
Abstract
Right ventricular failure (RVF) is the most important prognostic factor for both morbidity and mortality in pulmonary arterial hypertension (PAH), but also occurs in numerous other common diseases and conditions, including left ventricle dysfunction. RVF remains understudied compared with left ventricular failure (LVF). However, right and left ventricles have many differences at the morphological level or the embryologic origin, and respond differently to pressure overload. Therefore, knowledge from the left ventricle cannot be extrapolated to the right ventricle. Few studies have focused on the right ventricle and have permitted to increase our knowledge on the right ventricular-specific mechanisms driving decompensation. Here we review basic principles such as mechanisms accounting for right ventricle hypertrophy, dysfunction, and transition toward failure, with a focus on epigenetics, inflammatory, and metabolic processes.
Introduction
Pulmonary hypertension (PH) is a pathophysiological disease defined by a mean pulmonary artery (PA) pressure exceeding the upper limits of normal (i.e. 25 mmHg at rest). 1 Considering the diverse causes and mechanisms contributing to its pathogenesis, PH has been classified by the World Health Organization (WHO) into five categories depending on common clinical parameters, potential etiological mechanisms, as well as pathological, pathophysiological, and therapeutic characteristics. 2 One of these categories corresponds to pulmonary arterial hypertension (PAH), a proliferative vascular remodeling disease that leads to progressive obliteration of the pulmonary arterial lumen, a pressure overload-driven failure of the right ventricle (RV) and premature death. PAH is characterized by the proliferation and reduced apoptosis of pulmonary artery smooth muscle cells (PASMCs), inflammation, and vasoconstriction, which lead to an increased in pulmonary vascular resistance and ultimately to right heart failure. It is now known that the most important prognostic factor for both morbidity and mortality in PAH is not the degree of PA pressure elevation, but its consequences on the RV.1,3–5
The RV is a critical component of the cardiovascular system. Impairment of its functions occurs in numerous common diseases and conditions other than PAH, including heart failure with preserved6,7 or reduced8–11 ejection fraction,12–22 valvular heart disease,23–30 chronic respiratory disease such as pulmonary fibrosis31–36 and chronic obstructive pulmonary disease (COPD),37–41 and chronic thromboembolic PH (CTEPH), where right ventricular failure (RVF) is an important and independent predictor of patients’ outcomes. In PAH, RVF is the most important prognostic factor for both morbidity and mortality1,3,4 and is the cause of death of approximately 70% of PAH patients. 42 Indeed, the increase in pulmonary resistance, which is the distinctive characteristic of PAH, poorly predicts patient outcomes. Thus, the survival in PH is determined by the condition of the RV rather than the degree of the afterload increase.42–44 Unfortunately, the RV is less able to adapt to pressure overload compared to the LV. A patient with systemic hypertension can develop adaptive LV hypertrophy and live asymptomatically for decades. Conversely, a patient with increases in afterload due to PAH generally lead to RVF and die within three years from the time of diagnosis when left untreated, 1 although the RV compensatory response is highly variable among individuals.45,46
Thus, RV and LV responses to pressure overload are not similar, in addition to differences that appear at the level of the morphology or the embryologic origin. 47 Therefore, knowledge from the LV cannot be extrapolated to the RV.3,4 Despite that, only few studies have specifically focused on the RV without applying concepts from the LV, and it is not surprising that the concept of RV-specific therapies remains embryonic.
Here we review the basic principles of mechanisms accounting for RV hypertrophy (RVH), dysfunction, and transition toward the failure as well as potential RV-specific therapies. However, it remains to be established what are the cellular, molecular, and metabolic insights that differ between LV and RV, and what makes the RV so susceptible to fail in response to pressure overload. Therefore, it is now critical to open new avenues of investigation, to enhance our understanding of RVF, our ability to predict it, and to develop RV-specific therapies to eventually be able to reverse RVF.
Disease spectrum
The first manifestation of RVH is mainly an adaptive response, also called compensated state, as the RV is able to balance the increase in pulmonary pressure. This compensatory phase is significantly shorter in the RV compared with the LV, and explains why the mortality in PAH occurs relatively rapidly after disease onset compared with patients with systemic hypertension. As the disease progresses, the adaptive response becomes maladaptive and unable to compensate for the rise in pressure, eventually resulting in RV dilatation and RVF. 3
Adaptive hypertrophy
As described by Laplace in 1709, hypertrophy is an adaptive mechanism that follows the increase of intraluminal pressure to decrease the stress applied on the wall. Thickening of the wall and reduction of the internal radius of the chamber minimize the increase in wall tension required to withstand the increase in internal pressure, favoring RV compensation. In general, however, the RV adapts better to volume rather than pressure overload and to chronic rather than acute stressors. 4 Cardiac hypertrophy in response to pressure overload has been attributed to an increase of the cross-sectional area of myocytes48–51 due to the parallel addition of sarcomeres in myofibrils, 52 while their length is decreased. This differs from the response to volume overload where sarcomeres are added in series, resulting in an increase in the length without changes in the cross-sectional area of the cardiomyocytes.52–55
Mechanisms of mechano-transduction are strongly implicated in pressure overload-induced hypertrophy in the RV as in the LV. The mechanical forces created by an increase in afterload are sensed by cardiac cells and converted into biochemical or electrical signals that initiate structural and functional changes in cells and tissues. Integrins and focal adhesion complexes that are linked to both the extracellular matrix (ECM) and the cytoskeleton are specialized in the detection of extracellular stress and participate actively in signal mechano-transduction in ventricular hypertrophy56–58 by interacting with many signaling molecules, such as the focal adhesion kinase 59 and integrin-linked kinase 60 such as Src. This results in the activation of signaling pathways that are important for cardiomyocytes growth such as STAT3, ERK, and JNK. 61 Cardiomyocytes growth needs to be paralleled by ECM synthesis. The matrix scaffold of the heart is predominantly composed of collagen and small amounts of fibronectin, laminin, and elastin. 3 Pressure overload has been associated with the re-expression of fibronectin 48 and increased levels of β1-integrin. 62
Dysfunction in contractility
The α-myosin heavy chain (α-MHC) and the β-myosin heavy chain (β-MHC) isoforms are expressed in cardiomyocytes as critical components of the fundamental contractile unit, the sarcomere. As in LV hypertrophy (LVH), a proportional decrease in α-MHC and an increase in β-MHC have been observed in adult RVH. 63 In the normal adult human RV, the α-MHC isoform makes up to approximately 23–34% of the total MHC, and the β-MHC the remainder. Although the β-MHC is already the predominant isoform in human, it has been suggested that even a small shift can significantly alter cardiomyocyte power output. 64 The β-MHC is actually characterized by a lower adenosine triphosphatase activity and a lower filament sliding velocity compared to the α-MHC, two properties that contribute to cardiomyocyte contractile dysfunction. 63 On the other hand, the β-MHC can generate cross-bridge forces with less consumption of energy65–67 and can be an adaptive response in order to preserve energy. In the LV, miRNA-208 has been identified as one of the key regulators of this contractile protein switch, 68 but it remains to be established if this is the case in the RV as well. MHC gene expression is regulated in part by the thyroid hormone signaling. Interestingly, thyroid diseases are recognized as a predisposing condition for PH. 69 The Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) demonstrated that approximately 20% of patients have thyroid disease,70,71 predominantly hypothyroidism. 3,3′,5-Triiodothyronine (T3) in myocytes regulates the expression of many other proteins in addition to α- and β-MHC, such as voltage-gated potassium channels and calcium transport/regulatory proteins: actually, all of them are implicated in the regulation of myocytes’ contractility.72–75 Thus, impaired T3 signaling may be a key factor in contractile dysfunctions observed in RVF. In keeping with this, a tissue-specific regulation of T3 signaling seems to occur in RV during hypertrophy processes. The type 3 deiodinase (D3) converts T3 in inactive metabolites and reverses T3 (rT3) and 3,3′-diiodothyronine (T2) via inner-ring deiodination. It has been demonstrated that D3 is specifically induced in the RV wall during RVH in a hypoxia-inducible factor-dependent manner, reducing locally T3 content and action. 76
RV respond sub-optimally
Since they undergo the same type of stress (increase in afterload), common mechanisms must occur in both ventricles during the process of compensation, such as matrix remodeling, metabolic changes, and actin cytoskeletal alterations. Nonetheless, there are processes that diverge between the RV and LV in order to explain the RV suboptimal response. Some evidence can be found by carefully looking at the embryologic origin of the two chambers, which involve different pathways and expression of different proteins.
Evidence found in the embryologic origin
One of the featured theories enounced to answer the question of what are the mechanisms driving heart hypertrophy and failure is the possible re-expression of a fetal gene program. Contrary to the initial thoughts, the RV and LV do not originate from the same region of the primary heart field (or cardiac crescent) (Fig. 1a).
77
In fact, the two ventricles are already separated in the early stages of heart development, suggesting that if a fetal program is re-expressed in the stressed RV, it should not be the same as the one stimulated in the stressed LV. Cell tracing experiments have demonstrated that RV progenitors (as well as outflow tract and septum progenitors) are located in the widest region of the cardiac crescent called the anterior heart field,
78
while LV progenitors are present in the narrower region called the posterior mesoderm.
79
Those progenitors migrate to generate the primitive heart tube that is patterned to form along the anterior–posterior axis, the aortic sac, the outflow tract, RV, LV, and atria. At this stage of development, the identity of heart chambers is evident morphologically (Fig. 1d), but also with the presence of different contractile properties and the expression of specific genes.80–82 For example, expression of the LIM homeodomain transcription factor islet-1 (Isl-1) is required in progenitors of the anterior heart field to give rise to the outflow tract and a majority of cells of the RV and the atria, and some cells within the LV. At the opposite, progenitors that will give rise to the majority of cells of the LV and some atrial cells do not express Isl-1.
83
The role of Isl-1 in RVH/RVF has not yet been studied and may offer some important clues. The cardiac Isl-1-interacting protein (CIP) has been recently identified and characterized as a novel cardiac-specific nuclear protein that, as suggested by its nomenclature, interacts with Isl-1 and serves as a cofactor of Isl-1 to regulate its transcriptional activity. CIP has been described as repressed in LVH secondary to transverse aortic constriction in mice and, most importantly, had an inhibitory effect on cardiomyocytes hypertrophy.
84
It has also been described that the MADS domain transcription factor Mef2c is required for the proper formation of the cardiac outflow tract and RV, and that Mef2c is dependent on GATA and Isl-1 direct binding.
85
Interestingly, the predominant member of the MEF2 family of protein expressed in postnatal cardiac muscle is MEF2a. It has been shown that mice expressing a MEF2a transgenic deficiency had a susceptibility to die suddenly within the first week of life with pronounced dilation of the RV, myofibrillar fragmentation, mitochondrial disorganization, and activation of a fetal cardiac gene program.
86
Recently, a study established that MEF2a could be regulated by FAK (focal adhesion kinase) and conditional inactivation of FAK in the costameres of the embryonic heart compromised MEF2a expression. This impaired MEF2a expression in FAK knock-out (KO) hearts led again to death in the embryonic stage for the majority of mice, and the small fraction of mice able to survive to adulthood developed spontaneous eccentric RVF.
87
This suggests that FAK/MEF2a expression may be critical for the transition from RVH to RVF. Finally, Mef2 proteins appeared to be recruited by GATA4 for a synergic activation of atrial natriuretic peptide, as well as cardiac actin and MHC expressions,
88
which can also contribute to the contractile dysfunction seen in RVH/RVF.
Human heart development. The determination and differentiation patterns are mentioned at each concerned stage. (a) Formation of the cardiac crescent at day 15, showing the first and second heart field (also known as anterior and posterior heart field, respectively). (b) At days 18–19, endocardial tubes appear. RV progenitors are located in the anterior heart field, whereas LV progenitors are positioned in the posterior mesoderm. (c) Fusion to form the heart tube. (d) At day 21, identification of heart future regions is morphologically possible. (e) Reorientation of heart’s anterior portion (looping) along the left/right axis of the embryo. (f) The four-chamber heart is now entirely formed. LA, left atrium; LV, left ventricle; OFT, outflow tract; RA, right atrium; RV, right ventricle.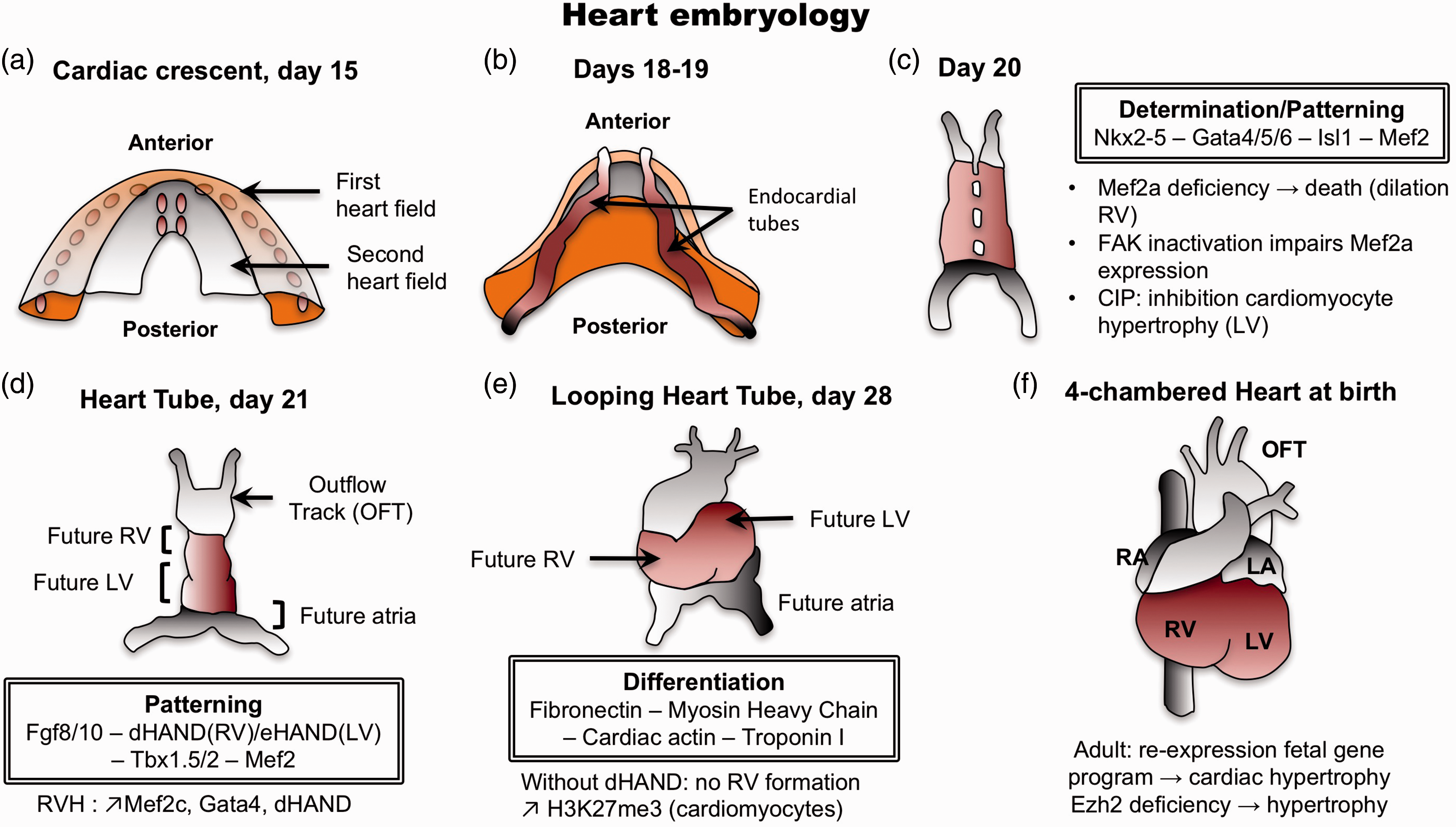
To align the presumptive structures and form the primitive heart that presages the definitive morphology, the heart tube undergoes looping to reorient its anterior portion along the left–right axis of the embryo at day 28 (Fig. 1e). The basic helix–loop–helix transcription factors dHAND and eHAND are expressed in the right and left ventricles, respectively, in a chamber-specific manner. dHAND-null mice have been shown to fail in the formation of the RV despite a correct looping, indicating that HAND genes are involved in the development of the chambers rather than controlling the looping. 89 Mef-2c, GATA-4, and dHAND expressions have been measured in the PA banding (PAB) model and found unregulated at the protein level. 90 These transcription factors with a specific role in the regulation of the RV development may also have a specific role in the regulation of RV hypertrophy and failure.
Evidence found in evolution
The emergence of an air-breathing living witnessed the need to improve circulatory systems. Thus birds, mammals, and crocodilians evolved to a four-chamber heart, with complete septation into the left and right sides, allowing the separation between pulmonary and systemic circulatory systems. However, fish, amphibian, lizard, and turtle species conserved a three-chamber heart with a single ventricle. Because the LV clearly demonstrates outstanding performances such as inflow and outflow valves that are superior to those of the RV, it has been believed that the RV has been added in order to handle the pulmonary circulation only. In the last decade, the discovery of the transcription factor Isl-1 has opened a new chapter: the morphological RV as the ancient heart. Indeed, as stated below, Isl-1 in mammals is required for the fashioning of the outflow tract and RV. Intriguingly, analysis of Isl-1 homologs expression in Xenopus laevis and Drosophila melanogaster revealed the presence of a cardiac Isl-1 progenitor pool in the developing heart despite the lack of an anatomically separated RV in adults.91,92 In keeping with these observations, it has been previously described that dHAND is the unique HAND gene expressed in the zebrafish Danio renio and driving the development of a single ventricle. 93 This suggests that lower vertebrates may be helpful in the study of RV hypertrophy and failure. But above all, as the RV is defined as the Isl-1-positive chamber proximal to the arterial trunk, it seems that the RV corresponds to the ancient heart, at least morphologically, and that the LV is the new evolutionary ventricle. Following this, and considering that the RV may have undergone several millions of years of refinement, it is maybe inappropriate to associate its poor performance during disease to its design.
Epigenetic mechanisms involved in RVF
Epigenetic processes are defined as changes in gene expression secondary to factors unrelated to changes in DNA sequence.94,95 Here we detail three epigenetic mechanisms that occur in RVF secondary to PH: DNA methylation; modification of histone proteins; and the role of specific non-coding RNAs, microRNAs (miRNAs).
DNA methylation
DNA methylation is defined as the addition of a methyl group to the carbon 5′ position of the nucleotide cytosine ring. Cytosine methylation in mammals is most commonly found in the context of the sequence 5′-CG-3′, which is also referred to as a CpG dinucleotide. The methylation occurs via DNA methyltransferases (DNMTs) and usually in silenced genes: 96 DNMTs can both repress and activate DNA transcription.97,98 In the mammalian genome, it is estimated that 70% of all CpGs are methylated. 99 Unmethylated CpG, on the other hand, are largely grouped in clusters called “CpG islands” in the 5′ regulatory region of many genes. CpG islands mark 70% of annotated mammalian promoters. 100 Also, CpG methylation is essential for proper gene expression, development, and genome stability. 101 Notably, differential methylation of CpG islands is part of the epigenetic variation found in humans.99,102
DNA methylation is highly dynamic during cardiomyocyte development and postnatal maturation: it is important for the perinatal switch in sarcomere protein isoforms and postnatal cardiomyocyte maturation and adaptation. A substantial number of CpGs are differentially methylated between newborn and adult cardiomyocytes. 103 For example, two prototypical cardiomyocyte genes, α- and β-MHC (Myh6 and Myh7), show a CpG demethylation in cardiomyocytes purified from newborns compared with adults. On the other hand, genes that are hypermethylated in cardiomyocytes after birth are involved in myocyte contraction, cardiac morphogenesis, cell differentiation, and other processes. Postnatal de novo DNA methylation is then required for the expression of several sarcomere components and is dependent on the DNA methyltransferases 3A/B. 103 Demethylated regions of adult cardiomyocytes were shown to be significantly enriched for motifs of known cardiac transcription factors, including MEF2C, GATA1-4, and others. Postnatal life is therefore accompanied by de novo methylation of fetal genes and demethylation of adult isoforms. 103
Recent studies have investigated DNA methylation in human cardiac tissue biopsies from patients with chronic heart failure of different etiologies.104,105 Both studies described distinct signatures of DNA methylation in failing versus non-failing hearts. Disease-associated differentially methylated regions were adjacent to genes involved in cardiomyocyte development, cardiac morphogenesis, and energy metabolism, indicating adaptation of DNA methylation in disease-relevant regions. 103 Therefore, it will be important to uncover the role of DNA methylation during RVH and RVF in the different cardiac cell types (e.g. fibroblasts, endothelial cells, immune cells) to determine their epigenetic contribution.
Modification of histone proteins
Histones are the building blocks of the nucleosomes and, as such, alter the exposure of the DNA to the transcription machinery. In addition to DNA methylation, there are histone modifications that alter the structure of chromatin and thus DNA transcription. 4 The histone acetylation and deacetylation are forms of post-translational modifications that lead to increased and decreased transcription of genes, respectively. The acetylation occurs via histone acetyltransferases (HATs), whereas deacetylation is accomplished through histone deacetylases (HDACs).95,96
Several reports have addressed the effects of HDAC inhibitors in models of RV remodeling with discordant results. The valproic acid has been shown to block RV cardiac hypertrophy in response to PAB and monocrotaline (MCT)-induced PH, 106 and to reduce established hypoxia-induced PH in another animal model. 107 As valproic acid has many additional effects, including regulation of ion channels, glycogen synthase kinase-3β, and mitogen-activated protein (MAP) kinase, it is still unclear if the observed effects are only due to HDAC inhibition. In contrast, another study showed that trichostatin A—a potent pan-HDAC inhibitor—and valproic acid did not prevent the development of RV hypertrophy and was associated with RV dysfunction, capillary rarefaction, fibrosis, and increased cardiomyocytes death in the PAB model. 108 Other HDAC inhibitors, specifically focused on class I HDACs, can modulate the hypoxia-induced cardiopulmonary remodeling via anti-proliferative mechanisms. 109 The role of HDACs in RV remodeling still needs to be elucidated as previous studies primarily focused on LV remodeling. 110
Ezh2 is a histone methyltransferase (HMT) that trimethylates the histone H3 at lysine 27 (H3K27me3). Ezh2 is expressed in cardiomyocytes and H3K27me3 is increased in cardiac progenitor cells during differentiation to cardiomyocytes. Mice with specific inactivation of Ezh2 by Cre-mediated recombination in the anterior heart field developed normally structured hearts that became enlarged after birth in an RV-restricted manner. 111 This suggests that the presence of Ezh2 suppresses the expression of genes that promote cardiac hypertrophy. Indeed, Ezh2 has been shown to prevent cardiac hypertrophy by repressing the homeodomain transcription factor gene Six1, as genetically reduced Six1 levels rescued the pathology of Ezh2-deficient hearts. These results indicate that epigenetic mechanisms are critical for cardiac hypertrophy, but more importantly they show that embryonic epigenetic dysregulation can predispose to adult disease and maladaptive stress response. Because RV and LV have different embryonic origins, it is possible that RV possesses an embryonic epigenetic-associated predisposition that can explain its suboptimal respond to stress-induced hypertrophy.
Non-coding RNAs: microRNAs
miRNAs are short non-coding RNAs (∼22 nt) that are involved in post-transcriptional regulation of gene expression in multicellular organisms by affecting both the stability and translation of messenger RNAs (mRNAs). miRNAs are the most studied class of non-coding RNAs and control gene expression by binding to the 39-untranslated region of mRNA, which leads to either mRNA degradation or inhibition of protein translation.94,112
In order to define the molecular insights characterizing the transition from RVH to RVF, miRNA profile of expression has been studied in the PAB model
112
in different stages, including at day 2, during the RVH stage (day 4), and when the RV failed (RVF at day 10). First of all, it has been shown that most miRNAs are similarly expressed in the RVH/RVF compared with LVH/left ventricular failure (LVF), but some miRNAs such as miR-34 a, miR-28, miR-148 a, and miR-93
112
are upregulated in RVH/RVF while they have been reported downregulated in LVH/LVF.113–115 Moreover, the transition from RVH to RVF might be characterized by the expression of miR-34 (upregulated at day 10 only), miR-21 (upregulated at day 4 and to a lesser extent at day 10), and miR-1 (mostly downregulated at day 4).
112
They also assessed the miRNA profile in the RVH hypoxia model compared with the RVF Sugen/Hypoxia (Su/Hx) model where they observed that miR-133 was downregulated in the Su/Hx RVF (Fig. 2).
116
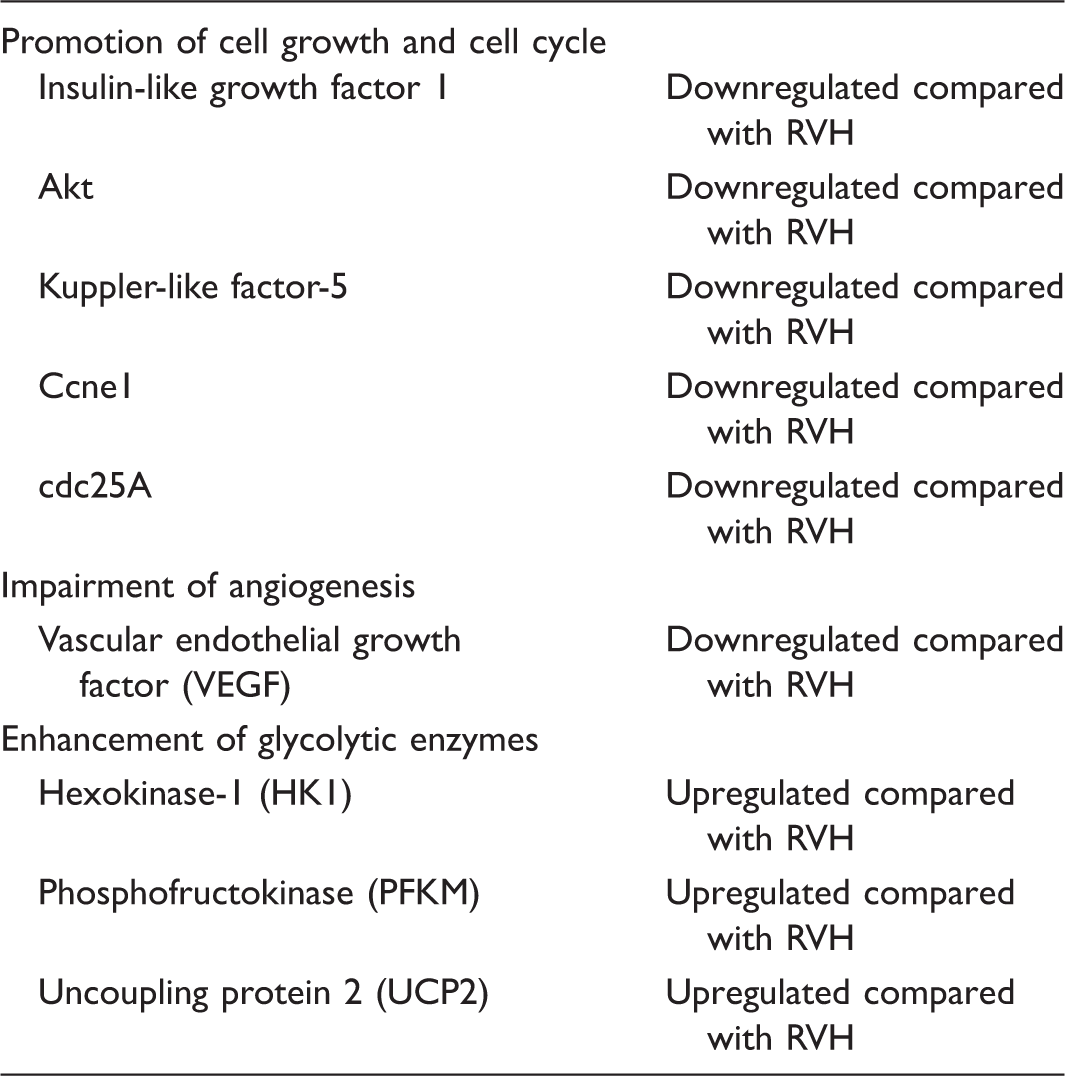
These two studies defined a molecular RVF program in which there is a loss of genes promoting cell growth and cell cycle (insulin-like growth factor-1, Akt, Kuppler-like factor-5, Ccne1, cdc25A), impairment of angiogenesis (vascular endothelial growth factor [VEGF] and Akt), and expression of genes encoding glycolytic enzymes (hexokinase-1, phosphofructokinase, uncoupling protein 2)112,116 (Table 1).
Differential miRNA expression between right ventricular hypertrophy and right ventricular failure. Genes characterizing right ventricular failure.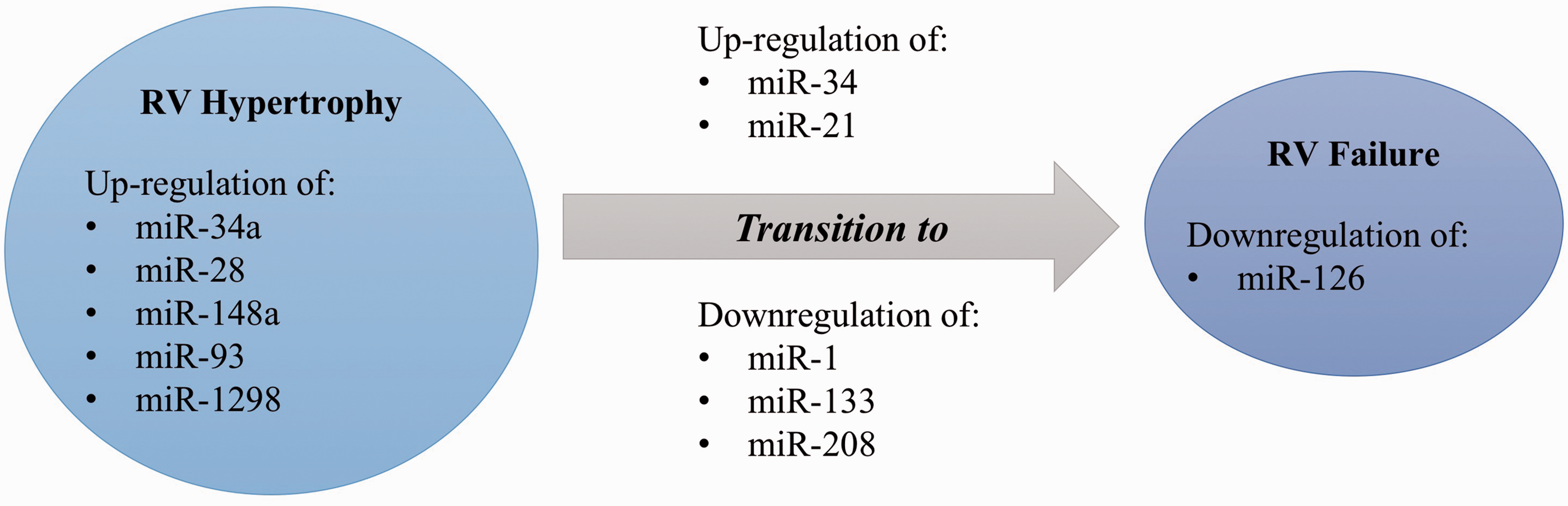
Later, the miRNA expression profile between RVH and RVF has been studied in the MCT model. In this model, RVF was defined as a state of ongoing RVH in which the RV systolic pressure decreases because of contractile failure (while the PA pressure remains elevated): the RV end-diastolic pressure increases; the cardiac output decreases; clinical evidence of heart failure (ascites, decreased appetite, and activity, > 20% weight loss) develops; and death occurs. In this model, Mef2c protein expression was sharply increased in late RVH stages, contributing to the adaptive nature of this stage as for the physiological RVH of the fetal circulation in utero, but was lost again at the RVF stage.
117
In contrast to what is known in LVH, levels of miR-208 (a myocardium-specific miRNA) were continuously decreasing as RVH progressed toward RVF
117
(Fig. 3). The subunit MED13 of the complex mediator of transcription has been identified as the miR-208 direct target gene in the heart.
68
The decreased expression of miR-208 was therefore associated with an increase in MED13 expression as well as an increased activity of its partner NCoR1, resulting in hypoacetylation of histones and a chromatin structure resistant to the transcription, allowing the repression of Mef2117 and the exit from a fetal-like compensatory phase in RVF. In this model, as in human tissues, it has also been shown that the transition from RVH to RVF is associated with a decreased in miR-126 expression. MiR-126 downregulation was associated with increased SPRED-1, leading to decreased activation of RAF (P-RAF/RAF) and MAP kinase (P-MAP/MAP), thus inhibiting the VEGF pathway and capillary density.
118
During RVF, miR-126 has been specified to contribute to a decrease in the RV vascular density, promoting the progression towards RVF.
MiRNAs and methylation patterns involved in right ventricular hypertrophy. Increase in miR-93 inhibits Sirt1 activity, which usually repress p53 known to induce apoptosis through the mitochondria. Mir-148 upregulation obstructs DNMT3b activity, and miR-1298 can both inhibit FAK and LAMB3 (Laminin subunit beta 3). Also, a diminution of miR-208 is responsible of Mef2c upregulation and inhibition of α-MHC expression. DNMT3b, DNA Methyltransferase 3 Beta; LAMB3, Laminin subunit beta 3; Sirt1, Sirtuin 1.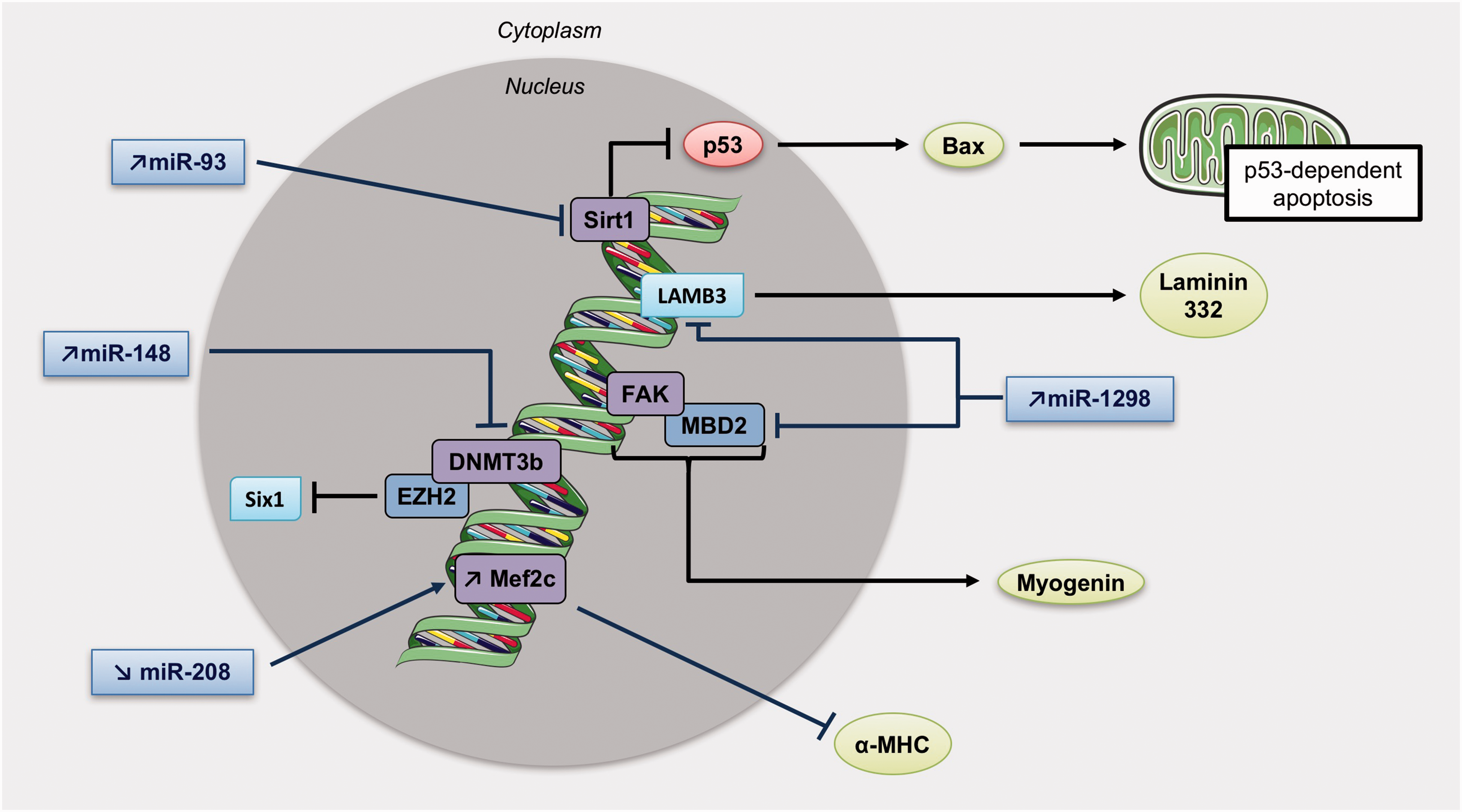
Finally, a recent study revealed that miR-1298 is increased in RVH but not in LVH. miR-1298 inhibits connexin-43 expression, 112 a gap junction protein required for maintenance of the normal cardiac rhythm, regulation of vascular tone, and endothelial function, 119 suggesting that miR-1298-mediated decrease of connexin-43 could be one of the causes of arrhythmia and impaired coronary artery function in the right heart of PH rats. 120
Inflammation in RVF
Inflammation is a complex set of interactions among soluble factors and cells that can arise in any tissue in response to traumatic, infectious, post-ischemic, toxic, or auto-immune injury. Inflammation can lead to persistent tissue damage by leucocytes, lymphocytes, or collagen. 1 Moreover, inflammation plays a significant role in the pathogenesis of PH. However, its role in RVH and RVF is still not well understood. RVF is less prevalent and occurs later in those with congenital heart disease (i.e. Eisenmenger syndrome) 121 and idiopathic PAH122,123 versus in those with scleroderma-associated PAH.121–124 suggesting that RVF is predominant in patients having an inflammatory burden. Overbeek et al. 125 confirmed that RV from scleroderma-associated PAH (n = 5) had more neutrophilic granulocytes, macrophages, and lymphocytes than in idiopathic PAH (n = 9) or controls (n = 4), whereas RV interstitial fibrosis was similar in all groups. Nonetheless, the participation of inflammation to RVF is also suspected in all types of PAH. Cardiomyocytes are believed to produce a trigger for autocrine, paracrine, and neuroendocrine signaling pathways leading to a vicious circle of RV inflammation and ischemia, leading to cardiomyocytes apoptosis and RVF. 3
Role of cytokines
Activation of cytokines may play an important role in patients with RVF. In patients with selected forms of congenital heart disease and RVF, elevated levels of tumor necrosis factor (TNF) and endotoxin were associated with more symptomatic disease. 126 Levels of pro-inflammatory cytokines like interleukin (IL)-6 and IL-1β, which production is mediated by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), 127 are linked to the severity of heart failure.128,129 In the pressure-overloaded RV, several chemokines are upregulated (CXCL10, CXCL6, CX3CL1, CCL5, CXCL16, and CCL2130) and stimulate the expression of small leucine rich-proteoglycans (SLRP), which are recognized to act as signaling molecules and directly regulate inflammatory processes.131,132
Tumor necrosis factor-alpha
Tumor necrosis factor-alpha (TNF-α) is a polypeptide hormone produced by activated monocytes/macrophages and an effector molecule in various inflammatory processes. Growing evidence has implicated TNF-α in the pathogenesis of heart failure: the healthy heart does not produce TNF-α, but the insufficient myocardium does. 133 Experimentally, it has been demonstrated that transgenic mice that chronically overexpress myocardial TNF-α develop cardiac hypertrophy, fibrosis, both left and right dilated myocardiopathy, and premature death. 134 Also, TNF-α depletion in a banding pressure-overload model improves cardiac hypertrophy and remodeling. 135 TNF-α involvement in the pathogenesis of heart failure has been also suggested by demonstrating a correlation between patients’functional class and circulating TNF-α levels. 136 TNF-α binds to TNF receptors type 1 (TNF-R1) and 2 (TNF-R2) to exert its biological functions. These receptors are significantly increased in relation to the severity of heart failure, 137 as observed in 1200 patients randomized in the VEST study, in which the levels of TNF-α-R1s and TNF-α-R2s correlated with functional classes and were significant independent predictors of mortality. 138
Similarly, a strong correlation has been established between the level of TNF-α mRNA or protein in the donor heart and the development of RVF after transplantation, which is an important problem after heart transplantation. 139 The mechanisms behind the detrimental effect of TNF-α on the RV remain poorly understood. TNF-α seems to depress myocardial contractile function through uncoupling β-adrenergic signaling 137 responsible for increasing cardiac nitric oxide and peroxinitrite140,141 and altering intracellular calcium homoeostasis. 142 In vitro, stimulation of adult human cardiomyocytes by TNF-α provoked a hypertrophic growth response. 143 It could also impair the coupling of the components of the beta-receptor-G protein system, thus leading to a negative ionotropic effect. 144 It can also enhance nitric oxide (NO) production, increasing ionotropy. 141 Also, we recently demonstrated that TNF-α may act as a second trigger along with the decrease in miR-208 to enhance MED13/NCoR1 upregulation and Mef2 suppression. 117 Therefore, the role of TNF-α in heart failure appears to be important, although it still needs to be investigated.
Interleukin-6
IL-6 is known to regulate innate and acquired immune responses 145 and is mainly produced by macrophages. 146 This cytokine has pleiotropic effects, such as stimulation of T- and B-cell differentiation and macrophage activation, 147 protection against cardiac ischemia-reperfusion injury, 148 and regulation of cardiac lipid metabolism through its effect on peroxisome proliferator-activated receptors (PPARs). 149 More specifically in the heart, IL-6 mediates fibrosis and cardiac hypertrophy, and promotes diastolic dysfunction. 150 IL-6 plasmatic levels also correlate with systolic function 151 and mortality 152 in heart failure with reduced ejection fraction. In vitro, pretreatment with IL-6 activates the phosphatidylinositol-3-kinase (PI3K)/Akt pathway and induces activity of inducible NO synthase (iNOS), which results in protection of cardiomyocytes. In pressure-overload LVH, genetic deletion of IL-6 attenuates Ca2+-calmodulin protein kinase II (CaMKII)-dependent activation of STAT3, which then attenuates hypertrophy. 151
Interleukin-1
IL-1 is a pro-inflammatory cytokine also implicated in heart failure that contains two active ligands, IL-1α and IL-1β. 153 This cytokine can be produced by cardiomyocytes themselves in response to injury 137 and its plasmatic levels are elevated in chronic heart failure. 154 IL-1β induces intracellular reactive oxygen species (ROS) production and modifies L-type Ca2+ current in cardiomyocytes, 155 but also decreases collagen synthesis in cardiac fibroblasts in vitro. 156
Toll-like receptors
Toll-like receptors (TLRs) are an essential family of pattern recognition receptors (PRRs) that triggers innate immune responses. 157 This family comprises ten members in humans (TLR1–TLR10) and each member recognizes a specific pathogen-associated molecular pattern (PAMP). 158 Following the recognition of PAMPs, TLRs recruit Toll/IL-1 receptor (TIR) domain-containing adaptor proteins such as TRIF and MyD88, which initiate signal transduction pathways that end in the activation of NF-κB, interferon regulatory factors (IRFs), or MAP kinases. This activation aims to regulate the expression of chemokines, cytokines, and type I interferons (IFNs) that protect the host from microbial infection. 159 Then, there are two TLRs-signaling pathways: TRIF-dependent 158 and MyD88-dependent pathways. 159
TLR2 and TLR4 are mostly expressed in cardiomyocytes, 160 with TLR4 being upregulated in the failing human heart161,162 and TLR2 playing a role in the cardiac adaptive response to pressure overload. 163 TLR4 acts as a signal-transducing receptor for lipopolysaccharide, while TLR2 distinguishes gram-positive bacterial lipoproteins and peptidoglycans. 164 Both TLR2 and TLR4 can activate NF-κB in order to recruit inflammatory cytokines in the myocardial tissue. 165 In addition, Boyd et al. 166 demonstrated that TLR3, TLR5, TLR7, and TLR9 are constitutively expressed in cardiomyocyte cell lines and murine heart tissue. Interestingly, TLR9 has been associated with cardiac inflammation and heart failure.167,168 A recent experimental study of LVF in mice generated a novel concept of a TLR9-dependent failure component. 169 In this model of chronic severe LV pressure overload, mitochondrial DNA from damaged heart cells generated a cardiac inflammatory response via TLR9, resulting in myocarditis and dilated cardiomyopathy. 169
Animal studies
Maybe one of the best models to investigate inflammation in RVF is the PAB model. PAB is usually performed by placing a suture, clip, or inflatable ring around the main pulmonary trunk at proximity from the RV, thus representing a “pressure overload model.” This model preserves the pulmonary vasculature so that RV remodeling, and its potential for reversal, is independent from the changes in pulmonary vasculature. 170 The banding can go to 21 days and RVH becomes maximal at 14 days.171–173 This model causes RVH and depression of myocardial contractility. 171 Dewatche et al. 174 observed that persistent RVF following PAB was associated with increased myocardial expression of IL-1β, IL-6, monocyte chemoattractant protein 1, pro-inflammatory IL-6/IL-10, concomitantly with neutrophil and macrophage infiltration; expression of IL-33 decreased, whereas macrophage inflammatory protein-1α expression remained unchanged. Thereby, they concluded that an acute afterload-induced RVF is associated with the activation of inflammatory processes. 174 It has also been shown in mice that levels of IL-1, IL-6, granulocyte colony-stimulating factor (G-CSF), and monocytes induced by gamma interferon (MIG) were increased after PAB. 175
Metabolic changes occurring during RVH and RVF
Switch toward glycolysis
Fatty acid oxidation (FAO) is the major source of ATP production (60–90%) in a normal adult heart, whereas glycolysis (GLY) or glucose oxidation (GO) are considered a secondary source, accounting for 10–40% of energy production.
176
Changes in cardiac metabolism have long been recognized as problematic in chronic LVF.177,178 Akin to LVF, it has been postulated that RVH is characterized by abnormal energy metabolism and a certain degree of metabolic remodeling.179,180 Studies in Su/Hx and MCT rat models have demonstrated that RVH exhibits an increased expression of glycolysis-related genes
116
and increased enzymatic glycolysis rate,
181
suggesting that the primary energy source becomes GLY (which takes place in the cytoplasm) as opposed to the FAO (which takes place in the mitochondria) (Fig. 4).
182
RV mitochondria become more hyperpolarized.
183
There is also evidence of decreased ROS production and activation of nuclear factor of activated T-cells (NFAT) in cardiomyocytes from compensated RV. In addition, this remodeling appears to promote hypoxia inducible factor (HIF)-1α activation and to stimulate angiogenesis, since increased blood/oxygen supply is needed to support the growing myocardial mass.
Metabolic mitochondrial switch. In right ventricular hypertrophy, the primary energy source becomes glycolysis, as opposed to the fatty acid oxidation. Some miRNAs interact with metabolism enzymes: an increase of miR-34 a can repress the activity of the LDH, miR-93 can block Mgst1 activity, and miR-28 prevent mitochondrial aldehyde dehydrogenase activity. ALDH2, mitochondrial aldehyde dehydrogenase; LDH, lactate dehydrogenase; Mgst1, microsomal glutathione S-transferase 1.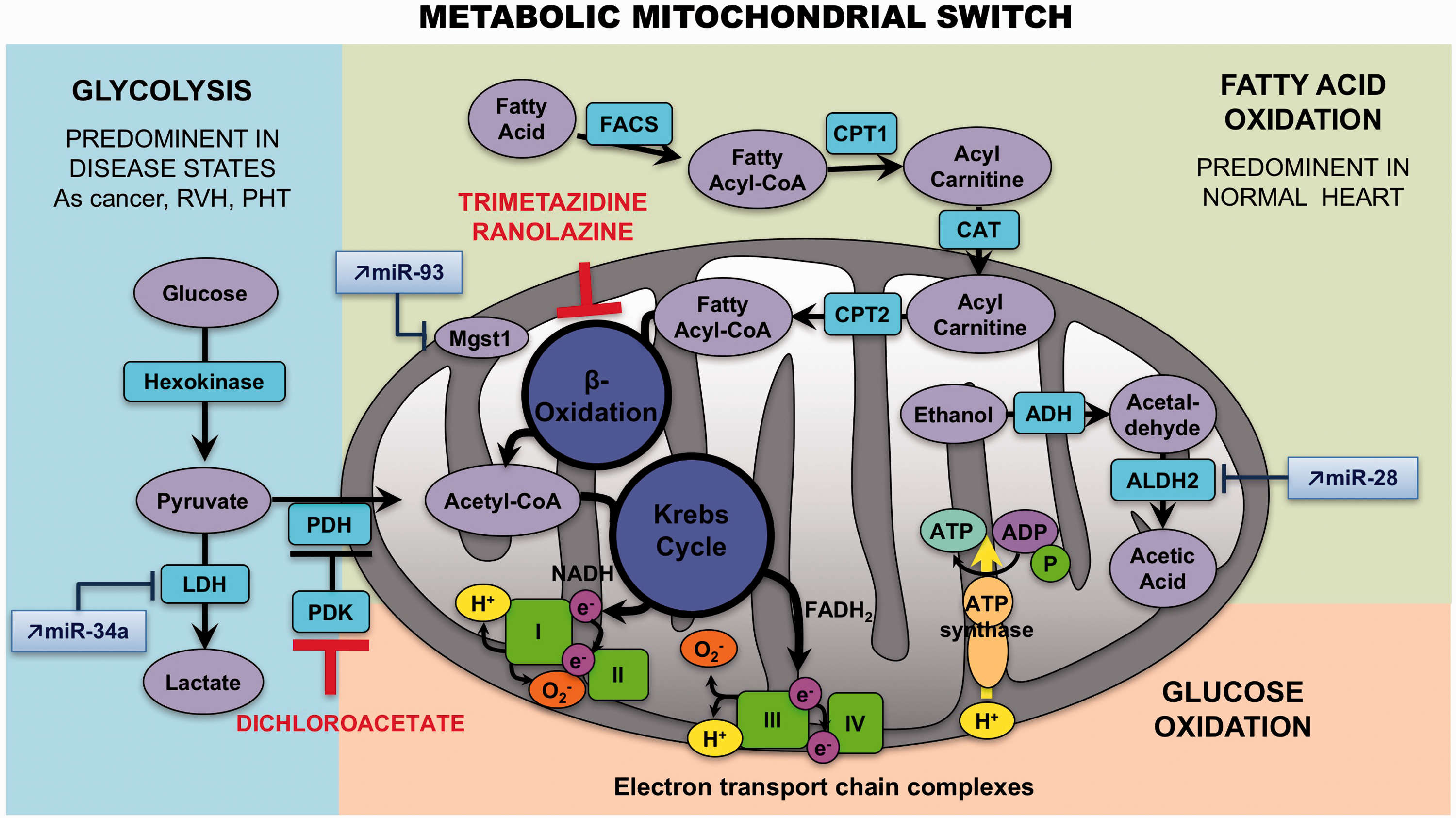
Positron emission tomography (PET) studies have shown increased accumulation of the radioisotope-labeled glucose analogue 18F-2-deoxy-2-fluoro-D-glucose (18-FDG) in the RV of PAH patients,184–186 confirming the results found in animal models. Glucose transporters (GLUTs) authorize the entrance of 18F-FDG, which will be phosphorylated by hexokinase (HK) to 18F-FDG-6-P. Then, in the cell, it is impossible for the 18F-FDG-6-P to be metabolized via the glycolytic pathway, and because of its polar nature, becomes confined. Since this metabolic remodeling can be detected and quantified in vivo by FDG-PET and is restricted to the RV in PAH, RV-specific metabolic targeting therapies might be feasible. However, 18-FDG uptake studies have multiple limitations and the physiological interpretation is not straightforward (i.e. 18-FDG uptake studies do not directly measure glycolysis). RV oxygen consumption can also be measured by [11C]-acetate PET scanning. 187 However, such studies are limited to a few specialized centers and are difficult to perform. 185
Recent evidence suggests that this potentially adaptive switch appears to be reversed in RVF, proposing that the mitochondrial suppression was a selective and reversible event in RVH. The increase in glucose uptake (measured by PET imaging) and the activation of HIF-1α as well as the mitochondrial hyperpolarization are lost at this point. 188 The trigger for the loss of this adoptive molecular program is unknown but it seems associated with the loss of the mitochondrial remodeling. The metabolic regulator dichloroacetate (DCA) has been shown to enhance oxidative phosphorylation through inhibition of the mitochondrial pyruvate dehydrogenase kinase, 189 restoring electrical remodeling in RVH181 and K+ channel expression/function. 190
On the other hand, little is known about the role of FAO in RVH and RVF. Multiple studies have shown that the rate of FAO is preserved or increased in LVH, and that it decreases only during the progression toward failure. 191 Similar results have been reported in rats with PAB-induced RVH, which exhibit high rates of FAO. 192 Due to the reciprocal relationship between FAO and GO, called the Randle cycle, inhibiting FAO might increase GO 193 and enhance metabolic efficiency (FAO uses 12% more oxygen than GO to generate the same amount of ATP). 194 Partial inhibitors of FAO (pFOXi) decreased FAO and restored pyruvate dehydrogenase (PDH) activity and GO in PAB, thereby increasing ATP levels. Fang et al. 192 demonstrated that pFOXi such as trimetazidine and ranolazine restore PDH activity and GO in PAB, thereby increasing ATP levels, but also increase cardiac output and exercise capacity. Chronic administration of ranolazine reduces RVH and RV collagen deposition in MCT-induced PH and also improves PH. 195 Besides, it diminishes levels of B-type natriuretic peptide and prevents ventricular arrhythmias. 195 Trimetazidine inhibits apoptosis by increasing miR-21 expression in cardiomyocytes during hypoxia-reperfusion injury 196 and reduces cardiac fibrosis by diminishing collagen accumulation and ROS production. 197
Finally, glutaminolysis has been shown to be amplified in MCT induced-RVH. The use of a glutamine antagonist (6-diazo-5-oxo-l-norleucine or DON) 198 caused an increase in GO in this model, restored PDH activity, and reduced RVH. Coherent with the increased glutaminolysis, expressions of glutamine transporters (SLC1A5 and SLC7A5) and mitochondrial malic enzyme were improved in the RV. 198
Alterations in mitochondrial structure and dynamics
There is evidence to suggest important alterations of mitochondrial structure and dynamics during RVH and RVF. First, expressions of critical transcription factors involved in the regulation of mitochondrial biogenesis, including the mitochondrial transcription factor A (TFAm) and the PPAR-γ coactivator-1α (PGC-1α), have been described significantly downregulated in RVF. 199 In accordance, RVF has also been described as exhibiting a net loss in the mitochondrial number per gram of RV tissue, 200 the remaining mitochondria having an abnormal ultrastructure on electron microscopy. 200 Another study showed in a PH animal model that the hypertrophied RV presents a diminution in mRNA expression of mitochondrial markers like sirtuin1 (SIRT1), PGC-1α, nuclear respiratory factor 1 (NRF1), and citrate-synthase. 199
It has also been exposed that the dysregulation of mitochondrial dynamics occurs in RVH.
Increased levels of mitophagy have been suggested in PAB-induced RVH with enhanced expression of autophagy/mitophagy marker Light Chain 3 (LC3)A/B and p62, 201 whereas mitochondria biogenesis may be impaired because of the decreased expression of PGC-1α. 199
Angiogenesis
The growing RV needs to be neo-vascularized in order to be adequately supplied and nourished, and it seems to be the case early during the progression of the disease. However, there is some evidence that the hypertrophied RV becomes relatively ischemic, 202 not because of coronary disease, but potentially because of suppressed angiogenesis. Also, it has been described that while exposed to similar degrees of afterload, 3 the RV has less susceptibility to fail in the PAB rat model3,203,204 compared with PH rat models (MCT or Sugen). This increased susceptibility to failure has been associated to a dysfunctional angiogenic signaling. 3 This raises the intriguing hypothesis that the trigger for RVF may be an inability to sustain angiogenesis as the myocardial mass increases. Because angiogenesis is regulated by variations in O2 tension and metabolic factors, 205 the use of metabolic modulators for the treatment of RVF in PAH is of potential interest. However, their efficiency could be limited by underlying mitochondrial abnormalities.
Conclusion
RVF is now a priority for preclinical and translational research in PAH, as well as numerous conditions for which RV function independently predicts morbidity and mortality. It is critical to improve our knowledge on the mechanisms underlying the transition from compensated RVH to maladaptive RVF in order to improve our ability to predict it and develop RV-specific therapies. The pathophysiology and pathobiology of RVF has to be approached in a comprehensive and specific manner in which its embryonic origin, its response to overload, its plasticity, and its relation with the pulmonary circulation are analyzed, avoiding systematic extrapolation of findings from the LV.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.