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
Tumor cells can suppress immune cells leading to tumor escape from host immune surveillance. Cancer-allocated increased programed cell death (PD or apoptosis) of circulating lymphocytes is observed in multiple forms of cancer and have led to target the receptor on T cells, PD1, using monoclonal neutralizing antibodies, which are successfully used in cancer therapy, including lung cancer. However, it has also been found that not all patients benefit from these antibodies. This proposal aims to study an alternative mechanism, T cell apoptosis induced by EMAP II and its receptor CXCR3, which has not been considered yet of clinical applicability in cancer. By quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blotting, we analyzed whether CXCR3 expression correlated with phytohemagglutinin (PHA) - induced sensitization of T cells towards EMAP II-induced apoptosis . Additionally, using recombinant EMAP II and/ or tumor cells, we quantified the amount of apoptosis by Annexin V staining. Finally, we assessed EMAP II expression in section from human tumors in relation to T cell apoptosis using IHC and TUNEL Using qRT-PCR and western blotting analysis, we found that CXCR3 is upregulated T cells after treatment with PHA, which can sensitize lymphocytes to undergo apoptosis in response to EMAP II. EMAP II and CXCR3 neutralizing antibodies abrogate EMAP II-induced T cell death either induced by tumor cells or by recombinant EMAP II. Finally, EMAP II expression in tumor cells significantly correlated with T cell apoptosis. EMAP II and CXCR3 mediated T lymphocyte cell death could be a mechanism of immune suppression caused by tumors. Thus, targeting this pathway could be an alternative for tumors unresponsive to PD-1 antibodies.
Rapid regulation of the G-protein coupled receptor GPR55 by 2-oxoglutarate dependent oxygenases in pulmonary endothelium
Vanderbilt University Medical Center, Nashville, TN, USA; University of Florida Medical Center, Gainesville, FL, USA
The pathogenesis of pulmonary arterial hypertension (PAH) and the associated endothelial dysfunction is complex. Oxidative stress and dysfunctional signaling through bone morphogenic protein receptor type 2 (BMPR2) have been shown to be central pathogenic mechanisms in PAH. We have previously shown that relative expression level of the G-protein coupled receptor GPR55 exerts a significant modifier effect on the penetrance of BMPR2 mutations in heritable PAH in humans. We have also previously reported that levels of isofurans – products of free radical lipid peroxidation – are significantly elevated in PAH, and that stimuli that increase oxidative stress and isofuran production in the murine lung – namely, brief daily hyperoxia – also enhance the development of PAH. We thus initially hypothesized that GPR55 might be regulated by oxidative stress. Consistent with this hypothesis, we found that GPR55 is upregulated in the murine lung following hyperoxia (average 5.2-fold increase). However, GPR55 was also found to be upregulated in the lungs of mice treated with Sugen/hypoxia (average 1.9-fold increase), a condition that disfavors isofuran formation. Oxidative stress and hypoxia will both independently stabilize and activate hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2). In human lung endothelial cells, treatment with dimethyloxalylglycine (DMOG) or CoCl2, chemical inducers of HIF1 and HIF2 that work via inhibition of 2-oxoglutarate dependent oxygenases (2OGDOs), dose- dependently increased GPR55 by western blot (1.2-fold increase at 0.1 mM DMOG up to 6.2-fold increase with 1 mM DMOG). Time course experiments revealed that GPR55 increased with kinetics similar to that of HIF-1, suggesting direct regulation of GPR55 by 2OGDOs as opposed to regulation by HIF (which would require a time lag for gene transcription and new protein synthesis). GPR55 thus appears to be a novel GPCR involved in the pathogenesis of pulmonary vascular disease and PAH through HIF-independent, but 2OG-dependent injury response pathways.
Concentric RV remodeling represents an adaptive phenotype only in patients with advanced pulmonary arterial hypertension
University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA; Department of Medicine, Department of Cardiology, Division of Translational and Regenerative Medicine, Department of Pulmonary, Critical Care, Sleep, and Allergy Medicine, University of Arizona, Tucson, AZ, USA
Group 1 had lower PVR than Groups 2 and 3.
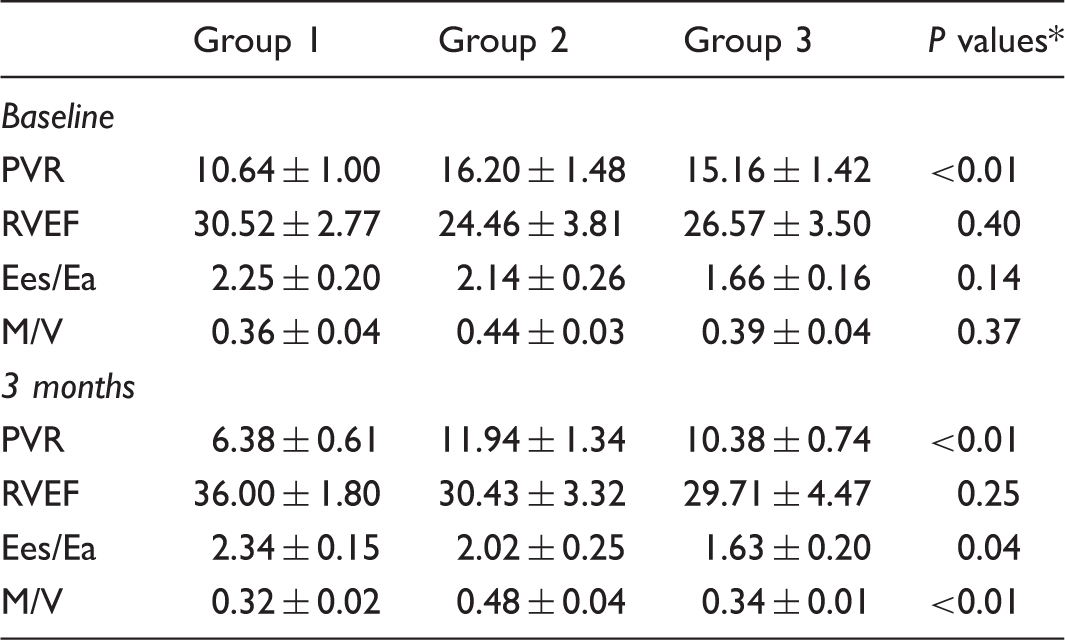
P values represent between group mean differences at baseline and three months, respectively.
MicroRNA biomarkers in persistent pulmonary hypertension of the newborn
Children’s Hospital of Michigan and Hutzel Women’s Hospital, Detroit, MI, USA
Persistent pulmonary hypertension of the newborn (PPHN) is a rapidly progressive and potentially fatal vasculopathy stemming from a failure of the pulmonary vascular resistance (PVR) to drop at or shortly after birth. Recent studies suggest that microRNAs (miRNAs) are dysregulated in adult and experimental PH. The goal of this study was to identify potential miRNAs involved in the pathogenesis of PPHN. In a case-control study, expression profiles of miRNAs were investigated in de-identified formalin fixed paraffin embedded (FFPE) postmortem lung specimens from 12 infants with PPHN and 12 infants who died of other causes. We have completed microarray analysis of miRNA expression in archived (FFPE) samples from 12 PPHN and 12 control participants. MiRNAs that were detected in at least eight of 12 samples for either the PPHN or control groups were selected reducing the total set from 2570 to 1202. Statistical analysis was performed with a moderated t-test and correction of P values with the Storey bootstrap method. Stringent criteria were used to select 12 miRNAs significantly different between the two groups: (1) q-values ≤0.05 (5% false discovery rate); and (2) minimum 1.5-fold change between PPHN and control. We then focused on three of the differentially expressed miRNAs that had predicted target genes associated with the disease etiology: miR-379-5p, miR-7977, and miR-455-3p. Among the relevant predicted target genes of these miRs are prostaglandin E synthase, endothelin, and cardiac t-box factor TBX20. The latter has been associated with cardiac defects and PH. We used quantitative reverse transcription polymerase chain reaction to confirm that these three miRNAs were repressed (lower expression) in the PPHN cases. This is the first study to identify miRNAs associated with PPHN in archived postmortem lung specimens. These may contribute to the pathogenesis of PPHN and may provide potential novel therapeutic approaches for the treatment of PPHN.
Feasibility of determining right ventricular diastolic stiffness from a standard right heart catheterization
Division of Translational and Regenerative Medicine, BIO5 Institute, Department of Medicine, Department of Pulmonary, Critical Care, Sleep, and Allergy Medicine, Department of Cardiology, University of Arizona, Tucson, AZ, USA; Department of Pulmonary and Critical Care Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Diastolic right ventricular (RV) function has recently been shown to be of high clinical and prognostic relevance in patients with pulmonary arterial hypertension (PAH). However, current methods to determine accurate measures of diastolic stiffness require the same-day completion of a right heart catheterization (RHC) combined with cardiac magnetic resonance imaging (CMR) and rely on assumptions about RV volume at zero pressure (V0). We investigated the feasibility of determining a clinically meaningful measure of RV diastolic stiffness from a standard RHC without advanced imaging. Treatment-naïve PAH patients (n = 31; 26 women, 5 men; age = 55 ± 12 years) were initiated on parenteral treprostinil therapy and after three months underwent same-day RHC and CMR. Diastolic stiffness coefficient, β, was estimated by fitting the end-diastolic pressure-volume relationship (EDPVR), P = α(eβV-1) to both pressure points (0 = beginning diastolic pressure, end-diastolic pressure) and volume points (V0 = end-diastolic volume [EDV]–stroke volume [SV], and EDV). SV was calculated from cardiac output and heart rate (SVRHC) or the difference between EDV and end-systolic volume (SVCMR). Bland–Altman analysis was used to compare β when V0 = 0 (Fig. 1a) or estimated using single-beat analysis (Fig. 1b).
Using CMR-derived stroke volumes, average β was similar under conditions when V0 equals zero (0.047 ± 0.022 mL–1; SVCMR: V0 = 0) as well as when V0 was calculated (0.049 ± 0.021 mL–1; SVCMR: V0). The bias and confidence intervals (CI) were 0.002 mL–1 and −0.002–0.006 mL–1, respectively (Fig. 1c). Using SVRHC, average β was also similar whether V0 = 0 (0.043 ± 0.019 mL–1) or was calculated (0.045 ± 0.018 mL–1). The latter bias and CI were 0.002 mL–1 and (−0.002–0.006 mL–1). SV significantly influenced estimates of β whether V0 = 0 (Fig. 1D) or was calculated (bias = –0.005, CI = −0.03–0.02). In contrast to V0, the current study suggests that stroke volume contributes to the precision but not the accuracy of calculating RV diastolic stiffness.
Right ventricular (RV) diastolic stiffness (β) was calculated using two different methods: A) RV volume at zero pressure (V0) was assumed to be zero and B) V0 was determined using the end systolic end systolic elastance (Ees). C) When calculated using the same stroke volume (SVCMR), the value of V0 had no effect on β. D) The average β is similar whether stroke volume is measured using cardiac magnetic resonance imaging or thermodilution cardiac output (Bias: −0.005) but there is a decrease in precision between the two methods (CI: −0.03 to 0.02).
An estrogen receptor α (ERα)-BMPR2-apelin axis mediates 17β-estradiol (E2)’s protective effects on right ventricular function in experimental pulmonary hypertension
Indiana University School of Medicine, Indianapolis, IN, USA; University of Laval, Quebec City, QC, Canada; Richard L. Roudebush VA Medical Center, Indianapolis, IN, USA
Women with pulmonary arterial hypertension (PAH) exhibit superior right ventricular (RV) function and survival compared to men, a phenomenon attributed to poorly understood cardioprotective effects of E2. E2, through ERα, attenuates pulmonary hypertension (PH)-induced RV dysfunction by upregulating the pro-contractile and pro-angiogenic peptide apelin via a bone morphogenetic protein receptor 2 (BMPR2)-dependent mechanism. ERα, BMPR2, and apelin were measured (western blot, reverse transcription polymerase chain reaction [RT-PCR]) in RVs from PAH patients with compensated or decompensated RV hypertrophy (CRVH or DRVH) and in RV homogenates from male or female Sprague Dawley rats with sugen/hypoxia (SuHx)-induced PH. H9c2 rat cardiomyoblasts were stressed with TNF-α (10 ng/mL) or staurosporine (50 nM) ± E2 (100 nM, 24 h). ERα-, BMPR2-, and apelin-dependence were evaluated by siRNA (5 pM). Apelin downstream signaling was assessed by measurement of ERK1/2 activation. ERα binding to the BMPR2 promoter was assessed by ChIP. P < 0.05 by ANOVA was considered significant. ERα correlated with BMPR2 and apelin expression in SuHx-RVs and human RVs. Treatment of SuHx-PH rats with E2 or ERα agonist increased RV BMPR2 and apelin, whereas RV apelin was decreased in E2-treated hypoxic ERα (P < 0.05), but not ERβ knockout mice. In H9c2 cells, E2 or ERα agonist attenuated TNF-α- or staurosporine-induced decreases in BMPR2, apelin, and phospho-ERK1/2 (P < 0.05). E2 protection was lost after knockdown of ERα, BMPR2, or apelin (P < 0.05). ERα was necessary for E2-mediated increases in BMPR2, apelin, and ERK1/2, and BMPR2 was required for the E2-mediated increase in apelin (P < 0.05 for siRNA vs. scramble). ERα interacted with the BMPR2 promoter. ERα, BMPR2, and apelin were unchanged in CRVH (vs. control), but increased in DRVH (accompanied by an unexpected decrease in ERK1/2 activation). E2, via ERα and BMPR2, increases apelin in the failing RV and in stressed cardiomyoblasts. Resistance to protective E2-ERα-BMPR2-apelin signaling may contribute to progression from CRVH to DRVH.
CT measures of intraparenchymal arterial and venous volume distribution in pulmonary hypertension
Brigham and Women’s Hospital and Harvard School of Medicine, Boston, MA, USA
CT-derived quantification of vascular morphology may be useful for detection, classification, and understanding of pulmonary vascular disease (PVD). Changes such as pruning and proximal dilation of the vessels have been described in pulmonary hypertension (PH), but the differential effect on arterial and venous pulmonary circulation has not been well characterized. In this study, we attempted to quantify blood volume distribution in small- and large-sized vessels in the arterial and venous pulmonary vasculature using clinically acquired CT scans. A cohort of 700 patients evaluated for unexplained dyspnea at Brigham and Women’s Hospital was retrospectively identified. In total, 179 patients had CT angiography within one year of right heart catheterization (RHC). Forty-nine patients with PH (15 Group II, 16 PAH, and 18 CTEPH) and 15 individuals with no PVD were identified. Lung segmentation and 3D reconstruction of parenchymal vasculature was generated as previously described. Arteries and veins were manually labeled in the right lung. Primary analysis focused on the small vessel fraction, the fraction of volume in vessels with cross-sectional area <5 mm2, divided by the total vessel volume. Medians, interquartile range, and Wilcoxon rank-sum test (SAS 9.3) were used for group comparisons. Arterial small vessel fraction was significantly lower in all groups of PH: Group I (0.45 [0.40–0.52], P = 0.001); Group II (0.42 [0.37–0.48], P = 0.0002; Group IV (0.49 [0.40–0.58], P = 0.001); compared to controls (0.60 [0.54–0.66]). In the venous pulmonary vasculature, however, the difference reached statistical significance only in Group II patients (0.43 [0.41–0.50], P = 0.008) but not in Group I (0.49 [0.40–0.55], P = 0.09) or group IV (0.51 [0.44–0.58]), P = 0.09) participants compared to controls (0.54 [0.49–0.58]). Arterial remodeling can be observed and quantified using CT imaging in patients with both pre-capillary and post-capillary PH compared to controls without any PVD. Venous volume redistribution measured by small vessel fraction is additionally present in post-capillary disease.
Examples of CT-based 3D reconstruction of the vasculature from three participants (a–c) with red color indicating smaller vessels. The lower panels (d–f) show the arterial and venous labeling of the same; blue represents arterial and red represents venous vasculature. The profiles to the left show the relative distribution of blood volume.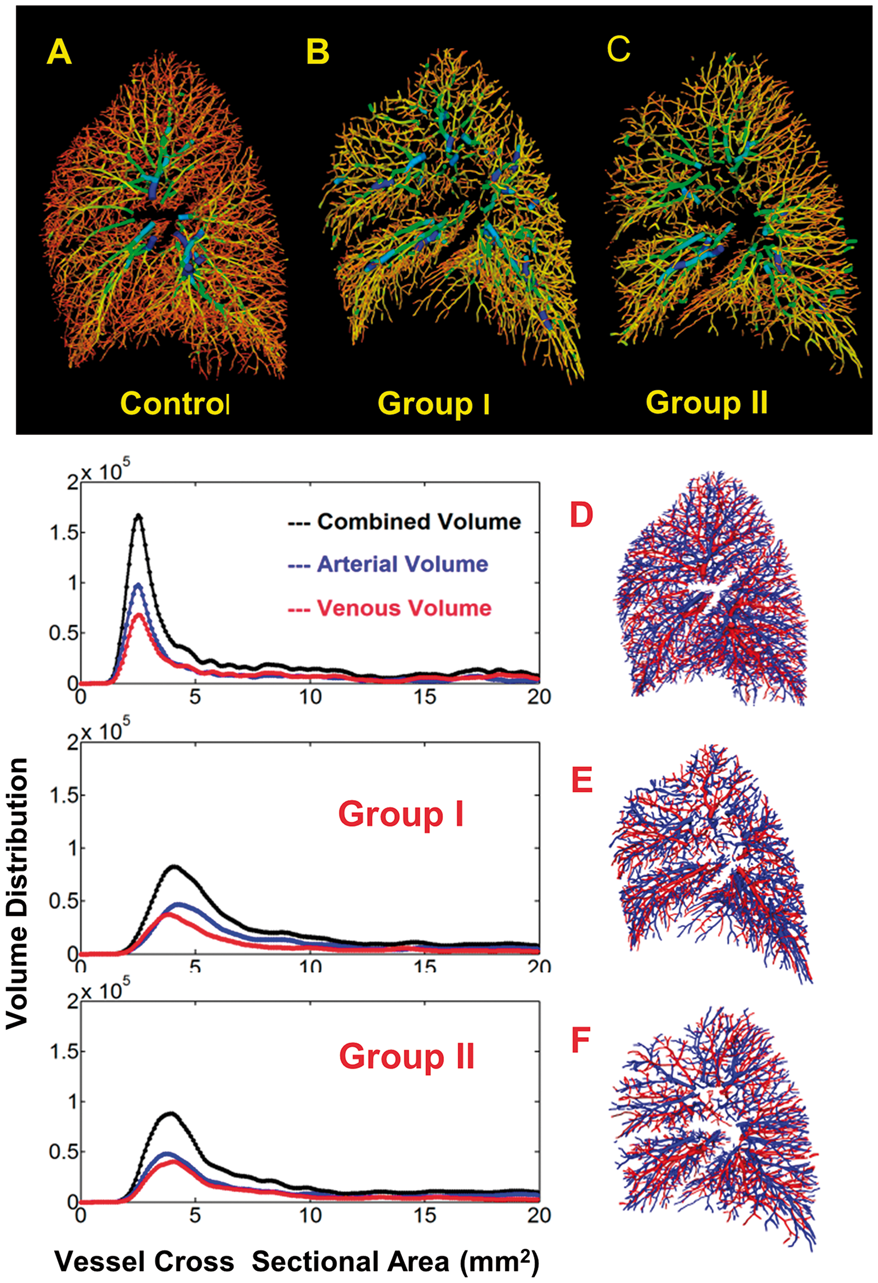
Improvement in right ventricular dispersion after inhalation of iloprost in pulmonary hypertension
Turkiye Yuksek Ihtisas Hospital, Ankara, Turkey; Bursa Yuksek Ihtisas Hospital, Bursa, Turkey; Bingol State Hospital, Bingol, Turkey; Ataturk Pulmonary Disease and Thorax Surgery Research Hospital, Ankara, Turkey
Chronic prostanoid therapies demonstrate functional, hemodynamic, and mortality benefits in patients with pulmonary hypertension (PH). Right ventricular (RV) function is an important predictor of morbidity and mortality in PH. The aim of this study is to investigate acute RV mechanical dispersion (RV-MD) response to inhalation of iloprost in patients with PH on chronic iloprost inhalation therapy. Two-dimensional speckle-tracking strain analysis was used to calculate RV-MD before and after inhalation of iloprost in 16 patients with PH on chronic iloprost therapy (11 idiopathic pulmonary arterial hypertension [IPAH] and five chronic thromboembolic PH; ten women, six men; mean age = 38.5 ± 10.2 years). Contraction duration was measured as the time in milliseconds from onset R wave on ECG to peak RV shortening by strain. RV-MD was defined as the standard deviation of contraction duration a six segment RV model (three RV free wall plus three septal segments). Before iloprost inhalation tricuspid annular plane systolic excursion, mean pulmonary artery pressure (mPAP) and 6-min walk distance (6MWD) were also recorded. Comparison of RV-MD before and after iloprost inhalation revealed a significant improvement in RV-MD after the iloprost inhalation (58.2 ± 5.4 ms vs. 51.3 ± 6.4 ms, respectively; P < 0.01). The mean difference in RV-MD (ΔRV-MD) was 6.9 ± 3.4 ms. There were significant positive correlations between ΔRV-MD and 6MWD (rs = 0.95, P < 0.001) and tricuspid annular plane systolic excursion (rs = 0.84, P < 0.001), and significant negative correlation with mPAP (rs = –0.65, P < 0.01). RV-MD significantly improves in patients with PH on chronic iloprost inhalation therapy after each inhalation, and ΔRV-MD is significantly correlated with tricuspid annular plane systolic excursion, 6MWD and mPAP.
Identifying components of the mitochondrial electron transport chain that mediate oxygen sensing in the pulmonary circulation: potential therapeutic targets for pulmonary hypertension
Department of Medicine, Queen’s University, Kingston, ON, Canada
The aim of the study is to identify the subunit(s) of the mitochondrial electron transport chain responsible for oxygen-sensing in the pulmonary vasculature. The ability to respond to changes in oxygen is mediated by the homeostatic oxygen sensing system, consisting of resistance pulmonary arteries (PA), ductus arteriosus, carotid bodies, and neuroepithelial bodies, functioning together to improve oxygen uptake and delivery. Within the PA, hypoxic pulmonary vasoconstriction (HPV) matches lung perfusion to ventilation, whereas systemic arteries dilate during hypoxia. Mitochondria in pulmonary artery smooth muscle cells (PASMC) serve as redox-sensitive O2 sensors, modulating vascular tone by releasing reactive oxygen species (ROS) in proportion of alveolar O2 levels. These ROS control the function of potassium channels (e.g. Kv1.5) and enzymes (e.g. rho kinase). However, the identity of the specific mitochondrial protein(s) responsible for oxygen-sensing remains elusive. Because HPV is unique to PASMC (systemic ASMC dilate during hypoxia), we compared these mitochondria, reasoning the oxygen-sensing components of the mitochondria might be differentially expressed. Mitochondria from rat pulmonary and renal artery (RA) SMC were compared via activity assay, micropolarimetry, mass spectrometry, reverse transcription polymerase chain reaction (RT-PCR), and western blot. Hydrogen peroxide (H2O2) and calcium flux were measured using H2O2- and Ca2+-specific probes and confocal microscopy. Relative to RASMC, PASMC exhibit increased Complex I activity and oxidative metabolism. Seven candidate proteins/subunits have been identified as differentially expressed in PASMC and RASMC, including iron-sulfur subunits of Complexes I and III, and mitochondrial uncoupling proteins. Upon hypoxia, PASMC H2O2 levels decrease prior to calcium influx, supporting the theory that decreased H2O2 during hypoxia is the cellular signal mediating HPV. Further experiments aim to confirm the identity of the mitochondrial oxygen sensor via knockdown/silencing of candidate proteins. Identification of specific components of the oxygen-sensing system may identify therapeutic targets for diseases of impaired oxygen-sensing, such as pulmonary hypertension and chronic altitude sickness.
Phosphoproteomic analysis of lung tissue in pulmonary arterial hypertension reveals activation of immune modulatory, angiogenic, and cell proliferation pathways
The Rensselaer Center for Translational Research, Rensselaer, NY, USA; Pulmokine Inc., Rensselaer, NY, USA; Yale University, New Haven, CT, USA
Increased kinase signaling may play an important role in the abnormal cellular proliferation, angiogenesis, and immune dysregulation associated with pulmonary arterial hypertension (PAH). We performed an unbiased phosphoproteomic analysis of lung tissue obtained from individuals with idiopathic PAH (iPAH) and compared it to control donor lung tissue using UPLC MS/MS on an LTQ Orbitrap ELITE System. Data were analyzed with Progenesis LCMS software. NetworKin and Phosphonet were used to predict kinases responsible for phosphorylation. STRING was used to build regulatory kinase networks implicated in PAH. Immunohistochemistry and western blot analyses were performed to confirm the presence of the most upregulated phosphoproteins in iPAH lungs. Several dramatically increased and decreased phosphoproteins were identified in iPAH lungs compared to controls. The most increased phosphoproteins included S378-IKZF3, S73-HMHA1, S709- BCAS3, T442-RHG25, S2047-NUMA1, Y900-AQR, S710-ASPP1, T180/Y182-MK14, T231-ZN404, and S107-HDGF. The most decreased phosphoproteins included S119-PTPRB, Y48-SNG2L, S624-HTSF1, S275-PSIP1, S294-ANXA2, S422-HDAC2, and T113-S10A9. Kinase networks identified from phosphopeptide analysis revealed CSNK2A1 (casein kinase 2 subunit alpha), CDK1, MAPK12, MAPK14, PRKs, PDKs, DYRK2, AKT1, AurkA, CAMK2A, CAMK2B, CAMK2D, CAMK2G, CAMK4, GSK3A, GSK3B, HIPK2, HYRC, and PAKs as potentially important kinases in PAH. In vitro phosphorylation assays indicated that GSK3α and/or GSK3β could phosphorylate IKZF3 (Aiolos) at S378. Western blot analysis confirmed that pIKZF3 (S378), a zinc finger transcription factor that plays a key role in lymphocyte regulation, was increased in iPAH compared to control lung lysates. Immunohistochemistry demonstrated pIKZF3 in lymphocytes surrounding severely hypertrophied pulmonary arterioles which corresponded predominantly to perivascular T cells. An unbiased phosphoproteomic analysis demonstrated several novel targets regulated by kinase networks, and reinforced the role of dysregulated immune, angiogeneic, and proliferative pathways in iPAH. The identified up- and downregulated phosphoproteins have potential as biomarkers to guide targeted therapy for PAH and provide new insights for therapeutic strategies.
Incremental efficacy of an inhaled PDGFR inhibitor in combination with tadalafil and ambrisentan for the treatment of pulmonary arterial hypertension
Pulmokine Inc., Rensselaer, NY, USA
Many patients with pulmonary arterial hypertension (PAH) fail to respond to combination therapy with PDEV inhibitors and endothelin receptor antagonists. We tested the hypothesis that a novel inhaled PDGFR kinase inhibitor, PK10571, in combination with ambrisentan and tadalafil would provide incremental benefit in an animal model of severe PAH. AAV5/6.2CMV PDGF B was administered by pulmonary insufflation to male Sprague Dawley rats in order to overexpress PDGFBB in the lung. Four weeks later, SU5416 was given and rats housed in a hypoxia chamber at FiO2 10% for three weeks. After PAH had developed, PK10571 was administered either alone (0.25 mg/kg twice daily via inhalation route) or in combination with ambrisentan and tadalafil (10 mg/kg, oral gavage; once daily) for 28 days. AAV-PDGFB alone did not result in PAH. Overexpression of PDGF B resulted in a decrease in total PDGFRβ. However, subjecting these animals to SU5416/hypoxia dramatically increased pPDGFR β/PDGFR. Right ventricular systolic pressure (RVSP) was significantly decreased in rats treated with ambrisentan + tadalafil (TA-V) or PK10571 (V-PK) alone compared to vehicle (V-V). Combination treatment (TA-PK) provided additional benefit (V-V = 62.64 ± 4.07; TA-V = 42.89 ± 2.23; V-PK = 44.30 ± 1.88; TA-PK = 33.24 ± 1.15 mm Hg, P < 0.01 TA-V, V-PK, or TA-PK vs. V-V; P < 0.01 TA-PK vs. TA-V or V-PK). RV hypertrophy (RV/(LV + IVS)) was significantly decreased in rats treated with TA-V, V-PK, or TA-PK compared to V-V, and TA-PK provided additional reduction in RVH (V-V = 0.505 ± 0.03; TA-V = 0.376 ± 0.02; V-PK = 0.36 ± 0.01; TA-PK = 0.277 ± 0.01, P < 0.01 TA-V, V-PK, or TA-PK vs. V-V; P < 0.05 TA-PK vs. TA-V or V-PK). The incremental efficacy of combination therapy (TA-PK) was associated with a decrease in pPDGFR β/PDGFRβ. These data show that inhaled PK10571 was as effective as treatment with tadalafil + ambrisentan in the AAV-PDGFB SU5416/Hypoxia rat model of PAH. Combination treatment of inhaled PK10571 plus tadalafil and ambrisentan provided additional benefit compared to tadalafil + ambrisentan.
Reduction in CD68 macrophage number causes a spontaneous and gender-specific pulmonary arterial hypertension phenotype in mice
Department of Infection, Immunity & Cardiovascular Disease, University of Sheffield, Sheffield, UK
Perivascular macrophages are well documented in patients and animal models of pulmonary arterial hypertension (PAH). The exact role for macrophages and whether their presence or absence is required for the vascular remodeling seen in PAH is unclear. Using a novel inducible macrophage (CD68) depletion model (MacLow), we aimed to determine the requirement for macrophages in pulmonary arterial remodeling associated with PAH. After macrophage depletion (∼50% of F4/80 positive cells) for six weeks, mice were phenotyped for PAH by echocardiography, closed chest cardiac catheterization, and immunohistochemistry (IHC). To investigate the origin of the effector cells, male chimeric mice were generated, and to study gender-specificity of the disease phenotype, mixed gender chimeric MacLow/wild type (wt) mice were also produced. Male but not female MacLow mice developed a PAH phenotype compared to controls (RVSP 66 vs. 24 mmHg, P < 0.0001, n = 5–8) that was associated with right ventricular hypertrophy (RVH) and pulmonary vascular remodeling. Immunohistochemistry analysis of demonstrated increased iNOS- |CD206+ |F4/80+ macrophages suggesting a M2-like macrophage population drive the PAH phenotype in these mice. Studies on mixed gender chimeric mice demonstrated that neither male or female MacLow BM transferred into wild-type was sufficient to induce PAH, and female MacLow BM failed to protect male MacLow mice, suggesting that in a male gender-specific manner, resident lung M2-like macrophages drive the development of disease in this model. Studies are currently underway to determine whether 17 beta-estradiol treatment can prevent development of PAH in male MacLow mice.
Preliminary ambrisentan tadalafil combination experience
Cerrahpasa Medical Faculty, Pulmonary Disease and Cardiology Departments Istanbul University, Instanbul, Turkey

As presented above, the combination of A + T in the treatment of PAH shows improvement in certain important parameters. Although statistical analysis could not be performed due to the small number of patients, these findings give the impression of the results of the clinical study are translated to real-life experience.
The obesity paradox in pulmonary hypertension: observations from a Nationwide Inpatient Sample database 2003–2011
Department of Internal Medicine, University of Tennessee Health Science Center, Memphis, TN, USA; Division of Cardiovascular Medicine, Department of Internal Medicine, St. Luke University Medical Center, Bethlehem, PA, USA; Division of Cardiovascular Medicine, Department of Internal Medicine, Lehigh Valley Medical Center, Allentown, PA, USA; Division of Cardiovascular Medicine, Department of Internal Medicine, John Ochsner Heart and Vascular Institute, New Orleans, LA, USA
Many studies and meta-analyses have reported lower mortality rates in obese patients than non-obese patients with various cardiovascular disorders, a phenomenon known as the “obesity paradox.” However, very few data exist regarding the impact of obesity status and prognosis in patients with pulmonary hypertension (PH). We used the 2003–2011 National Inpatient Sample databases to identify all patients aged ≥18 years with a primary diagnosis of PH (both primary and secondary PH). Univariate and multivariable analyses were used to compare in-hospital mortality, length of stay, and hospitalization cost between obese and non-obese PH patients. In total, 140,382 patients with a primary diagnosis of PH were identified. The overall PH group had a mean age of 63.2 ± 16.3 years, were predominantly women (67.3%), white (67.7%), Medicare insurers (59.3%), and lean, with obesity present in 22.8% patients. Obese patients had higher prevalence of diabetes, hypertension, dyslipidemia, heart failure obstructive sleep apnea, and chronic pulmonary disease than non-obese patients. Additionally, they were less likely to have atrial fibrillation, prior myocardial infarction, carotid artery disease, peripheral vascular disease, valvular heart disease, acquired immunodeficiency disease, and connective tissue disorders . In the overall cohort of PH patients, obesity was associated with lower in-hospital mortality (2.7% vs. 5.5%; unadjusted odds ratio [OR] = 0.47, 95% confidence interval [CI] = 0.43–0.50, P < 0.001). This unadjusted mortality difference was reduced markedly, but remained significant statistically, after risk adjustment for demographics, hospital characteristics, and baseline co-morbidities (adjusted OR = 0.77, 95% CI = 0.70–0.84, P < 0.001). Obesity was also associated with shorter mean length of hospital stay (3.5 vs. 4.5 days, P < 0.001) and lower average hospitalization charges. In this nationwide cohort of patients with PH, we observed significantly lower risk adjusted in-hospital mortality in obese patients, suggesting that the obesity paradox also applies to PH patients.
Transcriptional repressive and activation programs regulate alveolar epithelial cell fate and plasticity for lung alveolar generation
Division of Pediatric Cardiology, The Children’s Hospital of Philadelphia, PA, USA; Department of Medicine, Department of Cell and Developmental Biology, Penn Cardiovascular Institute, and Penn Center for Pulmonary Biology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
Generation and regeneration of the neonatal lung is critical in the treatment of pediatric pulmonary hypertension (PH) associated with developmental lung disease. In order to generate diverse lung lineages, multipotent lung progenitor cells must simultaneously express genes for multiple specific lineages. For progenitor cell commitment to occur, cell-specific transcriptional networks must be activated, and simultaneously, alternative fate gene expression must also be switched off, processes that are currently poorly understood for alveolar epithelial cell fate. Formation of the alveolus, the functional unit of gas exchange in the lung, entails appropriate differentiation of the alveolar epithelium into a type 1 (AT1) or type 2 (AT2) alveolar epithelial cell and maturation of a juxtaposed capillary plexus. Of note, disruption of this process, results in developmental lung disease such as bronchopulmonary dysplasia, a disease frequently associated with PH. Therefore, an understanding of the mechanisms of alveolar epithelial cell fate may provide critical insight into reinitiating lung growth in developmental lung disease and PH. Recently, we prospectively identified a pool of AT2 cells that primarily contributes to lung growth and regeneration. Separately, we have also determined that commitment to the alveolar epithelial lineage occurs much earlier in development than previously recognized. Furthermore, we have demonstrated that this cell fate is regulated by a transcriptional repressor, called Hopx, and the activating transcription factor, β-catenin. These preliminary results suggest a new paradigm for lung alveolar development, where alveolar epithelial cells are specified in the early stages of lung development. In addition, these findings suggest an additional layer of regulation in switching off alternative fate gene expression in cell commitment. Unraveling these mechanisms will provide critical insight into reinitiating lung growth in developmental lung disease. Similarly, understanding the regulation and coordination of alveolar epithelial plasticity will help drive new potential therapies in neonatal and adult lung regeneration.