
Abstract
Bone morphogenetic protein (BMP) signaling regulates vascular smooth muscle maturation, endothelial cell proliferation, and tube formation. The endogenous BMP antagonist Follistatin-like 1 (Fstl1) is highly expressed in pulmonary vascular endothelium of the developing mouse lung, suggesting a role in pulmonary vascular formation and vascular homeostasis. The aim of this study was to investigate the role of Fstl1 in the pulmonary vascular endothelium. To this aim, Fstl1 was conditionally deleted from endothelial and endothelial-derived cells using Tie2-cre driven Fstl1-KO mice (Fstl1-eKO mice). Endothelial-specific Fstl1 deletion was postnatally lethal, as ∼70% of Fstl1-eKO mice died at three weeks after birth. Deletion of Fstl1 from endothelium resulted in a reduction of right ventricular output at three weeks after birth compared with controls. This was associated with pulmonary vascular remodeling, as the percentage of actin-positive small pulmonary vessels was increased at three weeks in Fstl1-eKO mice compared with controls. Endothelial deletion of Fstl1 resulted in activation of Smad1/5/8 signaling and increased BMP/Smad-regulated gene expression of Jagged1, Endoglin, and Gata2 at one week after birth compared with controls. In addition, potent vasoconstrictor Endothelin-1, the expression of which is driven by Gata2, was increased in expression, both on the mRNA and protein levels, at one week after birth compared with controls. At three weeks, Jagged1 was reduced in the Fstl1-eKO mice whereas Endoglin and Endothelin-1 were unchanged. In conclusion, loss of endothelial Fstl1 in the lung is associated with elevated BMP-regulated genes, impaired small pulmonary vascular remodeling, and decreased right ventricular output.
Pulmonary vascular development and maturation is a tightly controlled process that is orchestrated by multiple signaling pathways, including bone morphogenetic protein (BMP), which control spatio-temporal gene expression patterns. BMP signaling is important in embryonic vascular development and adult vascular homeostasis.1,2 Dysregulation of BMP signaling has been strongly associated with the pathogenesis of hereditary vascular diseases, including familial pulmonary arterial hypertension (PAH), hereditary hemorrhagic telangiectasia, atherosclerosis, and cerebral cavernous malformation.3–11 BMP signaling regulates vascular smooth muscle maturation, endothelial cell proliferation, and tube formation.9,12 Tight spatio-temporal control of BMP signaling and biological availability is therefore crucial for the normal development of vascular structure and function, including in the lung.
BMP ligands elicit their activity by binding to type I BMP receptors (ALK1, 2, 3, and 6) and by recruiting type II BMP receptors (BMPRII, ACVRIIA, and ACVRIIB). After forming a heterotetrameric complex, the signal propagates to the nucleus by specifically phosphorylating the Smad proteins Smad1/5/8. Phosphorylated Smad1/5/8 proteins form a complex with Smad4, which results in accumulation of this complex in the nucleus. This directly and indirectly influences transcription of target genes, among which are Endoglin, Gata2, and Jagged1. 13
Follistatin-like 1 (Fstl1) is an endogenous BMP antagonist that is highly expressed in pulmonary vascular endothelium of the developing mouse lung, suggesting a role in pulmonary vasculature development.14,15 Fstl1 is important for normal lung development. In fact, multiple organ defects were observed in whole-body knockout of Fstl1 (Fstl1-KO).15,16 Fstl1-KO mice showed increased thickness of alveolar walls and increased numbers of immature cuboidal alveolar epithelial cells in the lung.15,16 In addition, Fstl1-KO mice exhibited impaired tracheal cartilage formation, alveolar maturation, and lung morphogenesis, resulting in respiratory failure and in postnatal lethality in mice.15,16 Major defects in cardiac and skeletal development were also reported in Fstl1-KO mice.15,16
Given the complex nature of BMP signaling and the predominant endothelial expression of the BMP antagonist Fstl1 in the developing lung, this study aimed to explore the role of endothelial Fstl1 signaling in pulmonary vascular development using Tie2-cre mediated endothelial-specific deletion of Fstl1 in vivo. Specifically, we aimed to investigate the impact of Fstl1 deletion on the morphology of the pulmonary vasculature, on gene expression associated with BMP signaling, and its implication on pulmonary function.
Methods
Antibodies and reagents
Polyclonal rabbit anti-Fstl1 (#HPA035251), polyclonal rabbit anti-Jagged1 (#HPA021555), and monoclonal mouse anti-β-actin (#A5441) were purchased from Sigma Aldrich (St. Louis, MO, USA). Polyclonal rabbit anti-α-smooth muscle actin (α-SMA, #AB5694) and polyclonal rabbit anti-CD31 (#AB28364) were obtained from Abcam (Cambridge, UK). Polyclonal goat anti-mouse Endoglin (#AF1320) was procured from R&D Systems (Oxford, UK). Polyclonal rabbit anti-phospho-Smad1/5/8 (#AB3848) and monoclonal mouse anti-Troponin I (TnI) (#MAB16910) were obtained from Millipore (Molsheim, France) and polyclonal rabbit anti-Smad1 (#9743) from Cell Signaling (Danvers, MA, USA). Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG (#711-035-152), donkey anti-goat IgG (#705-035-003), and donkey anti-mouse IgG (#715-035-150) were purchased from Jackson Immunoresearch (West Grove, PA, USA). Alexa Fluor 647 donkey anti-mouse IgG (#A-31571) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals were of analytical grade.
Tie-2cre mediated endothelial-specific deletion of Fstl1
Conditional deletion of Fstl1 from vascular endothelial and endothelial-derived cells was achieved by crossing floxed homozygous Fstl1fl/fl mice with double heterozygous mice carrying one Fstl1 knockout allele and the Tie2-cre cassette (Fstl1WT/KO Tie2-cre) on a FVB background.15,17 Within this cross, endothelial-specific knockout mice (Fstl1fl/KO Tie2-cre) and littermate controls (Fstl1fl/WT Tie2-cre, Fstl1fl/KO, and Fstl1fl/WT) were generated. Fstl1fl/KO Tie2-cre mice are denoted as Fstl1-eKO and their littermate controls are denoted as controls. The breeding lines were maintained in the animal facility of the University of Amsterdam. All experimental procedures complied with institutional and national ethical guidelines regarding animal experimentation. Mice were sacrificed at one and three weeks after birth (one week: n = 12 Fstl1-eKO mice and n = 16 controls; three weeks: n = 11 Fstl1-eKO mice and n = 30 controls). The lung tissue was collected. Lung lobes (right superior, middle, and inferior lobes) were inflated with 50% Tissue-Tek (Sakura Finetek; Alphen-aan-den-Rijn, The Netherlands) in 0.9% NaCl (Braun; Kronberg, Germany) and fixed with 10% v/v formalin for paraffin-embedded sections. The non-inflated lobes (left and post caval lobes) were snap-frozen in liquid nitrogen for mRNA and protein analysis. The pups were genotyped using polymerase chain reaction (PCR) with Fstl1 and cre-recombinase primer sets. Fstl1 forward (5’-GCCAGAATCCCACTCCATCG-3’); Fstl1 reverse (5’-TCGGAGCCTGGTGATAAGCG-3’); cre-recombinase forward (5’-GGTTCGCAAGAACCTGATGGACAT-3’); cre-recombinase reverse (5’-GCTAGAGCCTGTTTTGCACGTTCA-3’).
In situ hybridization
In situ hybridization was performed as previously described. 18 In short, lung sections were deparaffinized and rehydrated in a graded series of alcohol, followed by 15 min incubation at 37°C in 10 mg/mL proteinase K dissolved in phosphate buffered saline (PBS). The sections were post-fixed for 10 min in 4% paraformaldehyde (PFA) and 0.2% glutaraldehyde in PBS, followed by rinsing in PBS. Pre-hybridization was done for at least 1 h at 70°C in hybridization mix (50% formamide, 5 × SSC (20 × SSC; 3 M NaCl, 0.3 M tri-sodium citrate, pH 4.5)), 1% blocking solution, 5 mM EDTA, 0.1% 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate (Sigma Aldrich; St. Louis, MO, USA), 0.5 mg/mL heparin (BD Biosciences; Erembodegem, Belgium), and 1 mg/mL yeast total RNA (Roche Applied Science; Penzberg, Germany). A digoxigenin (DIG)-labeled probe (1 ng/mL) was added to the hybridization mix. Probes specific to Fstl1 were used. After overnight hybridization, the sections were rinsed with 2 × SSC, followed by two washings (50% formamide, 2 × SSC, pH 4.5) at 65°C, and rinsing with TNT (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.05% Tween-20) at room temperature. The sections were incubated for 1 h in MABT-block (100 mM maleic acid, 150 mM NaCl, pH 7.4, 0.05% Tween-20, 2% blocking solution), followed by 2 h incubation in MABT-block containing 100 mU/mL alkaline phosphatase-conjugated anti-DIG Fab fragments (Sigma Aldrich; St. Louis, MO, USA). After rinsing in TNT and subsequently in NTM (100 mM Tris, pH 9.0, 100 mM NaCl, 50 mM MgCl2), probe binding was visualized using nitro blue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate (Roche Applied Science; Penzberg, Germany). The sections were dehydrated in a graded alcohol series, rinsed in xylene, and embedded in Entellan (Millipore; Molsheim, France). Images were captured using a Leica DFC320 camera mounted on an AxioPhot microscope (Zeiss; Oberkochen, Germany).
Pulmonary physiological measurement
Pulmonary physiological functions were measured in mice at one week (n = 4 Fstl1-eKO mice and n = 14 controls) and three weeks (n = 8 Fstl1-eKO mice and n = 19 controls) after birth using transthoracic echocardiography as previously described. 19
RNA isolation and real-time quantitative PCR
Total RNA was extracted from lung tissue (post caval lobe) using the NucleoSpin® RNA isolation kit according to the manufacturer’s instruction (Macherey Nagel; Düren, Germany). The total RNA concentration was determined using the NanoDrop® ND1000 spectrophotometer (Thermo Fisher Scientific; Waltham, MA, USA). Equal amounts of total RNA were reverse transcribed using the Reverse Transcription System (Promega; Madison, WI, USA) to generate cDNA. Diluted cDNA was mixed with FastStart Universal SYBR Green Master Mix (Roche Applied Science; Penzberg, Germany) and gene of interest primer sets (Biolegio; Nijmegen, The Netherlands). Primer sequences are listed in Supplementary Table 1. Real-time quantitative PCR (qPCR) was performed using the Illumina Eco Personal qPCR System (Westburg; Leusden, The Netherlands). The qPCR reaction was started by denaturation at 95°C for 15 min followed by 45 cycles of denaturation at 94°C for 30 s, annealing at 59°C for 30 s, and elongation at 72°C for 30 s. Final elongation was for 5 min at 72°C. Real-time PCR data were analyzed using LinRegPCR software version 2013.1. 20 Data were expressed in arbitrary units as ratio of the starting concentration (N0) of each gene of interest corrected to the geometric mean of the N0 value of two reference genes (B2m and Hprt). Comparing the expression levels of the genes of interest between the three non-conditional knockout groups (Fstl1fl/WT Tie2-cre, Fstl1KO/fl, or Fstl1WT/fl mice) did not reveal significant differences, allowing pooling of the data and using them as age-matched controls.
Western blot
Total protein was extracted from lung tissue (left lobe) using RIPA lysis buffer (RIPA lysis buffer 65 mM Tris, 155 mM NaCl, 1% Igepal CA-630, 0.25% sodium deoxycholate, 1 mM EDTA, pH 7.4) supplemented with protease inhibitors (1 µg/mL aprotinin, 1 µg/mL leupeptin, 1 µg/mL pepstatin A, 1 mM Na3VO4, 1 mM NaF, 1 mM β-glycerophosphate). Equal amounts of protein lysate were subjected to SDS PAGE electrophoresis and transferred to nitrocellulose or Polyvinylidene difluoride (PVDF) membranes to evaluate the expression level of non-phosphorylated (β-actin, Jagged1, Endoglin, total Smad1) and phosphorylated (pSmad1/5/8) proteins, respectively. Proteins of interest were detected using primary antibodies for overnight at 4°C in in TBST (50 mM Tris-HCl, 150 mM NaCl, 0.05% [w/v] Tween-20, pH 7.4). The following day, membranes were incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. Protein bands were subsequently visualized using enhanced chemiluminescence substrate (Perkin Elmer; Groningen, The Netherlands) using the G-box gel documentation system (Syngene; Cambridge, UK). Protein band intensities were quantified using Image Studio Lite version 5. Data are presented as ratio of non-phosphorylated protein band intensity corrected to β-actin as a reference protein. For phosphorylated proteins, data are presented as ratio of phosphorylated protein corrected to total protein.
Immunostaining
Paraffin-embedded lung tissues were sectioned at 5 µm thickness and stained for α-SMA (Abcam; Cambridge, UK), which was visualized by staining with HRP-conjugated secondary antibody and diaminobenzidine as a substrate (Sigma Aldrich; St. Louis, MO, USA). To determine the actin content in large vessels (>50 µm in diameter), 21 the α-SMA staining surrounding muscular and elastic vessels was digitally captured in two lung tissue sections per animal and quantified using Image J (National Institute of Health, USA) in a blinded fashion. Data are expressed as ratio of the actin-positive area over the square of the length of the tunica intima. To determine the total number of small vessels (<50 µm in diameter), 21 CD31 staining was performed in serial sections to stain for endothelial cells. Data are expressed as the total number of CD31-positive small vessels corrected to lung surface area (cm2). Furthermore, the number of actin-positive small vessels as well as the total number of CD31-positive small vessels was quantified in two lung tissue sections per animal, in a blinded fashion. Data are expressed as the number of actin-positive small vessels corrected to the total number of CD31-positive small vessels. To evaluate muscularization of the right heart, paraffin-embedded heart tissues were sectioned at 10 µm and stained using the myocardial marker TnI which was visualized by staining with Alexa Fluor 647-conjugated secondary antibody. The images were digitally captured in a blinded manner using a fluorescent microscope (Leica Dm6000; Wetzlar, Germany). The right ventricular free wall was outlined in each section and the mean fluorescence intensity of the outlined structure was measured using 3D Amira software (Version 5.4.3).
Endothelin-1 enzyme-linked immunosorbent assays (ELISA)
The Endothelin-1 protein concentration in lung homogenates was determined using ELISA according to the manufacturer’s protocol (R&D system; Oxford, UK) in duplicate. The sample absorbance was determined at 450 nm and at 570 nm to correct for optical imperfections using Gen5an software using a plate reader (BioTek; Winooski, VT, USA). The lower and upper detection limits for Endothelin-1 were 0.39 pg/mL and 25 pg/mL, respectively.
Hematoxylin and eosin staining
Paraffin-embedded lung tissue sections of 5 µm thick were deparaffinized and rehydrated in a graded series of alcohol, followed by hematoxylin staining. After washing with flowing water, sections were counterstained with eosin. The sections were dehydrated in a graded series of alcohol, rinsed in xylene, and embedded in KP-mounting medium (Klinipath BV; Duiven, the Netherlands). Images were captured using Cell^D imaging software using a light microscope (Olympus BX41; Zoeterwoude, the Netherlands).
Data analysis
Data are presented as medians per genotype-group except otherwise stated. To determine the normality of data distribution, a Shapiro–Wilk normality test was performed prior to further statistical analysis. The statistical significance of differences of normally distributed data was performed using an independent samples two-tailed t-test for comparing two groups or a two-way ANOVA followed by a post hoc Tukey multiple comparisons test for comparing more than two groups. For non-Gaussian distributed data, the statistical significance of differences was determined using a non-parametric Mann–Whitney U test for comparing two groups or non-parametric one-way ANOVA with a post hoc Kruskal–Wallis multiple comparisons test for comparing more than two groups. The Bonferroni correction was used to correct for multiple testing. Differences were considered to be statistically significant at P < 0.05.
Results
Loss of Fstl1 from endothelial cells is postnatally lethal in mice
We and others have previously demonstrated that the homozygote global Fstl1 knockout mice die at birth due to respiratory distress. Here, we generated a mouse line in which Fstl1 is conditionally impaired in endothelial and endothelial-derived cells using Tie2-cre targeted gene deletion, which will be referred to as Fstl1-eKO. Fstl1-eKO knockout pups were born alive at the expected Mendelian ratio (25%). However, ∼70% of Fstl1-eKO mice had died by three weeks after birth whereas all their littermate controls, Fstl1fl/WT Tie2-cre, Fstl1KO/fl, and Fstl1WT/fl mice survived (Fig. 1a). In contrast to Fstl1-KO mice, which display abnormal tracheal cartilage formation and thickening of alveolar septa, Fstl1-eKO mice had macroscopically normal tracheal cartilage formation and normal alveolarization (Supplementary Figure 1). In view of the postnatal lethality of Fstl1-eKO mice and the marked differences with the global Fstl1-KO mice, we quantified Fstl1 mRNA and protein in the lungs of these animals. Fstl1 was highly expressed in the control mice at one week after birth and significantly declined three weeks after birth (mRNA P < 0.005; protein P < 0.05; Fig. 1b and 1c), suggesting Fstl1 is crucial in the early stages of postnatal lung development. The expression of Fstl1 mRNA and protein in lung homogenates of Fstl1-eKO mice were significantly reduced compared with age-matched controls at one week after birth (mRNA P < 0.005; protein P < 0.05; Fig. 1b and 1c), whereas at three weeks after birth the expression of Fstl1 mRNA and protein were not significantly different in Fstl1-eKO mice compared with age-matched controls (Fig. 1b and 1c). In situ hybridization on sections of control mice showed expression of Fstl1 mRNA both in blood vessels and lung parenchyma at one week. At three weeks after birth, the pattern of expression was not different, though the staining intensity was markedly less, which is in line with the qPCR findings (Fig. 1d). In the Fstl1-eKO mice, the expression pattern of Fstl1 mRNA was similar to the control mice, except that Fstl1 mRNA was not or hardly detectable in the endothelium of the blood vessels both at one and three weeks after birth (Fig. 1d).
Endothelial deletion of Fstl1 in mice is postnatally lethal. (a) Kaplan–Meijer survival curve of Fstl1-eKO mice compared with controls. Approximately 70% of Fstl1-eKO mice died at three weeks after birth compared with controls. (b) Gene expression analysis of Fstl1 in lung homogenates using real-time qPCR. Horizontal line represents the median of 16–30 mice per group. (c) Immunoblot analysis of Fstl1 protein in lung homogenates. Data are expressed as means ± SEM of the ratio of Fstl1 protein corrected to β-actin as a reference protein from four to nine mice per group. (d) Representative images of Fstl1 in situ hybridization in lung tissue of Fstl1-eKO mice compared with controls. Red arrowheads point to the absence or presence of Fstl1 probes. AW, airway; V, blood vessel. Each data point represents an individual animal. *P < 0.05; **P < 0.01; ***P < 0.005 compared with the indicated group; ns = not significant.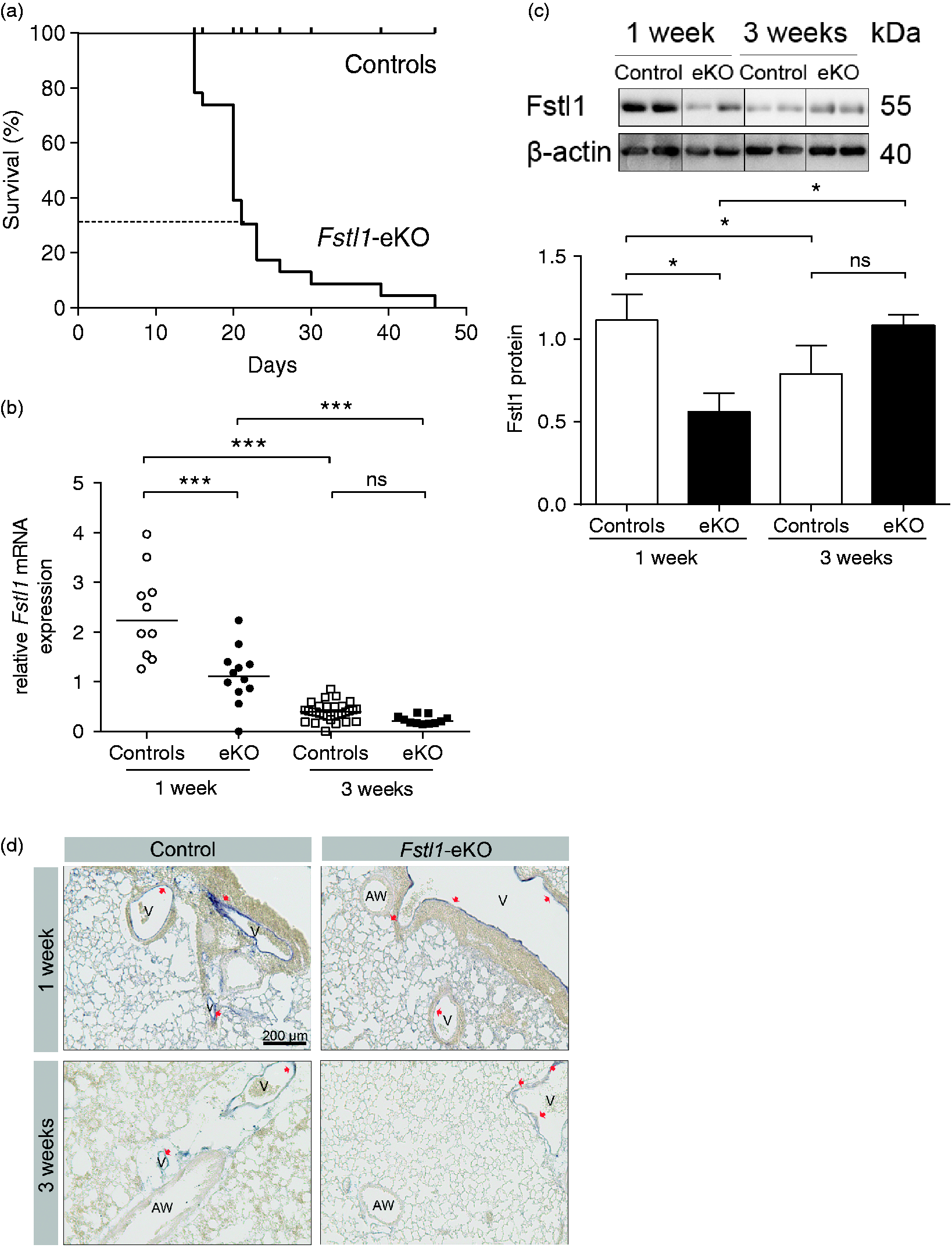
Right ventricular output is reduced in Fstl1-eKO mice
To shed light on the cause of neonatal lethality of Fstl1-eKO mice, we determined the physiological function of the pulmonary vasculature using echocardiography. During normal postnatal development, a significant increase in right ventricular output was observed at three weeks compared with one week as shown by the pulmonary valve (PV) and velocity-time integral (VTI) parameters (P < 0.05; Fig. 2a). A significant reduction in right ventricular output in Fstl1-eKO mice was observed at three weeks compared with age-matched controls (P < 0.05; Fig. 2a). Other physiological parameters, including PV peak gradient, PV mean gradient, PV peak velocity, and PV mean velocity, were significantly increased at three weeks compared with one week after birth both in control and in Fstl1-eKO mice (P < 0.05; Fig. 2b–e). No differences between Fstl1-eKO mice and age-matched controls were observed for these parameters (Fig. 2b–e). A validated non-invasive method to asses pulmonary vascular resistance22,23 was performed by measuring pulmonary acceleration time (PAT) corrected for the pulmonary ejection time (ET). There was no significant difference in pulmonary vascular resistance during normal postnatal lung development as reflected by the ratio of PAT/ET in control mice at three weeks compared with one week after birth (Fig. 2f). A significant change was also not observed between Fstl1-eKO mice compared with age-matched controls either at one week or three weeks after birth (Fig. 2f). Moreover, we evaluated the muscularization of the right ventricle using myocardial marker TnI, as an indicative of right ventricle hypertrophy by measuring the volume of the right ventricle. There were no changes in the right ventricle volume at three weeks compared with one week after birth in the control mice (Fig. 2g). At three weeks, there was a trend towards increased RV volume in Fstl1-eKO mice compared with age-matched controls (Fig. 2g). A significant increase in RV volume was observed in Fstl1-eKO mice at three weeks compared with one week after birth (P = 0.005; Fig. 2g).
Right ventricular output is reduced in Fstl1-eKO mice. Pulmonary functions were measured using echocardiograph. (a) Pulmonary valve (PV)-velocity time integral (VTI) was significantly decreased in Fstl1-eKO mice compared with age-matched controls. (b) PV peak gradient, (c) PV mean gradient, (d) PV peak velocity, (e) PV mean velocity, and (f) the ratio of Pulmonary Acceleration Time (PAT) corrected by the pulmonary ejection time (ET) in percentage were unaltered in Fstl1-eKO mice compared with age-matched controls. Data are expressed as the mean ± SEM of 4–19 mice per group. (g) The right ventricle (RV) volume of Fstl1-eKO mice was unaltered compared with age-matched controls. A significant increase in RV volume was observed in Fstl1-eKO mice at three weeks compared with one week after birth. Data are expressed as the mean ± SEM of three mice per group. *P < 0.05; **P < 0.01; ***P < 0.005 compared with the indicated group. ns, not significant.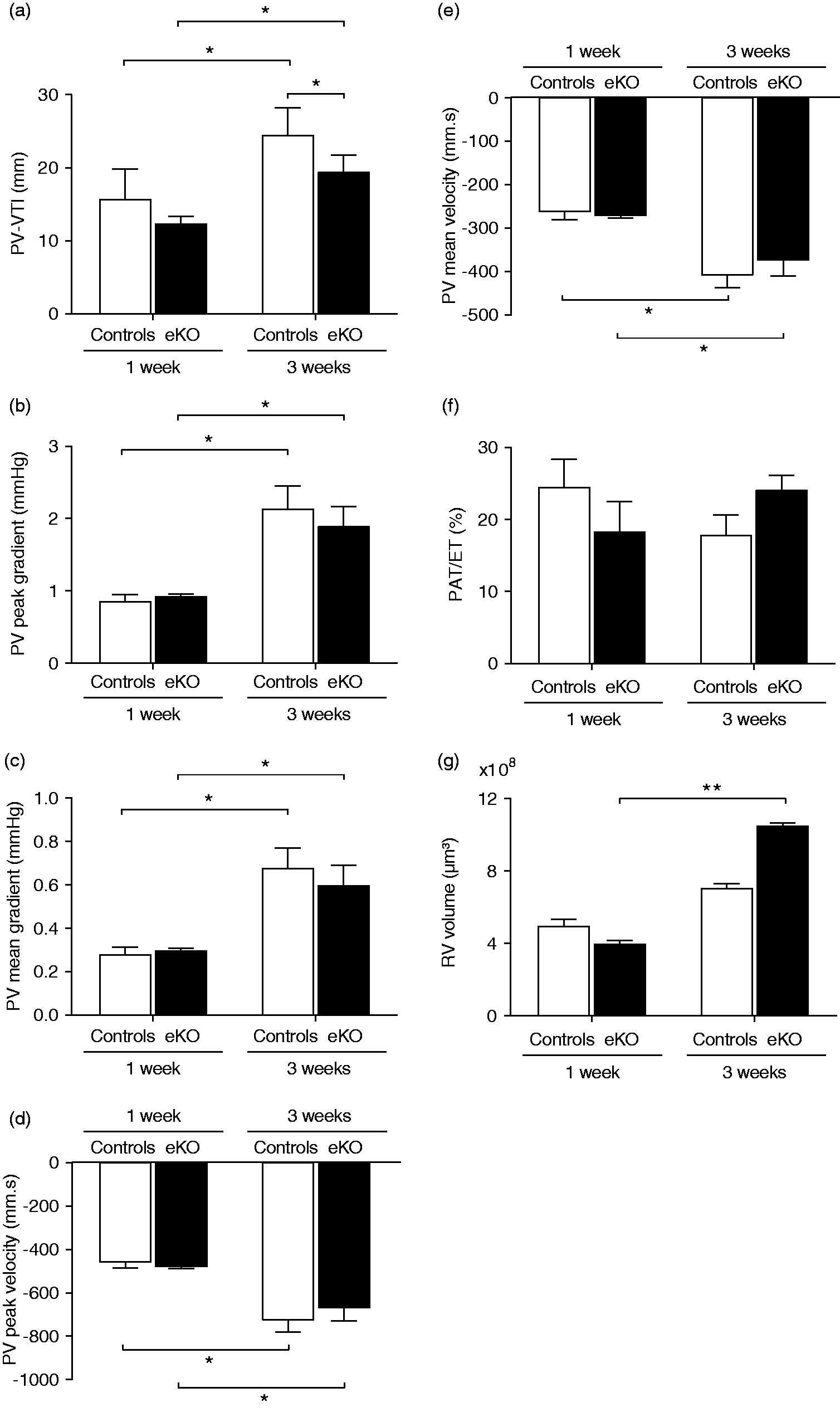
Pulmonary vascular remodeling in Fstl1-eKO mice
In view of these results, we examined the effect of endothelial deletion of Fstl1 on pulmonary vascular phenotypes and quantified the morphological changes and actin content in large and small pulmonary vessels. During normal development, the thickness of the large muscular arteries was unaltered in lung tissue of control mice at three weeks compared with one week after birth (Fig. 3a), whereas the actin content was reduced in large elastic (P < 0.005) and muscular arteries (P < 0.005) in lung tissue of control mice at three weeks compared with one week after birth (Fig. 3b and 3c). In Fstl1-eKO mice, the thickness and the actin content in large muscular arteries were unaltered at three weeks compared with one week after birth (Fig. 3a and 3c), whereas the actin content in large elastic arteries was significantly reduced at three weeks compared with one week after birth (P < 0.005; Fig. 3b). No significant differences were observed in the thickness of the large muscular arteries (Fig. 3a) and in the actin content in large elastic and muscular arteries in lung tissue of Fstl1-eKO mice compared with age-matched controls at either one or three weeks after birth (Fig. 3b and 3c). Reduction in actin content at three weeks after birth seems to be limited to vascular smooth muscle as there were no temporal changes in the actin content in the airway smooth muscle bundles and there was no observed difference between Fstl1-eKO mice compared with age-matched controls (Fig. 3d).
Number of actin-positive small pulmonary vessels is increased in the lung tissue of Fstl1-eKO mice at three weeks. (a) The thickness of large muscular arteries in lung tissue was quantified and expressed as ratio of total area of tunica media over the square of the length of the tunica intima. Horizontal lines represent the median of n = 9–16 vessels from N = 6–8 mice per group. (b) The α-SMA content in large elastic arteries and (c) in large muscular arteries in lung tissue was quantified and expressed as ratio of the actin-positive area over the square of the length of tunica intima. Horizontal lines represent the median of n = 9–26 vessels per group from N = 5–12 mice per group. (d) The α-SMA content in airway smooth muscle bundles surrounding the airway in lung tissue was quantified and expressed as ratio of the actin-positive area over the square of the length of basement membrane. Horizontal lines represent the median of n = 9–34 vessels per group from N = 5–13 mice per group. (e) Total number of vessels in lung tissue was quantified using CD31 staining and expressed as ratio of total vessel number/lung surface area (cm
2
). Horizontal lines represent the median of the vessel number/cm2 of N = 7–12 mice per group. (f) The percentage of actin-positive small pulmonary vessels in lung tissue was quantified and expressed as percentage of actin-positive small vessel number/total vessel number. Horizontal lines represent the median of the percentage of actin-positive vessels of N = 7–12 mice per group. Representative images of tissue sections taken at three weeks after birth were shown. AW, airway; V, blood vessel. Scale bars represent 100 µm. Magnification 200×. *P < 0.05; **P < 0.01; ***P < 0.005 compared with the indicated group.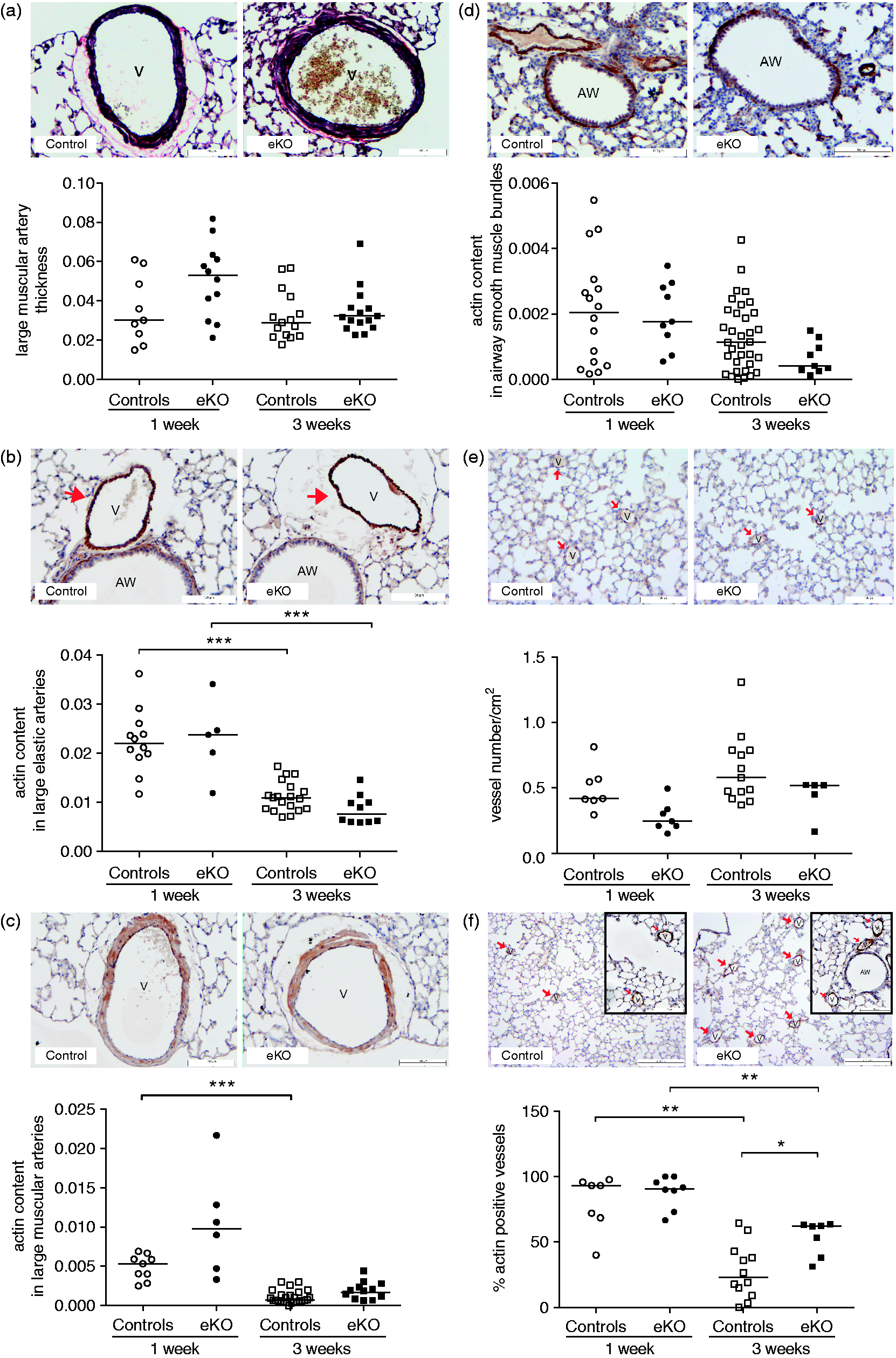
We also explored the number of small blood vessels in lung tissue as these may contribute to an important extent to changes in pulmonary blood pressure. In normal development, no significant temporal change in the CD31 positive total number of vessels was observed, whereas the percentage of actin-positive vessels was significantly reduced at three weeks compared with one week after birth (P < 0.01; Fig. 3e and 3f). Although the CD31 positive total number of vessels was not significantly different between Fstl1-eKO mice compared with age-matched controls (Fig. 3e), the percentage of actin-positive small pulmonary vessels was significantly higher in Fstl1-eKO mice at three weeks after birth compared with age-matched controls (P < 0.05; Fig. 3f).
Increased BMP/Smad signaling in the lung of Fstl1-eKO mice
To unravel whether endothelial Fstl1 modulates BMP signaling during postnatal development, we investigated the activity of BMP/Smad signaling in lung homogenates using pSmad1/5/8 antibodies. During normal postnatal development, Smad1/5/8 phosphorylation was higher at three weeks compared with one week after birth (P < 0.05; Fig. 4a). In addition, we found increased levels of pSmad1/5/8 in lung homogenates of Fstl1-eKO compared with age-matched controls at one week after birth (P < 0.05; Fig. 4a), whereas, the level of pSmad1/5/8 at three weeks was unaltered in Fstl1-eKO mice compared with age-matched controls (Fig. 4a).
Increased activation of Smad1/5/8 and Smad1/5/8-regulated genes in the lung of Fstl1-eKO mice. (a) Immunoblot analysis of pSmad1/5/8 in lung homogenates. Data are expressed as means ± SEM of the ratios of pSmad1/5/8 corrected to total Smad1 of four to nine mice per group. Pulmonary expression of the pSmad1/5/8-regulated genes, Jagged1 (b) and Endoglin (c) in lung homogenates. Data are expressed as starting concentration N0 in arbitrary units corrected to B2m and Hprt as reference genes. Horizontal lines represent medians of 11–30 mice per group. (d) Immunoblot of Jagged1 protein in lung homogenates and its quantification. Data are expressed as means ± SEM of the ratios of Jagged1 over the reference protein β-actin of four to nine mice per group. (d) Immunoblot of Endoglin in lung homogenates and its quantification. Data are expressed as means ± SEM of the ratios of Endoglin over the reference protein β-actin of four to nine mice per group. *P < 0.05; **P < 0.01; ***P < 0.005 compared with the indicated group.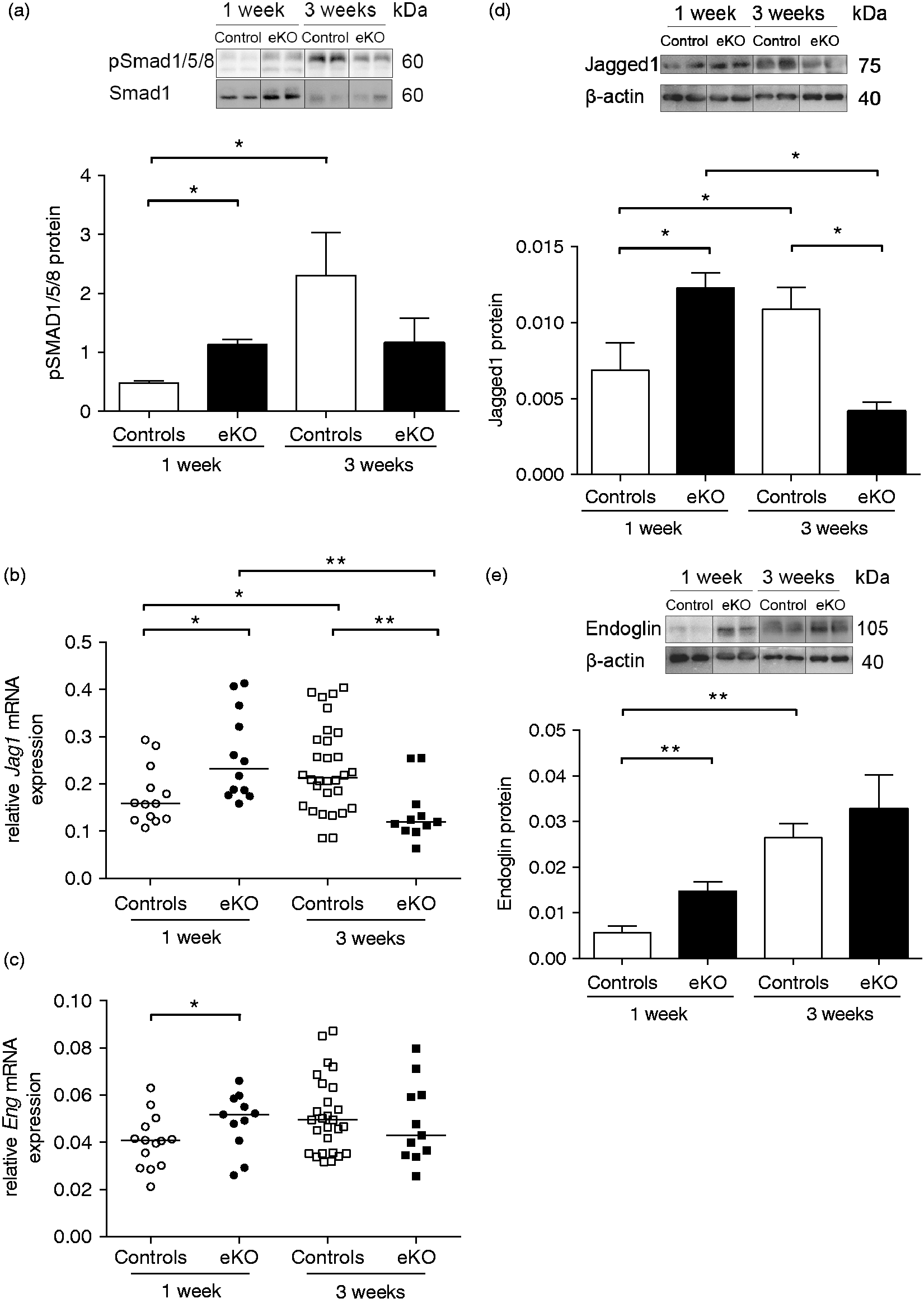
To study the role of BMP signaling further, we selected previously identified pSmad1/5/8 target genes which relate to vascular remodelling: 24 Id3, Epas1, Zeb2, Zfp423, Ephb4, Klf4, Flt1, Jag1, Eng, Gata2, Vegf. Of these, we found that the expression of Jagged1, Endoglin, and Gata2 was altered in Fstl1-eKO mice compared with age-matched controls with a nominal P value < 0.05, whereas the expression level of the other genes was not different. Therefore, Jagged1, Endoglin, and Gata2 were included for further analysis. Consistent with the increased Smad1/5/8 activation, Jagged1 and Endoglin mRNA and protein were increased in lung tissue of control mice at three weeks compared with one week after birth (Jagged1 mRNA P < 0.05; protein P < 0.05; Fig. 4b and 4d; Endoglin protein P < 0.01; Fig. 4c and 4e). In line with increased phosphorylation of Smad1/5/8, the expression of Jagged1 and Endoglin mRNA and protein was also increased in lung homogenates of one week Fstl1-eKO mice compared with age-matched controls (Jagged1 mRNA P < 0.05; protein P < 0.05; Fig. 4b and 4d; Endoglin mRNA P < 0.05; protein P < 0.01; Fig. 4c and 4e). At three weeks, Jagged1 mRNA (P < 0.01) and protein (P < 0.05) were reduced in the Fstl1-eKO mice compared with age-matched controls (Fig. 4b and 4d), whereas Endoglin was not different compared with age-matched controls (Fig. 4c and 4e).
The mRNA expression of the transcription factor Gata2 was higher in the lung homogenates of control mice at three weeks compared with one week after birth (P < 0.01; Fig. 5a). Accordingly, the level of mRNA and protein expression of the Gata2 target gene Endothelin-1, a potent vasoconstrictor, was also increased in lung homogenates of control mice at three weeks compared with one week after birth (mRNA P < 0.005; protein P < 0.05; Fig. 5b and 5c). Gata2 mRNA and Endothelin-1 mRNA and protein levels were increased in lung homogenates of Fstl1-eKO mice compared with age-matched controls at one week after birth (Endothelin-1 mRNA P < 0.05; protein P < 0.01; Fig. 5b and 5c). These levels did not further increase at three weeks after birth and were no longer different compared with age-matched controls (Fig. 5a–c).
Increased Endothelin-1 in the lung of Fstl1-eKO mice. (a) Pulmonary mRNA expression of Gata2 transcription factor and (b) Endothelin-1 in the lung homogenates. Data are expressed as starting concentration N0 in arbitrary units corrected to B2m and Hprt as reference genes. (c) Endothelin-1 protein concentration in lung homogenates is expressed as Endothelin-1 concentration in pg/ml per 1 µg protein lysates. Each data points represent an individual animal. Horizontal line represents the median of Endothelin-1 concentration of 8–12 mice per group. *P < 0.05; **P < 0.01; ***P < 0.005 compared with the indicated group.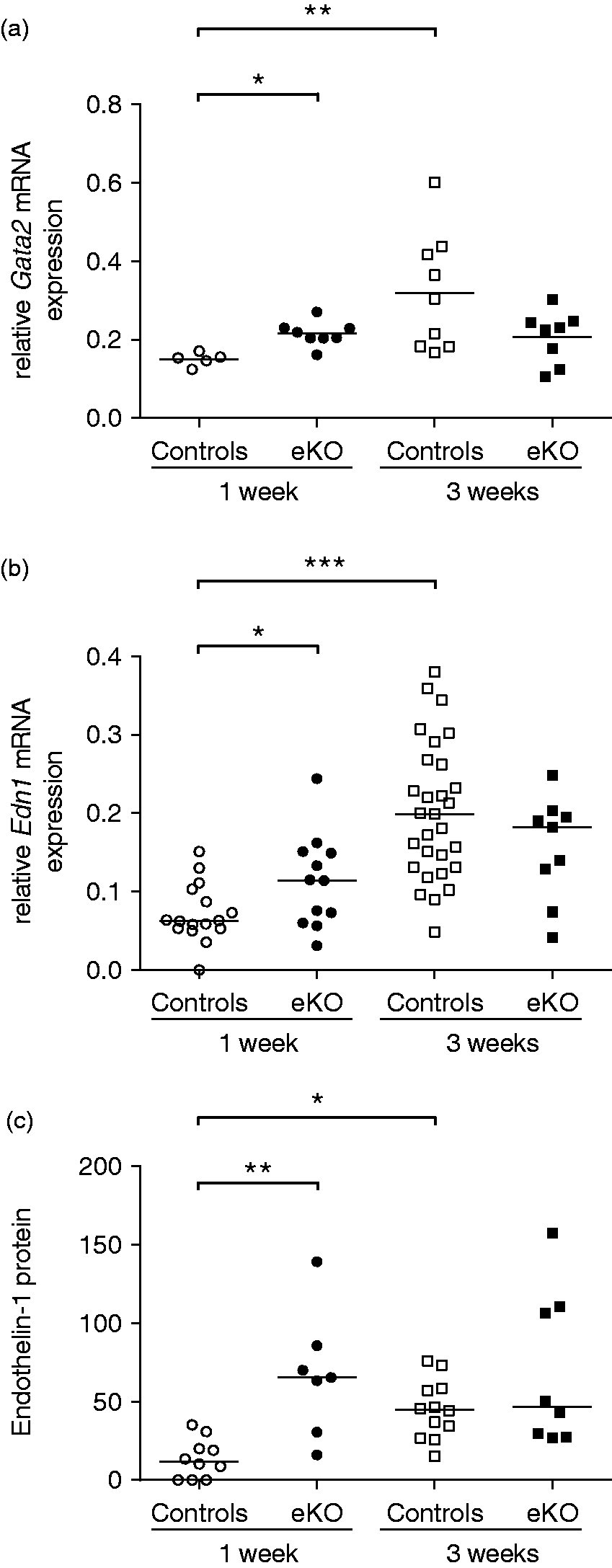
Discussion
Proper formation and maturation of the pulmonary vasculature is essential to support normal lung development and function. 25 BMP signaling is crucial in dynamic processes of vessel growth and vessel maturation.9,12,26–29 Maturation of immature vessels proceeds according to the developmental steps: (1) stabilization of the immature vessels; (2) vessel branching, remodeling, and pruning; and (3) vessel specialization. 30 Deregulation of molecules involved in vessel growth and maturation leads to vascular abnormalities and dysfunction.
In this study, the role of endothelial Fstl1 in postnatal maturation of the pulmonary vasculature was investigated in vivo using Tie2-cre targeted endothelial-specific deletion. Conditional deletion of Fstl1 from endothelium was postnatally lethal, and resulted in small pulmonary vessel changes, pointing to a critical role of endothelial Fstl1 in postnatal lung development. In line with the idea that Fstl1 is a BMP antagonist, phosphorylation of BMP/Smad signaling showed the inverse pattern of Fstl1 expression level during normal postnatal development. In the early stages of postnatal development, high Fstl1 expression is associated with low phosphorylated Smad1/5/8 at one week after birth whereas at later stages of postnatal development, decreased in Fstl1 expression is associated with increased phosphorylation of BMP/Smad signaling in lung tissue of control mice.
We also demonstrate that during early stages of postnatal development, high Fstl1 expression coincides with high actin content in the large muscular and elastic arteries and high percentage of actin-positive small pulmonary vessels in lung tissue of control mice. In later stages of postnatal development, low Fstl1 expression coexists with low actin content in the large muscular and elastic arteries and low percentage of actin-positive small pulmonary vessels in lung tissue of control mice. Recently, it was reported that Fstl1 mediates TGF-β-induced α-SMA expression by antagonizing BMP signaling in lung fibroblasts. 31 We speculate that Fstl1 antagonizes BMP signaling to facilitate TGF-β-induced α-SMA expression from vascular smooth muscle cells in the early stages of normal vascular development. Reduced Fstl1 levels in later stages of development in turn result in reduction in actin content, suggesting that Fstl1 possibly mediates endothelial-mural cell communication by modulating BMP/TGF-β signaling during normal pulmonary vascular development.
During normal postnatal vascular development, transient reduction of actin content in pulmonary vessels is normal and an indication of maturation of pulmonary vessels by reducing contractility of pulmonary arteries. 32 Our data indicate that loss of Fstl1 in endothelium prevents this normal reduction of actin in small pulmonary vessels and as such delays pulmonary vascular maturation.
Mechanistically, Fstl1-eKO mice have increased Smad1/5/8 phosphorylation and expression of pSmad1/5/8-regulated genes in the lung, including Gata2, Endoglin, and Jagged1. Previous studies demonstrated the important role of Jagged1 and Endoglin in vascular specialization and vascular remodeling, respectively, in the later stages of vessel maturation.10,33,34 In the early stages of normal development, high levels of Fstl1 inhibit BMP signaling and its regulated genes, Gata2, Jagged1, and Endoglin, indicating that modulation of BMP signaling by Fstl1 is important in early stages of postnatal lung development. On the other hand, low levels of Fstl1 in later stages of development lead to increased BMP-mediated Smad phosphorylation and its regulated genes Gata2, Jagged1, and Endoglin. Endoglin is a type III TGF-β receptor which is predominantly expressed in proliferating endothelial cells and triggers endothelial cell proliferation and vascular remodeling.34–37 The importance of Endoglin for normal vascular formation and homeostasis is evident in Endoglin null (Eng–/–) mice, which exhibit vascular deformities.37–40 Increased Endoglin expression in Fstl1-eKO mice at the early stages of postnatal pulmonary vascular development might shift the balance towards endothelial cell migration and proliferation instead of maturation. This supports the contention that Fstl1-eKO mice have delayed pulmonary vascular maturation.
The Notch ligand Jagged1 is an important regulator of cell fate in embryonic development 41 and vessel fate in pulmonary vascular maturation.10,33 In addition, it has a significant role in angiogenesis and vascular homeostasis.42,43 Ablation of Jagged1 in mice is embryonically lethal and leads to vascular abnormalities and defects in vascular remodeling. 33 The aberrant Jagged1 expression during early stages of postnatal pulmonary vascular development in Fstl1-eKO mice might lead to deregulation of vascular remodeling and disruption of the vessel specialization programs.
BMP/Smad increases expression of the transcription factor Gata2, subsequently promoting the expression of the most potent vasoconstrictor Endothelin-1.44–46 We demonstrate that Endothelin-1 levels are low at the early stages of normal development and increase at the later stages of normal development. We found that endothelial deletion of Fstl1 is associated with increased Endothelin-1 mRNA and protein expression at the early stages of development in the lung. Increased Endothelin-1 has been reported to promote proliferation and migration of endothelial cells and contribute to increased pulmonary vascular resistance and subsequent right ventricle hypertrophy,47,48 suggesting pivotal roles of Endothelin-1 in vessel formation, vascular remodeling, and vascular tone maintenance. Our data suggest that Endothelin-1 expression is negatively regulated by Fstl1 via inhibiting Smad1/5/8 and the Gata2 transcription factor, presumably by antagonizing BMP signaling. These findings are in line with previous studies demonstrating that Endothelin-1 expression is regulated by BMP via Smad1/5/8 in endothelial cells.44–46
Lower right ventricular output and small vascular remodeling were visible at three weeks after birth, whereas elevation in BMP/Smad phosphorylation and of its downstream targets Jagged1, Endoglin, Gata2, and Endothelin-1 were more prominent at one week after birth in Fstl1-eKO mice, indicating that molecular changes are more transient than their physiological and morphological consequences. As a consequence, the functional and morphological differences seen at three weeks after birth were initiated earlier during postnatal development. Alternatively, the activation of BMP/Smad signaling alone may not be sufficient to affect the pulmonary vasculature structurally and functionally at early stages of development, but instead becomes evident at a later time point. Despite of higher percentages of actin-positive small pulmonary vessels and notably decreased right ventricular output, there were minimum morphological defects of large pulmonary vessels, normal pulmonary vascular resistance as reflected by the ratio of PAT/ET in Fstl1-eKO mice, and a trend towards right ventricle hypertrophy. This may suggest very early stages of pulmonary vascular dysfunction in Fstl1-eKO mice. This phenotype is also commonly observed in patients with PAH, in which molecular changes/defects in BMP signaling manifest early in the disease followed by physiological and structural changes which are observed at later stages of the disease progression. 49 In addition, our data may imply that the molecular and morphological changes in the lung are possibly a secondary consequence of the right heart dysfunction as reflected by reduction in RV output. The morphological and physiological functions of the heart of Fstl1-eKO mice are currently under investigation.
In conclusion, our findings demonstrate that loss of endothelial Fstl1 in the lung is associated with increased BMP/Smad phosphorylation and elevations in its downstream targets Jagged1, Endoglin, Gata2, and Endothelin-1. These changes are associated with impaired small pulmonary vascular remodeling and decreased right ventricular output. Taken together, our findings suggest a key role of Fstl1 in titrating the level of BMP signaling for proper postnatal lung development.
Footnotes
Acknowledgements
The authors thank Jan M. Ruijter for expert advice on real-time PCR data analysis and Frank Ensink for technical assistance.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This study was financially supported by a grant from the Netherlands Lung Foundation (grant 3.2.12.083).