
Abstract
Pulmonary arterial hypertension (PAH) remains a mysterious killer that, like cancer, is characterized by tremendous complexity. PAH development occurs under sustained and persistent environmental stress, such as inflammation, shear stress, pseudo-hypoxia, and more. After inducing an initial death of the endothelial cells, these environmental stresses contribute with time to the development of hyper-proliferative and apoptotic resistant clone of cells including pulmonary artery smooth muscle cells, fibroblasts, and even pulmonary artery endothelial cells allowing vascular remodeling and PAH development. Molecularly, these cells exhibit many features common to cancer cells offering the opportunity to exploit therapeutic strategies used in cancer to treat PAH. In this review, we outline the signaling pathways and mechanisms described in cancer that drive PAH cells’ survival and proliferation and discuss the therapeutic potential of antineoplastic drugs in PAH.
Pulmonary arterial hypertension (PAH) is a multifactorial disease characterized by sustained elevation of pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) leading to right ventricular (RV) failure and death (Fig. 1). Despite our improved understanding of the mechanisms involved and treatment, current therapies focusing on vasodilators fail to reverse vascular remodeling and long-term survival remains poor, underscoring the need to identify new therapeutic targets.
1
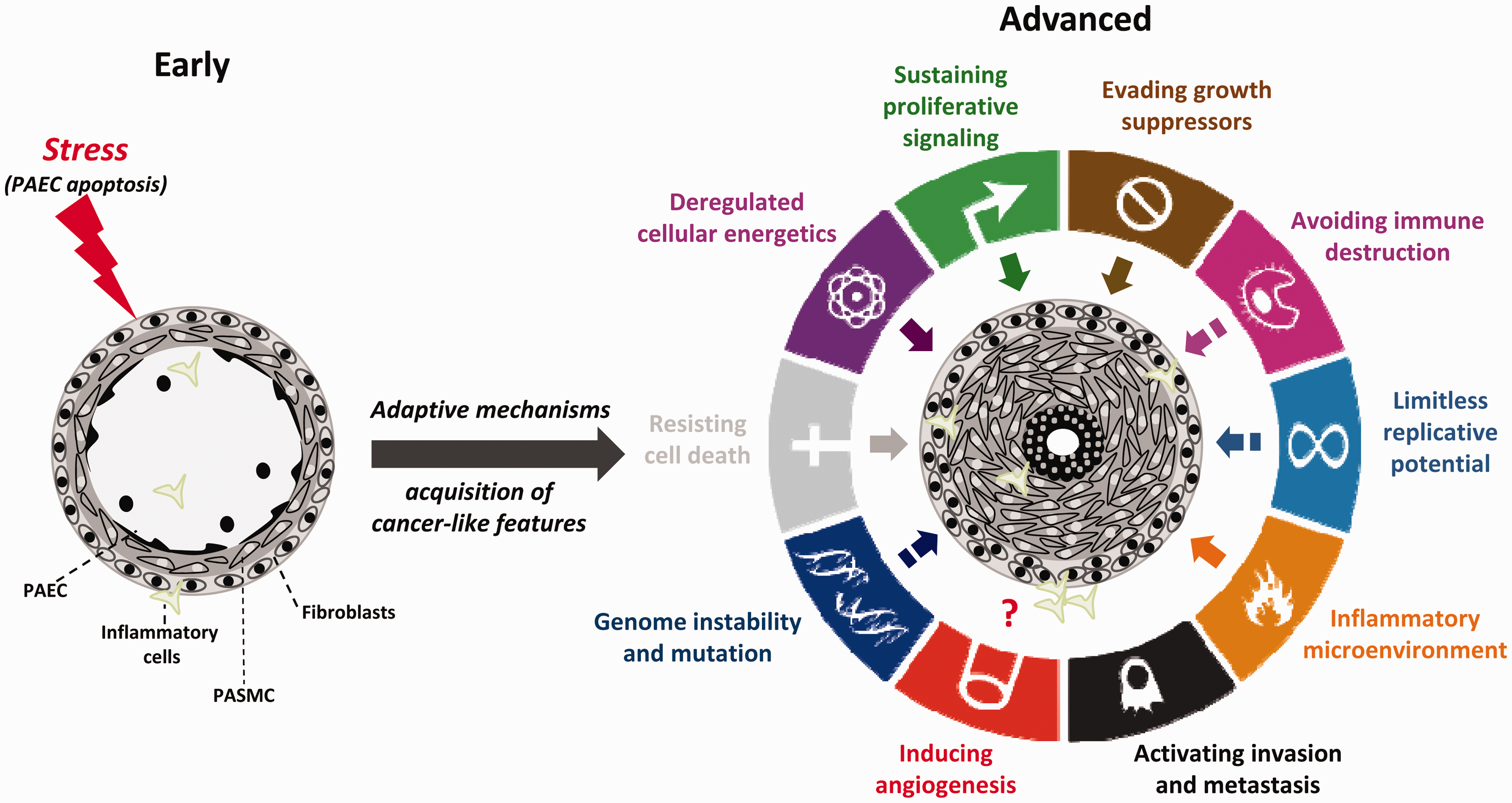
On the basis of many points of overlap, among which deregulated cellular metabolism, sustained proliferation, and escape from apoptosis are the most predominant, PAH has emerged as a cancer-like disease (Fig. 2), providing new therapeutic perspectives for scientists.2,3
Schematic representation of PAH. In an advance stage of the disease, obliteration of the vascular lumen due to vasoconstriction and excessive proliferation and resistance of apoptosis of resident cells (adventitial fibroblasts, pulmonary artery smooth muscle cells, pulmonary artery endothelial cells) increases PVR resulting in right ventricular adaptation (right ventricle hypertrophy). Initial compensation is followed by progressive and irreversible right ventricular enlargement leading to right ventricular failure. Acquisition of cancer traits by pulmonary vascular cells during PAH progression (figure adapted from Guignabert et al.
3
and Hanahan and Weinberg
41
). During disease progression, the vast majority of cancer hallmarks described by Hanahan and Weinberg
41
(excepted tissue invasion and metastasis) are also shared by pulmonary vascular cells from PAH patients. Solid arrows within the outer circle represent high degree of similarity between cancer and PAH. Hatched arrows represent low/medium degree of similarity between cancer and PAH.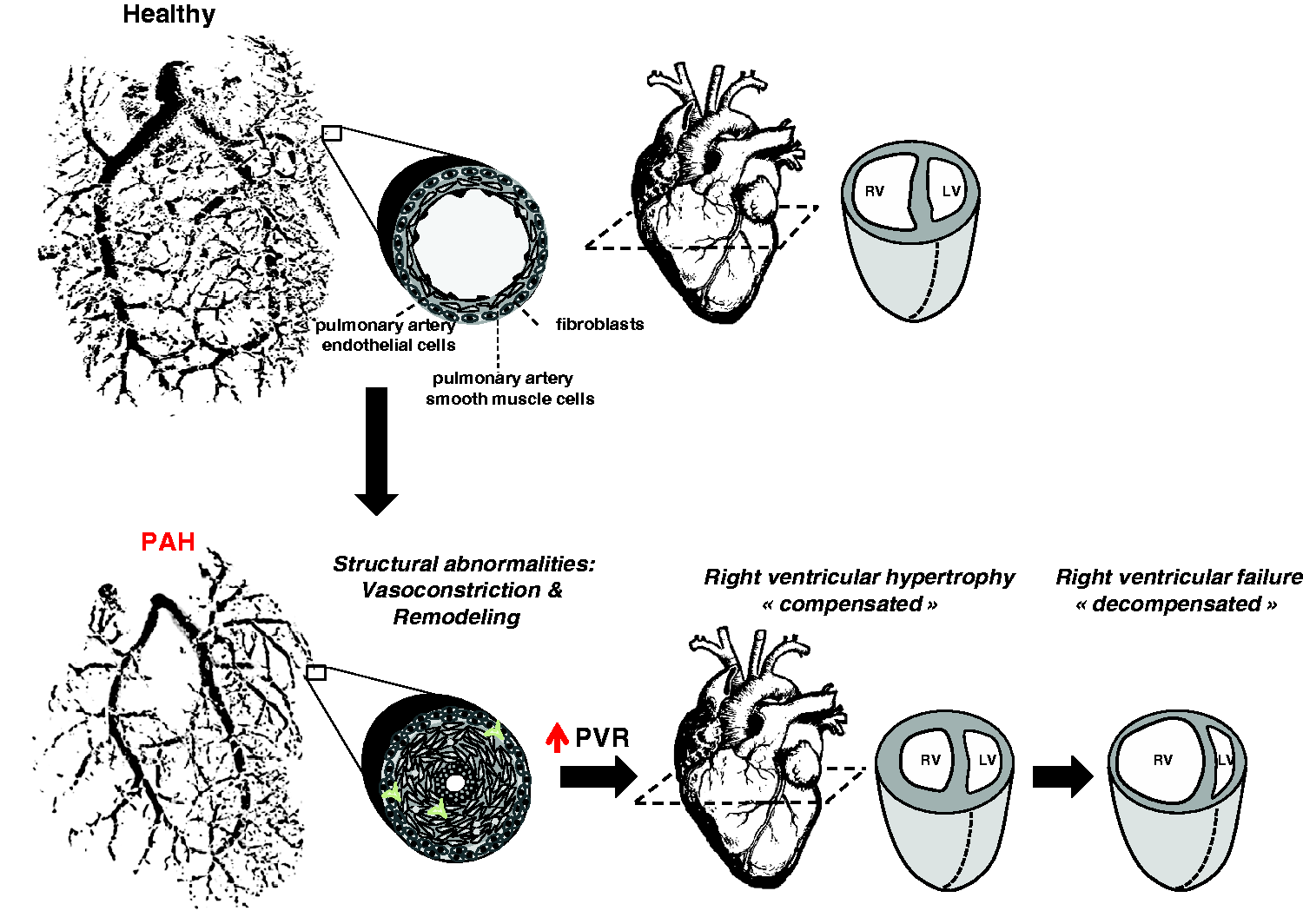
Hyperproliferative mechanisms in PAH cells
Growth factors and inflammatory mediators
A wide array of growth factors, cytokines, and chemokines has been documented to promote cancer and PAH development.4,5,6 Of these factors, interleukin 6 (IL-6) and platelet-derived growth factor (PDGF) have been the most studied. Infiltration of inflammatory cells such as macrophages, mast cells, leukocytes, and lymphocytes in pulmonary vascular lesions is a major hallmark of PAH. Indeed, inflammatory cells are a major source of pro-inflammatory mediators responsible for the accumulation of extracellular matrix components and directly implicated in the exaggerated proliferation of resident pulmonary artery (PA) cells. Therefore, elevated circulating levels of the pro-inflammatory cytokines including IL-6, monocyte chemotactic protein 1 (MCP-1), and tumor necrosis factor alpha (TNFα) have been found in PAH patients, and most of them correlate with a worse clinical outcome.7,8 The pathogenic relevance of IL-6 is illustrated by genetic studies in mice showing that knockout mice are protected against chronic hypoxia-induced pulmonary hypertension (PH),
9
whereas IL-6 over-expressing mice develop PH.
10
Like fibroblast growth factor-2 (FGF2), insulin-like growth factor 1 (IGF-1), and endothelin-1, PDGF also actively participates in mitogenic signaling and thickening of the pulmonary vascular media.11,12 The binding of these signaling molecules to their receptors activates various interconnected signaling pathways such as MEK/ERK, PI3K/AKT, and JAK/STAT3 to induce cell growth, cell cycle progression, and cell survival (Fig. 3). In view of its implications in cancer progression,
13
STAT3 has also been the focus of much attention in PAH.
14
In PA smooth muscle cells (PASMCs) from PAH patients, activation of STAT3 was shown to upregulate expression of proviral integration site for moloney murine leukemia virus-1 (PIM-1), a proto-oncogene with serine/threonine kinase activity regulating various oncogenic pathways.
14
The latter was reported to enhance nuclear factor of activated T-cells (NFAT)-mediated transactivation, leading to inhibition of voltage-gated potassium channels, increases in cytosolic-free calcium concentration, and subsequent stimulation of cell proliferation and contraction (Fig. 3).15–17 Consistently, in vivo inhibition of PIM-1 by nebulized siRNA reversed monocrotaline (MCT)-induced PAH in rats and Pim-1 deficient mice are resistant to development of PH.
14
Based on the current knowledge, inhibition of PIM-1 is likely to become an anti-proliferative strategy for patients suffering PAH. To this end, pharmacological inhibitors of PIM-1 currently undergoing evaluation in clinical trials as anticancer agents could be considered in a near future to treat PAH. Owing to its role in tumor angiogenesis,
18
vascular endothelial growth factor (VEGF) has also raised a great interest for attempting to explain the vascular remodeling events in PAH. High levels of VEGF were found in the plasma of PAH patients and VEGF as well as its receptor (VEGFR2) are robustly expressed in the complex pulmonary arterial lesions of PAH patients.19,20 However, somewhat counter-intuitively to the hypothesis that increased VEGF expression contributes to vascular lesions in PAH, treatment of rodents with a VEGF receptor/tyrosine kinase antagonist, Su5416, in combination or not to hypoxia was shown to induce PAH21 and VEGF over-expression was reported to attenuate the development of hypoxic PH.
22
With these findings, a two-step model can be proposed in which initial blockade of the VEGF pathway increases the susceptibility to PA endothelial cell (PAEC) apoptosis contributing to the selection of apoptosis-resistant cells followed by an increased expression of VEGF by resident cells promoting cellular expansion.
Scheme depicting the major intracellular signaling pathways implicated in the hyperproliferative phenotype of PAH cells. [Ca2+]cyt, cytosolic calcium concentration; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; AKT, protein kinase B; FGF2, fibroblast growth factor-2; ERK1/2, extracellular signal-regulated kinases 1/2; FOXO1, forkhead box protein O1; IGF, insulin growth factor; IL-6, interleukin 6; JAK, janus kinase; LATS1/2, large tumor suppressor kinases 1/2; MDM2, mouse double minute 2 homolog; MEK1/2, mitogen-activated protein kinase/ERK kinases 1/2; MST1/2, mammalian sterile 20-like kinases 1/2; mTOR, mechanistic target of rapamycin; NFAT, nuclear factor of activated T-cells; PDGF, platelet-derived growth factor; PDK1, 3-phosphoinositide-dependent kinase 1; PI3K, phosphoinositide 3-kinase; PIM-1, proviral integration site for Moloney murine leukemia virus-1; PIP3, phosphatidylinositol 3,4,5 triphosphate; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma; RTK, receptor tyrosine kinase; S6K1, ribosomal protein S6 kinase beta-1; STAT3, signal transducer and activator of transcription 3; TAZ, transcriptional coactivator with PDZ-binding motif; YAP, yes-associated protein.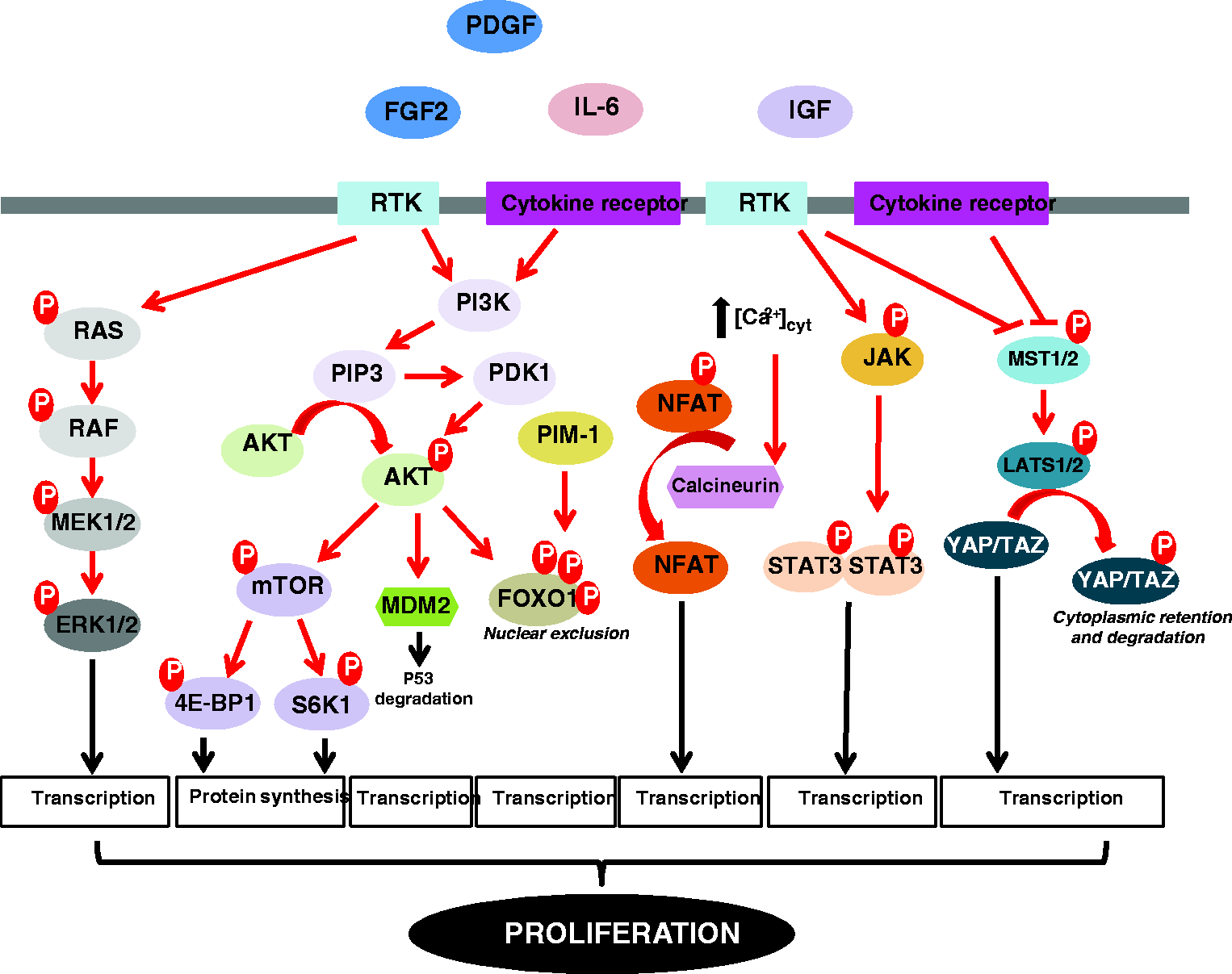
In response to growth factors and inflammatory cytokines, activation of the PI3K/AKT and JAK/STAT3 signaling pathways has been reported to converge on the transcription factor Forkhead box protein O1 (FOXO1) to induce its phosphorylation and inhibit its activity through nuclear exclusion (Fig. 3). 23 In agreement with studies indicating that FOXO1 functions as a tumor suppressor, 24 abrogation of FOXO1 transcriptional activity in PASMCs was associated with an upregulation of Cyclin B1 and D1 required for cell cycle progression and a reduced expression of p27 that inhibits Cyclin/Cdk complexes. Moreover, in vivo pharmacological inhibition or conditional inactivation of FOXO1 in smooth muscle cells was shown to induce PH, whereas constitutive activation of FOXO1 had the opposite effect. 23
mTOR signaling
The mechanistic target of rapamycin (mTOR) signaling is a pivotal regulator of cellular metabolism, cell proliferation, and survival.25,26 mTOR is a serine-threonine kinase that interacts with several proteins to form two independent complexes, mTORC1 (mTOR-Raptor) and mTORC2 (mTOR-Rictor), which have different sensitivity to rapamycin and downstream outputs. The mTORC1 pathway integrates inputs from many cues including growth factors, DNA damage, oxygen availability, amino acids, and energy status to activate ribosomal protein S6 kinase beta-1 (S6K1) and to inhibit eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), leading to enhanced anabolic cell growth and proliferation (Fig. 3). Conversely, mTORC2 mainly responds to growth factors and promotes cell survival. Due to its regulatory functions in cell proliferation and survival, the mTOR signaling generates considerable interest in the field of cancer and PAH. The importance of the mTORC2 signaling in PAH development was highlighted by in vitro studies demonstrating that Rictor knockdown reduced glycolysis-dependent proliferation and survival of PASMCs, while inhibition of Raptor led to a decrease in cell proliferation without effect on cell survival. 27 Consistently, selective hyperactivation of mTORC1 caused by inactivation of the negative mTORC1 regulator tuberous sclerosis complex 1 gene (TSC1) was sufficient to produce distal PA remodeling and PH in mice, a pathological state fully reversed following rapamycin treatment, emphasizing the relevance of this pathway in PAH. 28
Hippo signaling
The Hippo signaling pathway has recently emerged as an evolutionarily conserved signaling pathway that controls organ size.29,30 The backbone of the Hippo pathway consists of a cascade of tumor suppressive kinases mammalian STE20-like protein kinase 1/2 (MST1/2) and large tumor suppressor homolog 1/2 (LATS1/2) that regulate the activity of the downstream effectors yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). When the pathway is activated, LATS1/2 directly phosphorylate YAP and TAZ resulting in their cytoplasmic retention and cell growth inhibition (Fig. 3).29–31 In recent years, several studies have investigated the possible involvement of this pathway in PAH development and progression.32–34 Among these, Kudryashova and collaborators found that in response to mechanical cues or mitogenic factors, human PAH-PASMCs exhibit diminished expression of phospho-LATS1 and consistently YAP nuclear accumulation. To test the requirement of LATS1/YAP in cell proliferation, transfection of PAH-PASMCs with a LATS1 construct mimicking a constitutive phosphorylated state was done demonstrating a rescue of the abnormal pro-proliferative and apoptosis-resistant phenotype of these cells. Furthermore, the authors demonstrated that inactivation of LATS1 was associated with the over-expression and the activation of numerous proteins and pathways known to play a deleterious role in the pulmonary vasculature, including hypoxia inducible factor 1 alpha subunit (HIF-1α) and neurogenic locus notch homolog protein 3 (NOTCH3).34,35 Although Hippo signaling serves as a hub in regulating cell proliferation and represents a potential therapeutic target in PAH, the detailed characteristics of YAP/TAZ “oncogenic” properties remain unclear.
The cellular gatekeeper P53
Because more than 50% of human cancers contain loss of function mutations in the transcription factor p53, it is recognized as a master tumor suppressor that activates the expression of several genes that promote cell cycle arrest or apoptosis in response to stress. 36 Given its obvious role in cancer, p53 has received attention from the PAH community. Mizuno et al. found that p53 deletion exacerbates hypoxia-induced PH in mice. 37 In agreement with a protective role played by p53 in PH, Mouraret et al. demonstrated that daily intraperitoneal treatment of Nutlin-3a that inhibits p53-murine double minute2 (MDM2) interaction and activates p53 signaling improved PH, right ventricular hypertrophy and distal PA remodeling in mice exposed to chronic hypoxia or Sugen/hypoxia. Moreover, these beneficial effects were not observed in mice deficient for either p53 or p21 (cyclin-dependent kinase inhibitor), pinpointing the critical requirement of p53 stabilization and p21 expression in the Nutlin-3a effects. 38 Taking into account that p53 regulates many cellular processes such as metabolism, autophagy, and DNA repair, further studies are needed to delineate its precise role in PAH development and progression.39,40
Telomere length maintenance
Unlimited proliferation is one of the hallmarks of cancer (Fig. 2). 41 During each round of division, normal cells shorten their telomeres (a highly organized and dynamic nucleoproteic structure located at the ends of chromosomes) due to the end replication problems. In cancer cells, reactivation of the reverse transcriptase subunit of telomerase (TERT) stabilizes telomere length and thereby circumvents the senescence program and provides an endless replicative potential. 42 A similar mechanism was proposed in PAH, accounting for the exaggerated PASMC growth. Indeed, increased expression and activity of TERT was observed in lungs from PAH patients as well as in multiple PAH models.43,44 Moreover, PAH-PASMCs carry longer telomeres than control cells and the telomere lengths in PAH-PASMCs strongly correlates with PVR values. 45 Accordingly, chronically hypoxic mice deficient for TERT or the telomerase RNA component (TERC) exhibit reduced telomere length and develop less severe PH. 44 While over-expression of telomerase maintains telomere length and contributes to cell immortalization, accumulating evidence also indicates that telomerase increases capacity for DNA repair and renders cells more resistant to apoptosis. 46 Therefore, the over-expression of telomerase seen in PAH-PASMCs may also contribute to the sustained DNA repair capacity reported in these cells.47,48
Adaptive pro-survival mechanisms in PAH cells
Although exposed to stressful conditions, including a pseudo-hypoxic environment characterized by normoxic activation of HIF-1α, inflammation, shear stress, and oxidative stress that jeopardize their survival, PA cells from PAH patients exhibit a pro-proliferative and anti-apoptotic phenotype, indicating that cells have developed cytoprotective responses preserving homeostasis (Fig. 2).
DNA damage in pulmonary arterial hypertension
DNA repair processes are essential for maintenance of genomic integrity. Many DNA damaging events continuously challenge the genome integrity of dividing cells. In order to cope with DNA damage, cells trigger a signaling network termed as DNA damage response (DDR) that senses, signals, and repairs DNA lesions. Successful repair allows cells to continue proliferating. In contrast, unsuccessful DNA repair results in accumulation of DNA damage and activation of programmed cell death. 49 DNA damage and repair have recently been linked to the onset and progression of PAH. It is proposed that in early stages of the disease, persistent stress coupled to inefficient DNA repair mechanisms produces irreversible accumulation of DNA damage, PAEC apoptosis and subsequent selection of apoptosis-resistant, proliferative vascular cells. This is supported by findings showing that reduced bone morphogenetic protein receptor type II (BMPR2) signaling increases vulnerability of PAECs to DNA damage and apoptosis due to reduced expression of breast cancer 1 (BRCA1) and topoisomerase (DNA) II binding protein 1 (TOPBP1), two key players in DNA damaging signaling.50,51 Moreover, mice deficient for Ku70, a subunit of the Ku protein complex that plays an essential role in the non-homologous end-joining (NHEJ) pathway for DNA double-strand break (DSB) repair, display lung abnormalities reminiscent of plexiform lesions seen in human PAH. 52 As opposed to the early stages of the disease, sustained activation of the DNA repair machinery was identified as an adaptive response used by PAH cells to counteract the stress-induced DNA damage, allowing them to survive and proliferate and leading to the occlusion of small PA. 53 Indeed, increased expression and activation of the DNA repair protein poly (ADP-ribose) polymerase family, member 1 (PARP-1) due to miR223 downregulation, is a feature of PAH-PASMCs contributing to the cancer-like phenotype of these cells. Importance of PARP-1 in acquired resistance to apoptosis of PAH cells is exemplified by in vivo studies showing that pharmacological inhibition of PARP-1 reversed established PAH in two experimental models.47,48 Consistently, expression of the translationally controlled tumor protein (TCTP), implicated in cell growth as well as DNA damage sensing and repair, 54 was identified by proteomic screening as being upregulated in blood outgrowth endothelial cells from BMPR2 mutated PAH patients. 55 Based on these findings, exacerbating DNA damage in remodeled pulmonary arteries, in either exposing cells to DNA damaging agents or inhibiting factors that sense or respond to DNA damage offers a new therapeutic avenue for reversing PAH.
Adaptation of proteostasis network
In response to cellular stress, cancer and PAH cells accumulate misfolded proteins and aggregates, harmful to the cell. To deal with these cytotoxic agents, cells have developed a complex network of protein quality-control mechanisms. Central to these strategies are the activation of molecular chaperones to cope with the increasing number of misfolded proteins and the promotion of their clearance by the ubiquitin-proteasome system (UPS) and autophagy. The coordinated and complementary relationship between these two degradation pathways is exemplified by findings showing that inhibition of proteosomal activities induces autophagy.
Increased expression of molecular chaperones represents a defense mechanism against a wide range of proteotoxic conditions by inducing protein refolding or stabilizing new proteins. Heat shock protein 90 (Hsp90) is one of the most thoroughly characterized chaperones, whose over-expression is exploited by many cancers to stabilize numerous and essential pro-survival proteins. As a consequence, and owing to its nodal properties, Hsp90 has been extensively investigated as a target for cancer therapy. As observed in cancer cells, Hsp90 was found to be upregulated in remodeled distal pulmonary arteries of PAH patients. Moreover, inhibition of cytosolic Hsp90 was shown to alleviate PAH progression in the MCT model, as illustrated by lower pulmonary pressure and absence of right ventricular hypertrophy. 56 Nevertheless, since PAH is most frequently diagnosed at late stages when patients already present structural abnormalities, the efficacy and safety of Hsp90 inhibition in experimental models with established PAH remains to be investigated.
Autophagy, a stress-induced catabolic process characterized by the formation of double-membrane vesicles known as autophagosomes in which intracellular protein aggregates and damaged organelles are engulfed, degraded by fusion with lysosomes and recycled to sustain cellular metabolism, has been proposed to mediate apparently contradictory roles in cancer cells in stimulating both cell death and cell survival depending on the context. Although autophagy was repeatedly found to be increased in PAH cells,57,58 its role in PAH also remains uncertain. On the one hand, Long et al. demonstrated that inhibition of autophagy with Chloroquine exerts both preventive and ameliorating effects in the monocrotaline rat model of PAH, a beneficial effect accompanied with preserved BMPR2 expression. 58 On the other hand, Lee et al. found that loss of function of the autophagic protein microtubule-associated proteins 1A/1B light chain 3B (LC3B) increases susceptibility to hypoxia-induced pulmonary hypertension. 57 Based on these findings, more experimental studies are required before drawing any conclusion on the role of autophagy in both early and late stage of the disease. Intimately linked to autophagy, the UPS is the major mechanism of non-lysosomal proteolysis of intracellular proteins and as such is critical for the proliferation and survival of cancer cells. Based on antitumor activity in a broad range of preclinical cancer models, targeting the UPS has emerged as a rational apoptotic-based approach to reverse pulmonary vascular remodeling in PAH. Although proteasome inhibitors including Bortezomib59,60 and Carfilzomib 61 have demonstrated beneficial effects reducing the PA wall thickness, the potential toxicity of these inhibitors remains a strong concern. Since UPS and autophagy cooperate to remove abnormal proteins, coupling autophagy inhibition strategies with proteosomal inhibitors may provide synergistic benefits.
Metabolic reprogramming and mitochondrial dynamic
Cancer and PAH cells have been documented to undergo metabolic adaptations (Fig. 2).62–66 In response to hypoxia, endoplasmic reticulum stress, or inflammation, this metabolic reprogramming towards a glycolytic metabolism, known as the Warburg effect, has been shown to provide a selective pro-survival advantage to distal PA cells, irrespective of oxygen availability.63,64,67,68 This metabolic shift driven by HIFs overactivation (through the transcriptional upregulation of glucose transporters, hexokinase, pyruvate dehydrogenase kinase [PDK], and lactate dehydrogenase [LDH]) is directly associated with the hyperpolarization of the mitochondrial membranes. This leads to an increase in the opening threshold of the mitochondria transition pore (a channel through which pro-apoptotic factors move to the cytoplasm), thereby reducing the basal sensitivity to cell death inducers. In addition to augmented glycolysis, cancer and PAH cells rely on other metabolic fuels with increased fatty acid (FA) synthesis and boosted glutamine metabolism to sustain higher proliferation rate and resist to cell death signals.69,70 FA are essential components of biological membranes and are important substrates for energy metabolism. Mice deficient for the metabolic enzyme malonyl-coenzyme A decarboxylase (a key enzyme involved in FA biosynthesis) are resistant to the development of chronic hypoxia-induced PH. Consistently, treatment with trimetazidine (an antianginal drug that shifts metabolism from fatty acid oxidation to glucose oxidation) was shown to increase glucose oxidation/glycolysis ratio restoring mitochondrial functions and related molecular abnormalities in vitro and to decrease PA pressure, right ventricular hypertrophy and muscularization of small PAs in several PAH models. 71
Recent discoveries have highlighted that the morphology of subcellular distribution of mitochondria differs between PAH and control cells. Compared to control cells, mitochondria of PAH cells are often clustered perinuclearly. This subcellular organization of mitochondria may represent a major component of the stress response. Indeed, hypoxia was found to trigger microtubule-dependent accumulation of mitochondria in close proximity to the nucleus of PAECs, resulting in elevated levels of reactive oxygen species (ROS) in the nucleus and increased binding of HIF-1α to the VEGF promoter. 72 Moreover, this perinuclear positioning of mitochondria may facilitate the transfer of substrates, co-factors, and complex from the mitochondria to the nucleus leading to epigenetic modifications that remodel the chromatin, influence gene transcription and contribute to stress adaptation. 73
In healthy cells, mitochondria constantly divide and fuse to maintain mitochondrial functions. Mitochondrial fusion allows the exchange of contents including metabolites, proteins, and mitochondrial DNA. Such exchanges likely act as a defense mechanism buffering the effect of stress on mtDNA damage. However, mitochondrial fission is considered as a protective form of quality control mechanism by segregating damaged mitochondria from the healthy pool and their removal by mitophagy. 74 Indeed, accumulation of dysfunctional mitochondria is directly associated with augmented cellular toxicity. In response to various factors, including activation of HIF-1α, mitochondria of PAH cells undergo fission and show a fragmented pattern; a feature also observed in various cancer cells.75,76 Inhibition of fission was associated with reduced cell proliferation, cell cycle arrest, and induction of apoptosis in vitro and regression of PAH in experimental models. 77 Based on these data, it can be speculated that increased mitochondrial fission promotes mitophagy and that correction of the mitochondrial networking in PAH cells induces apoptosis secondary to inefficient clearance of dysfunctional mitochondria. However, further studies are needed to better understand the specific nature of the relationship between fusion/fission imbalance and PAH cell outcomes.
Increased expression of anti-apoptotic proteins
Acquisition of apoptosis-resistance of PAH cells is intimately linked to an imbalance between pro-apoptotic and anti-apoptotic proteins. As observed in cancer cells, upregulation of Survivin, a member of the inhibitors of apoptosis protein (IAP) family is a hallmark of PAH cells, while undetectable in healthy cells. 78 The deleterious role of the latter in vascular remodeling was illustrated by a study showing that intratracheal nebulization of an adenovirus carrying a phosphorylation-deficient Survivin mutant with dominant-negative properties reversed established MCT-induced PAH and prolonged survival. 78 Accordingly, pharmacological inhibition of Survivin using YM155 was reported to improve PA pressure and right ventricular hypertrophy in rats exposed to chronic hypoxia. 79 PAH-PASMCs are also characterized by an increased expression of the anti-apoptotic regulator B cell leukemia/lymphoma 2 (Bcl-2) and a diminished expression of the pro-apoptotic protein BIM.17,27 Similarly, over-expression of ARC (apoptosis repressor of CARD), an inhibitor of cell death, was also observed in remodeled vessels of patients with PAH as well as in rats in response to chronic hypoxia. 80 Mice deficient for ARC also displayed attenuated chronic hypoxia-induced vascular remodeling and resistance to PH, underscoring the critical role of suppressed apoptosis in the development and progression of vascular lumen obliteration. Therefore, this disequilibrium between pro- and anti-apoptotic proteins is a hallmark of PAH-PASMCs contributing to resistance to apoptosis in stressful conditions.
Role of ion channels
Potassium (K+) and calcium (Ca2+) channels are pore-forming transmembrane proteins that regulate flow of ions across biological membranes and thus govern a plethora of biological processes including cell proliferation and apoptosis. 81 Multiple studies have revealed that the deregulated expression of K+ and Ca2+ channels is associated with the development and progression of cancer and PAH.81,82 In addition to influence cell contractibility, the reduced K+ efflux in PAH-PASMCs secondary to suppression of voltage-gated potassium channels was accompanied by an elevation in the cytosolic Ca2+ concentration protecting cells from apoptosis and supporting proliferation.81,83,84 The significance of decreased K+ channels expression, particularly K+ voltage-gated channel, shaker-related subfamily, member 5 (Kv1.5) in terms of disease progression, is illustrated by findings showing that restoration of the function of Kv1.5 in PAH-PASMCs or cancer cells promotes apoptosis and reduced PA medial hypertrophy in rats exposed to chronic hypoxia.16,85–87 The sustained elevation of the intracellular Ca2+ concentration in PAH-PASMCs, due to K+ channels dysfunction coupled with excessive expression of transient receptor potential-canonical (TRPC) channels, has been documented to exert stimulating effects on the calcineurin signaling that boosts the capacity of the cells to resist to death.17,88 Interestingly, the loss of mitochondrial uncoupling protein 2 (UCP2) in control PASMCs was reported to cause a profound decline in the entry of Ca2+ from the endoplasmic reticulum to the mitochondria, that in turn led to a reduction in the activity of pyruvate dehydrogenase, suppression of mitochondrial function, activation of HIF1α and NFAT, and resistance to cell apoptosis. 67 In a similar fashion, increased expression of miR-25 and miR-138 in PAH-PASMCs was found to directly downregulate expression of mitochondrial Ca2+ uniporter (MCU) leading to reduced intramitochondrial concentration of Ca2+ and promotion of Warburg effect. 89 Based on these findings, reduced intramitochondrial Ca2+ concentration appears as one of the mechanism used by PAH-PASMCs to acquire a cancer-like phenotype.
Genetic predisposition/susceptibility: role of BMPR2
It has been recognized for a long time that some gene variants or mutations can cause a predisposition to cancer. 90 For example, mutations in BRCA1 or BRCA2 genes greatly increase a person’s risk of developing breast cancer. BMPR2 pathogenic variant is the most common genetic risk factor for PAH development, accounting for approximately 25% of idiopathic PAH and the majority of hereditary cases.91,92 Patients carrying a BMPR2 mutation are generally younger at diagnosis and have more severe disease. 92 Under basal conditions, mice heterozygous for Bmpr2 do not develop PH. Nevertheless, exposition to these mice to hypoxia or inflammatory stress leads to exacerbation of PH compared to wild-type mice.93,94 These findings, along with genetic studies reporting low penetrance of PAH among carriers of a BMPR2 mutation suggest that additional factors (the so-called “second-hit” hypothesis) required for the disease expression. In a similar way, loss of Bmpr2 function in mice expressing the mouse mammary tumor virus Polyoma virus middle T antigen (MMTV.PyVmT) was shown to increase tumor metastasis. 95 Mechanistically, reduced BMPR2 signaling in PAH cells was shown to promote the recruitment of inflammatory cells 96 and endothelial-to-mesenchymal transition, 97 and to impair mitochondrial function, 98 cooperatively contributing to endothelial cell dysfunction, cell demise, and occlusive vascular remodeling. 99 Importantly, Ataluren, a drug that permits ribosomal read through of premature stop codons and shown to induce full-length functional dystrophin in the mdx mouse model of Duchenne muscular dystrophy, 100 was able to correct BMP signaling in vitro suggesting that this compound could be proposed as a new personalized therapy for a subgroup of PAH patients harboring BMPR2 nonsense mutations. 101 It must keep in mind that contrary to cancer in which a wide array of mutations have been identified as predisposing factors, evidence on the contribution of genes to the hereditary predisposition to PAH is limited.
Epigenetic alterations
Major mechanisms of epigenetic regulation are DNA methylation, histone modification, and RNA interference. It is now widely accepted that epigenetic abnormalities are key drivers of both cancer and PAH development.102–104 While no mutation of SOD2 gene has been found in PAH, reduced expression of the latter is a common feature of PASMCs from PAH patients and animal models. Genomic bisulfite sequencing has demonstrated selective hypermethylation of CpG island in the intronic and promoter sequences of the SOD2 gene in PASMCs from Fawn-Hooded rats (FHR, a strain that spontaneously develops PH with age). 105 Importantly, the reversal of SOD2 hypermethylation by the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine was documented to restore both SOD2 expression and the ratio of PASMCs proliferation to apoptosis underscoring an epigenetic component in PAH pathogenesis. Along with DNA methylation changes, reversible acetylation of histones, governed by the opposite action of histone acetyltransferases (HAT) and histone deacetylases (HDAC), has emerged as an important post-translational modification that regulates chromatin structure and gene expression. In PAH, over-expression of nuclear-located HDAC1, 2, and 4 has been detected in human lung extracts from idiopathic PAH patients compared to controls, 106 suggesting that epigenetic changes that accompanied these differential expressions contribute to the structural anomalies seen in the disease. Intimately linked to proteins that incorporate histone marks are those implicated in their interpretation. A member of the bromodomain and extra-terminal motif (BET) protein family called bromodomain protein 4 (BRD4) was recently found to be upregulated in PAH-PASMCs affecting the expression level of Survivin, NFATc2 and Bcl-2. 107 The over-expression of BRD4 was shown to result from the downregulation of miR-204.107,108 Micro RNA (miRNA) are fundamental epigenetic components that repress translation and/or induce mRNA degradation in interacting with complementary sequences found in the 3’ untranslated transcribed region of target mRNAs. In addition to miR-204, the aberrant expression of a handful of miRNA has been related to PAH and cancer.109–111 For instance, several studies have provided convincing experimental evidence that the miR-17-92 cluster is a bona fide oncogene. 112 In PAH, BMPR2 was identified as a direct target of miR-17-5p and miR-20a 113 and genetic inactivation of miR-17-92 in smooth muscle cells conferred protection against hypoxia-induced PH in mice. 114 Consistently, pharmacological inhibition of miR-17 using antagomirs was demonstrated to improve lung and heart function in experimental PAH. 115 Although the contribution of epigenetic alterations in the development and progression of PAH is still poorly understood, epigenetic targets offer therapeutic potential.
Overview of therapeutic strategies used in cancer to treat PAH
Although great strides in treatment and care have been made, current therapies principally devoted to reverse the sustained vasoconstriction fail to reverse PAH and only result in a modest improvement in the morbidity and mortality. In the face of this reality, it is important to consider new therapies targeting alternative pathways such as those implicated in vascular remodeling. The numerous similarities between cancer cells and PAH cells have opened to the possibility of exploiting therapeutic strategies used in cancer to combat PAH (Fig. 4). Taking into account that PAH is most frequently diagnosed when patients have reached a more advanced stage of the disease, therapies aiming to stop the proliferation and to induce cell death in remodeled vessels appear to be fully justified.
Pathogenic concepts of PAH. Like cancer cells, PAH cells are exposed to stressful conditions. To deal with these insults causing initial cell death, PAH cells have developed adaptive mechanisms allowing them to survive and proliferate over time and leading to vascular remodeling of distal pulmonary arteries. Given that PAH and cancer cells share numerous similarities, therapeutic strategies used in cancer are exploited to treat PAH. “Anticancer” drugs showing beneficial effects in experimental PAH models and are tested in PAH clinical trials are highlighted within the dotted boxes.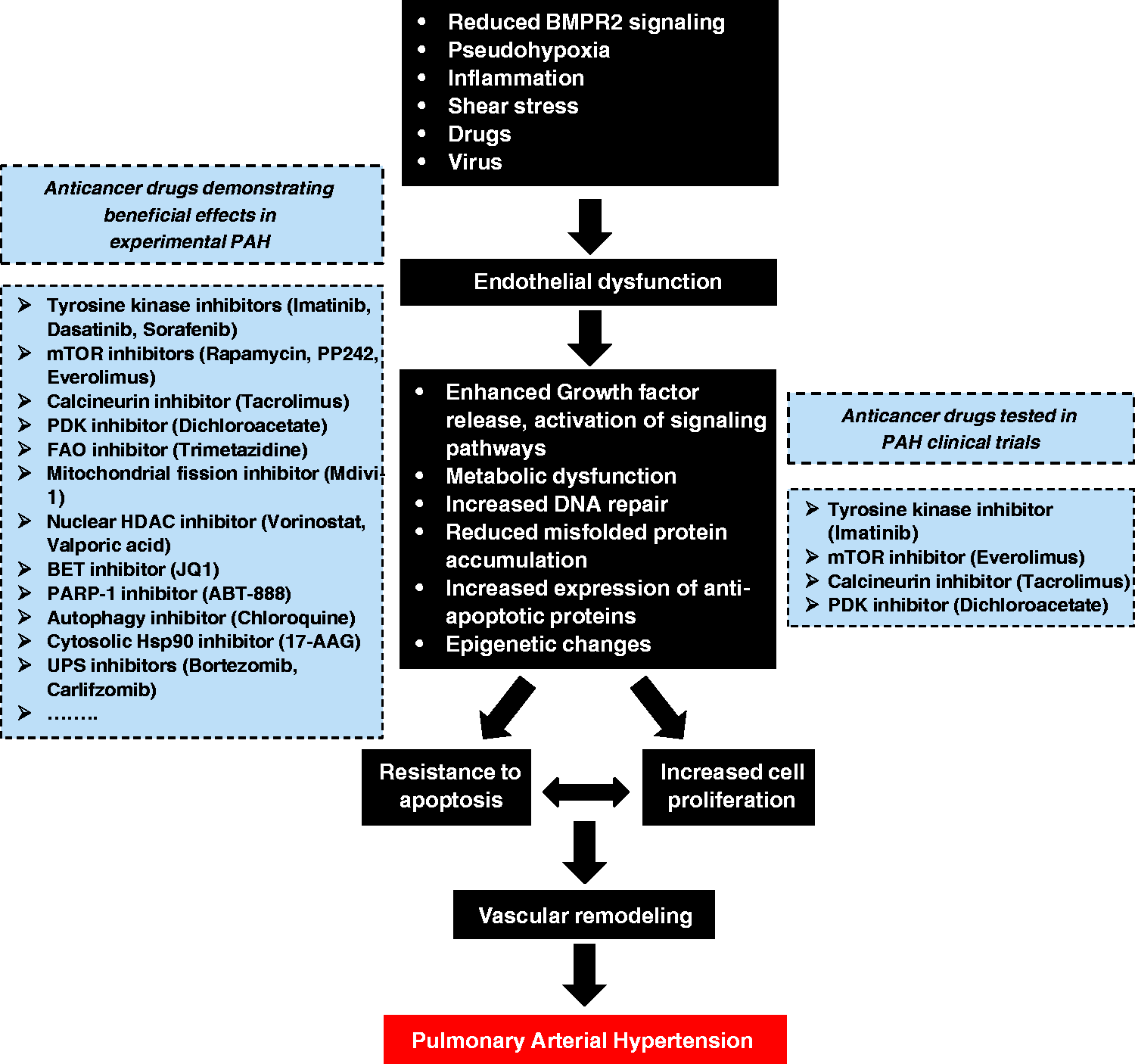
Tyrosine kinase inhibitors
Chemotherapy is commonly used as a front-line therapy for cancer. Tyrosine kinases inhibitors, including Imatinib and Dasatinib, have been successfully employed in the treatment of chronic myeloid leukemia. 116 In addition to inhibit the BCR-ABL kinase, these inhibitors target other tyrosine kinases including c-KIT and PDGF receptor. Because c-KIT-positive cells infiltrate remodeled vessels 117 and PDGF expression is elevated in lung biopsies of PAH patients,118,119 studies were undertaken to evaluate the therapeutic benefits of Imatinib and its derivative in preclinical models. Imatinib treatment demonstrated remarkable efficacy in several experimental models with established PAH, reducing right ventricular systolic pressure and hypertrophy, medial wall thickness, and mortality.119–121 Similar results were obtained with Dasatinib. 120 Despite these remarkable results achieved in pre-clinical PAH models, mixed results were obtained in humans. Results from clinical trials indicated that Imatinib is effective in improving exercise capacity, pulmonary hemodynamics, and echocardiographic measures as add-on therapy in patients with severe PAH.122–125 Nevertheless, the risk of clinical worsening was enhanced in Imatinib-treated patients likely because of adverse effects. Notably, cardiotoxicity and occurrence of subdural hematoma in PAH-treated patients has tempered the positive reports, discouraging the use of these compounds in PAH.126,127 Moreover, development of PAH was reported in some patients treated for Dasatinib for chronic myeloid leukemia. 128 In experimental models, treatment or rats with Dasatinib or Imatinib did not induce PAH. However, Dasatinib, but not Imatinib, -pretreated rats developed an exaggerated response to MCT or chronic hypoxia-induced PH, 129 indicating that Dasatinib can predispose to PAH development. Based on these data, tyrosine kinase inhibitors remain a promising tool in the rescue armamentarium for PAH. The beneficial hemodynamic effects of Imatinib in a patient subset support the need to identify individual factors that predict a positive or negative response to this drug.
Alkylating agents
Although Harbaum et al. reported a case in which PAH was completely resolved following cytotoxic treatment including cyclophosphamide, doxorubicin, and etoposide regimen, 130 exposure to these chemotherapeutic agents was identified as a risk factor for the development of PH in human.131,132 On the basis of these findings, the consequences of alkylating agents’ exposure on the pulmonary vasculature were explored in several species. In all the tested species, cyclophosphamide was shown to induce severe PH in mice associated with pulmonary venous remodeling reminiscent of pulmonary veno-occlusive disease (PVOD), an uncommon form of PH. 132 In direct connection with this, mitomycin-C therapy was also identified as a potent inducer of PVOD in humans and rats, 133 further supporting the fact that, in the early stage of the disease, DNA damaging agents pose a significant genomic stress contributing to endothelial cell apoptosis and PAH development, but in advanced stages of the disease, it may induce stress overload of the DDR pathway, improving vascular remodeling.48,53 Nevertheless, the current evidence suggests that there is no place for these agents in PAH therapy.
mTOR inhibitors
The evidence linking activated mTOR signaling to PAH have engendered significant interest in targeting this pathway for PAH therapy. Rapamycin (Sirolimus), a macrolide immunosuppressant and preferential mTORC1 inhibitor, was used in experimental PAH leading to mitigated results. Indeed, rapamycin failed to demonstrate beneficial effects in terms of survival, pulmonary hemodynamics, vascular remodeling, and right ventricular hypertrophy in the MCT model. 134 These results contrast with those reporting curative effects of high doses of rapamycin or PP242 (a more effective mTORC1 inhibitor than rapamycin) in MCT-induced PAH and chronic hypoxia-induced PH in rats, respectively.27,135 A pilot study was conducted to assess the safety and efficacy of Everolimus (an oral antineoplastic agent derived from rapamycin and approved for various conditions such as prevention of organ rejection after transplant) in patients suffering from PAH or chronic thromboembolic PH with very limited exercise capacity. Treatment with Everolimus was safe and tolerable and resulted in a significant decrease in PVR and an increase in 6-minute walk distance. 136 These preliminary results may justify its formal evaluation in future clinical trials.
Tacrolimus
Using a high-throughput assay to screen over 3500 FDA-approved drugs and bioactive compounds for induction of BMPR2 signaling, Spierkerkoetter et al. identified Tacrolimus (FK506), a calcineurin inhibitor and immunosuppressant used following solid organ transplantation. The therapeutic benefit of low-dose Tacrolimus was demonstrated in multiple experimental PAH models. 137 Interestingly, a clinical benefit was reported following the compassionate use of low-dose FK506 in three end-stage PAH patients. 138 In all patients, FK506 treatment was associated with stabilization in cardiac function, freedom from hospitalization for right ventricular failure, and upregulation of BMPR2 expression in peripheral blood mononuclear cells. On the basis on these findings, a randomized, double-blind, placebo-controlled trial is ongoing to evaluate the safety and tolerability of FK506 in stable patients with PAH.
Dichloroacetate
Molecules that stimulate mitochondrial respiration are expected to enhance the production of ROS in mitochondria and to induce mitochondria-dependent apoptosis in diseased cells, sparing control cells. In direct connection with this, it has been demonstrated that dichloroacetate (DCA), which inhibits pyruvate dehydrogenase kinase (PDK) causing elevation of oxidative phosphorylation and ROS production, directly induces cancer cells and PAH-PASMCs apoptosis without impacting control cells. 16 DCA was shown to regress established PAH in FHR rats 139 and MCT-induced PH rats, 140 as well as in mice over-expressing the serotonin transported targeted to smooth muscle cells. 141 Based on positive effects on animal models, the safety, tolerability, and effectiveness of DCA is currently studied in patients with moderate-to-severe PAH despite background therapy (NCT01083524).
Epigenetics-based therapies
Understanding epigenetic mechanisms arouses a growing interest in various diseases, as it may serve as a new therapeutic approach. Indeed, epigenetic modifications do not change the genetic program but just refer to a differential genome lecture that is reversible. Although the overall status of histone modification is largely unknown in PAH, class I and IIa HDAC inhibitors have also been exploited in PAH for their beneficial effects under several pathophysiological conditions like cancer and inflammation related pathologies. 142 These investigations have yielded mixed results. Indeed, studies conducted in rats exposed to chronic hypoxia have reported positive impact of class I or pan-HDAC inhibitors on pulmonary vascular remodeling.106,143 Conversely, suppression of HDACs had no therapeutic or beneficial effect in the SU5416/hypoxia-exposed rat model 144 or was accompanied by a deterioration of the right ventricular function in a rat model of right ventricular hypertrophy. 145 Given the unresolved controversies highlighted by these works and concerns regarding the unforeseeable pleiotropic epigenetic effects caused by these inhibitors, the potential utility of HDAC inhibitors in PAH need further evaluation by either genetic manipulation or more selective HDAC inhibitors.
The epigenetic sensor BRD4 that recognizes and binds acetylated histones influencing chromatin remodeling and gene expression has emerged as a promising drug target in multiple hyperproliferative disorders. 146 As documented in many cancer types, 146 BRD4 was found to be upregulated in PAH-PASMCs sustaining cell survival and proliferation. More importantly, molecular and pharmacological inhibition of BRD4 reversed established PAH in the Sugen/hypoxia rat model, suggesting that BRD4 inhibitors currently tested in clinical trials for cancer may offer a new therapeutic perspective for PAH patients.107,147 Finally, miRNAs represent an additional layer of regulation of gene expression. Among the miRNAs involved in the pathogenesis of PAH,109–111 many of them have been associated with cancer. Although many miRNAs with therapeutic value in PAH have been discovered, microRNA-based therapies are still in their infancy and potential adverse effects need to be evaluated.
Summary
In contrast to the earlier belief that vasoconstriction plays a key role in PAH pathogenesis, it is now clearly established that excessive proliferation and resistance to apoptosis of fibroblasts, PASMCs and PAECs are pivotal components of pulmonary vascular remodeling. Due to the long list of pathogenic analogies between cancer and PAH (Fig. 2), the use of antineoplastic drugs in combination with vasodilators seem to be a promising way to tackle established PAH. Nevertheless, although apoptosis-based therapies represent an attractive avenue to regress vascular lesions in PAH, their non-selectivity remains a major limitation, as they cannot differentiate healthy cells from diseased cells, leading to important side effects limiting their utilization. To circumvent this problem, identifying a biochemical feature specific to PAH cells (i.e. lacking in normal cells) with key network properties resulting in a combinatorial attack on multiple signaling pathways implicated in PAH pathogenesis and leading to programmed cell death is critical.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.