
Abstract
Ketamine, a drug introduced in the 1960s as an anesthetic agent and still used for that purpose, has garnered marked interest over the past two decades as an emerging treatment for major depressive disorder. With increasing evidence of its efficacy in treatment-resistant depression and its potential anti-suicidal action, a great deal of investigation has been conducted on elucidating ketamine’s effects on the brain. Of particular interest and therapeutic potential is the ability of ketamine to exert rapid antidepressant properties as early as several hours after administration. This is in stark contrast to the delayed effects observed with traditional antidepressants, often requiring several weeks of therapy for a clinical response. Furthermore, ketamine appears to have a unique mechanism of action involving glutamate modulation via actions at the N-methyl-D-aspartate (NMDA) and
Introduction
Major depressive disorder (MDD) remains a significant contributor to the global burden of disease and has been reported to place second for causes of worldwide disability. 1 In a 2012 epidemiological study of mental health in Canada, the lifetime prevalence of MDD was noted at 11.3%. 2 Unfortunately, the pathophysiology of depression remains largely unknown. While the monoamine hypothesis, postulating that depression is the result of a functional deficiency of serotonin and/or noradrenaline neurotransmitters in the central nervous system,3,4 has been useful for explaining the pharmacology of many antidepressant drugs, it is overly simplistic and does not seem to fully encompass the pathways underlying depression. One significant limitation is the discrepancy in the time frame of antidepressant drugs’ effects on monoamine neurotransmitters (hours to days) and on clinical symptoms (several weeks).3–5 Following treatment with antidepressant drugs, only half of patients are noted to have a significant clinical response.6,7 Furthermore, up to one-third of patients are considered to have treatment-resistant depression (TRD), defined as a lack of response to two or more adequate trials of antidepressant medications. 8 Given this large population of patients with TRD, there is a significant need for development of novel and more efficacious antidepressant treatments.
Ketamine is a non-competitive antagonist at glutamate N-methyl-D-aspartate (NMDA) receptors and has been traditionally used as a dissociative anesthetic. It was first reported to have antidepressant properties in 2000, when it was demonstrated that an intravenous (IV) administration of a sub-anesthetic ketamine dose resulted in a reduction of MDD symptoms rapidly and continuing to 72 h after treatment. 9 Subsequent randomized controlled trials (RCTs) replicated this finding, demonstrating a 60–70% response rate of ketamine in TRD populations.10–14 Of specific interest is the finding that ketamine has a rapid clinical effect within 2–4 h after administration. However, ketamine’s antidepressant properties are also transient, lasting an average of 1 week following a single infusion10–14 and 18–19 days following repeated infusions.15,16 Retrospective, real-world clinical data demonstrated a response rate of 44% after six intravenous ketamine treatments in a population of complex patients with multiple comorbidities and ultra-resistant depression.17,18 Furthermore, ketamine has been reported to also have anti-suicidal and anti-anhedonic actions.14,19,20 Not only does ketamine exert a rapid clinical effect within several hours and demonstrate efficacy in patients unresponsive to other antidepressants, it appears to have a novel mechanism of action that is distinct from conventional antidepressant drugs. This paper is a review of the current state of knowledge on the pharmacology/pharmacokinetics, status of clinical trials, adverse effects and postulated mechanisms of action of ketamine as an antidepressant. In addition, biomarkers including sleep, cognition, inflammation and metabolism and neuroimaging will be discussed.
As preparation for this review, the authors performed a literature search on each specific topic of interest using PubMed/MEDLINE and the Web of Science Core Collection, including papers in English produced from 2000 to 2019. Search topics included ‘ketamine as an antidepressant’, ‘mechanisms of action of ketamine as an antidepressant’, ‘biomarkers for antidepressant response to ketamine’, ‘enantiomers and metabolites of ketamine as antidepressants’, ‘inflammation and antidepressant actions of ketamine’, ‘ketamine and metabolism’, ‘sleep and antidepressant actions of ketamine’, ‘adverse effects of ketamine as an antidepressant’, ‘effects of ketamine on cognition’ and ‘neuroimaging studies on antidepressant effects of ketamine’.
Basic chemistry, pharmacology and pharmacokinetics of ketamine
Ketamine, an arylcyclohexylamine derivative (see Figure 1), is a racemate, that is a mixture of R and S enantiomers. Chiral forms (enantiomers) of a drug have the same number and type of atom groupings, but have different arrangements in space, analogous to right and left hands. Usually the chiral center consists of four different groups attached to a carbon atom, and when the compounds are synthesized there are often nearly equal quantities of the two enantiomers; that mixture is called a racemate or racemic mixture. Pairs of enantiomers differ in their optical activity, and rotate plane polarized light to the left (–, or levorotatory enantiomer) or the right (+, or dextrorotatory). Thus the prefixes (+)- and (–)-, dextro- and levo-, or d- or l- are used. The terms R (rectus) and S (sinister) are also often used, and describe the enantiomers based on their absolute configuration. While optical activity can be influenced by temperature and light wavelength, the absolute configuration can be modified only by breaking and reforming chemical bonds, and there is no relationship between absolute configuration and optical activity; for example, some drugs are R(+), S(–) while others are R(–), S(+).21,22
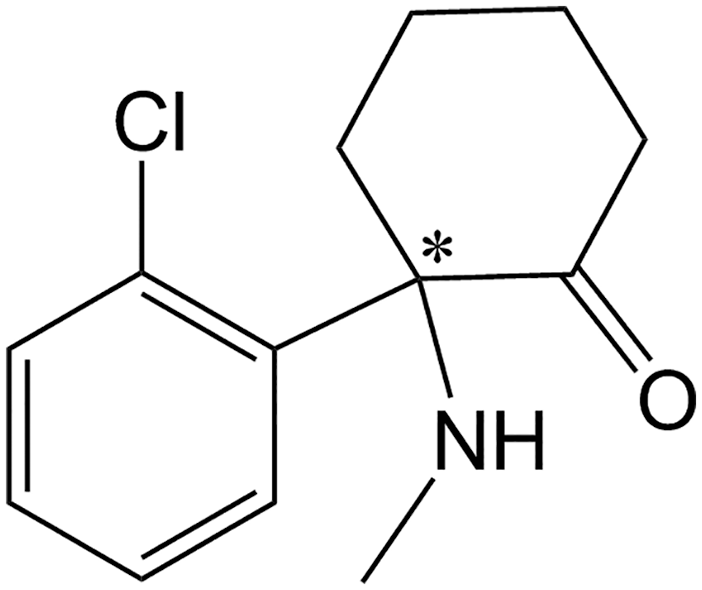
Chemical structure of ketamine, with the chiral center indicated by an asterisk.
There are many examples of antidepressants that contain a chiral center, 21 and often these drugs have been used as racemates. However, often the two enantiomers may differ from one another significantly with regard to pharmacokinetics and pharmacodynamics. Racemates have often been used instead of individual enantiomers because of the difficulty and expense involved in separating the enantiomers from each other. Often the pharmacological activity may reside primarily in one enantiomer, and the two enantiomers may influence each other’s pharmacokinetics. Complicating factors that may arise when using racemates include: (1) association of therapeutic actions and/or adverse effects with one enantiomer; (2) differences in absorption, protein binding and/or renal clearance between the enantiomers; (3) preferential metabolism of one enantiomer over the other by enzymes; (4) one enantiomer having an effect on the pharmacokinetics of the other enantiomer; and (5) differing extents of the enantiomers interacting with co-administered drugs. 21 However, there may also be cases in which the enantiomers produce complementary therapeutic effects or where one of the enantiomers counteracts adverse effects associated with the other enantiomer. Readers are referred to several review articles on this topic.21,23–25
Ketamine has been used for many years as an intravenous anesthetic, but has been the subject of intense interest in recent years in psychiatry after reports of its rapid-acting antidepressant and anti-suicidal effects.9,11,26 It contains a chiral center, and its R(–)- and S(+)-enantiomers have both been studied to varying extents. The S-enantiomer of ketamine (esketamine) is a more potent antagonist than R-ketamine at the phencyclidine site on the glutamate NMDA receptor,27–31 and has stronger analgesic potency than either R-ketamine or racemic ketamine. 32 Esketamine is now being investigated as an antidepressant by several research groups. 33 There is disagreement in the literature about the occurrence of adverse effects such as dissociation, psychoses and cognitive effects with the two enantiomers of ketamine.20,27,34 In animal studies, R-ketamine has been reported to have a rapid onset of antidepressant effects and a better side-effect profile than esketamine34–41; it has also been reported that R-ketamine improves phencyclidine-induced cognitive deficits in mice and that esketamine does not. 42 Importantly, large head-to-head clinical comparisons of esketamine with R-ketamine and racemic ketamine have not yet been reported.41,43
Ketamine is metabolized extensively in the body via CYP2B6- and CYP3A4-mediated N-demethylation to norketamine. Norketamine then undergoes further catabolism to hydroxynorketamines (HNKs) and dehyronorketamine. 20 Several researchers have investigated specific metabolites of ketamine for their antidepressant-like behavioral actions in animal models44–47; 2R,6R-HNK was reported to have antidepressant-like effects and no side effects in rodents. 44 However, the literature on the antidepressant effects of 2R,6R-HNK is controversial, with several contradictory reports.41,48–52 S-norketamine, a metabolite of S-ketamine, has been shown in animal models to have useful antidepressant-like properties and fewer adverse effects than esketamine.35,53 In these animal models, S-norketamine has been reported to be equipotent to S-ketamine with regard to antidepressant-like activity, but less potent than R-ketamine.34,35
Bioavailability of ketamine differs with route of administration. Intravenous administration provides the most predictable dosing with 100% bioavailability, and availability via other routes of administration including intranasal (45%), sublingual (30%), oral (20%), intramuscular (93%) and rectal (30%) is discussed in the literature.31,34,35,46,54
Overview of the status of clinical trials with ketamine and its enantiomers
Since Berman et al. 9 demonstrated a rapid antidepressant effect of intravenous ketamine, numerous studies have now replicated this finding. Multiple meta-analyses55–59 have now concluded that intravenous ketamine is effective as a rapid-acting antidepressant for major depressive episodes in both unipolar and bipolar depression, although one study suggested that there was a greater antidepressant effect size for unipolar depression than for bipolar. 57 While several of these studies have supported claims that ketamine’s antidepressant effect lasts up to 7 days,55,56 other authors have suggested this to be true only for unipolar depression. One meta-analysis found that ketamine loses its effect in bipolar depression after day 3 or 4. 58 Several RCTs have now looked at repeated infusions of six intravenous infusions over several weeks,60–62 but to date no long-term RCTs exist.
Studies on other modalities of ketamine administration are limited at this time. While one RCT on intranasal ketamine 63 suggested that intranasal administration may be a viable alternative, another study 64 was aborted early due to poor tolerance to the intranasal formulation. Other reports based on clinical experience65,66 have reported that although it remains experimental in nature, maintenance intranasal ketamine has been clinically useful in patients who have exhausted other treatment options. Studies on oral and sublingual ketamine have been the subject of a recent systematic review, 67 but the studies reviewed include wide variations in dosing and do not appear to take into account limited bioavailability of oral formulations, so may underestimate efficacy. 68 One previous pilot study has also suggested that intramuscular or subcutaneous routes may be viable options. 69
Intranasal esketamine was approved by the FDA in the United States in March 2019 for major depression that has failed treatment with two or more antidepressants. This approval was based on three acute-phase studies and two maintenance-phase studies. A phase III trial of over 200 patients using esketamine adjunctive to an antidepressant demonstrated significant improvement in depression at 4 weeks compared with those using a placebo nasal spray. 70 Two other phase III trials failed to meet primary endpoints.71,72 It has been suggested that the results of these studies were limited by a fixed dosing design in one 71 and an elderly, more treatment-resistant population in the other. 72 Of note, the esketamine acute studies have been conducted on populations more severely depressed than would be typical for FDA approval for antidepressant treatments of adjunctive medications. 73 Two maintenance studies followed patients on maintenance esketamine up to 88 weeks, and reported decreased risk of depressive relapse when patients used esketamine weekly or every second week, 74 and provided reassuring data on safety to over 1 year of esketamine use. 75 Janssen has ongoing trials in progress, including one that will track safety outcomes to 5 years. 33 It has been suggested that R-ketamine may confer antidepressant effects and greater tolerability than esketamine. 34 Perception Pharmaceuticals has a phase I clinical study with R-ketamine underway from 2019, but to our knowledge no results are available to date. 34
Potential adverse effects of ketamine
Ketamine administered at sub-anesthetic doses by infusion may result in several adverse effects, most of which occur during the infusion period and abate shortly thereafter. These acute and transient effects include an increase in blood pressure (usually asymptomatic), nausea and vomiting, perceptual disturbance, drowsiness, dizziness and dissociation.43,76–78 Blood pressure should be measured prior to ketamine administration and monitored after administration until it returns to normal values. 79 As mentioned previously in this review, there appear to be differences in the degree of adverse effects between the enantiomers of ketamine, although the two enantiomers have not yet been compared in a comprehensive head-to-head clinical study.27,34 Incidence and severity of adverse effects may vary with route of administration and length of time administered, although more investigation must be done on these matters. Swainson et al. 33 have provided a review of the adverse effects associated with intranasal administration of esketamine. It should also be remembered that ketamine is a potential drug of abuse, and high doses, particularly for long periods of time, can result in increased severity of the abovementioned side effects as well as severe urological side effects. 80 In addition, as pointed out in a recent paper by Talbot et al., 81 further studies on the possible risks associated with cessation of ketamine antidepressant treatment are warranted.
Antidepressant mechanisms of ketamine and potential biochemical biomarkers
Glutamate is the main excitatory neurotransmitter in the central nervous system, acting on NMDA and
There has been suggestion that glutamatergic neurotransmission is dysregulated in MDD.83,84 This is supported by findings of elevated serum and plasma glutamate levels in patients85,86 and reduction of plasma glutamate levels following treatment with selective serotonin reuptake inhibitors (SSRIs). 87 Of interest, the severity of depressive symptoms was found to be correlated with plasma glutamate levels. 88 Elevation of extracellular glutamate in MDD may, in part, be caused by loss of glial cells that are responsible for glutamate/glutamine cycling. 89 A consequence of increased extrasynaptic glutamate levels may actually be downstream suppression of glutamatergic neurotransmission via activation of metabotropic glutamate receptor 2 (mGluR2) autoreceptors.
However, ketamine’s mechanism of action is more complex than antagonism of NDMA receptors. This is supported by the finding that other NMDA receptor antagonists such as memantine, lanicemine and nitrous oxide do not exert a consistent antidepressant effect in RCTs. 90 Furthermore, a meta-analysis of single-infusion non-ketamine NMDA receptor antagonists, including traxoprodil, lanicemine and rapastinel (GLYX-13), showed smaller effect sizes in depressive symptom change in comparison with ketamine and non-superiority in remission compared with placebo for unipolar and bipolar depression. 55 It is likely that ketamine’s mechanism involves additional downstream targets, given that it is metabolized rapidly (within hours) but demonstrates longer-lasting antidepressant effects (days to weeks).
The mechanism of ketamine’s antidepressant action involves the following cascade of sequential events.41,91–93 Ketamine has a greater affinity for NDMA receptors on
The majority of the literature on ketamine’s neurochemical effects has involved animal models. In the rat prefrontal cortex (PFC), ketamine was demonstrated to activate glutamate release and neurotransmission. 95 AMPA receptor activation appears to be a critical step in ketamine’s mechanism of action as co-administration of an AMPA receptor inhibitor abolished its antidepressant effects.96–98 Ketamine administration was also shown to enhance AMPA-evoked electrophysiological responses in the rat hippocampus and medial PFC, suggesting that ketamine may augment AMPA receptor transmission.99,100 Furthermore, ketamine increased expression of AMPA receptor GluA1 and GluA2 subunits in the mouse hippocampus.44,101 Within 30 min of treatment, ketamine increased rat brain levels of BDNF 96 and mTOR. 102 This was further substantiated by observations that ketamine increased BDNF and mTOR expression in the rat hippocampus 103 and that pre-treatment with the analgesic tramadol enhanced the antidepressant effects of ketamine in the forced-swim test and potentiated the upregulation of mTOR in the rat PFC and hippocampus. 104 The increase in hippocampal and PFC BDNF and mTOR levels appears to be mediated by AMPA receptors as pre-treatment with an AMPA receptor antagonist increased forced-swim test immobility time and reduced levels of BDNF and mTOR, whereas pre-treatment with an AMPA receptor agonist reduced forced-swim test immobility times and increased levels of BDNF and mTOR. 105
Several studies have reported that ketamine’s antidepressant effects are abolished when animals were pre-treated with rapamycin, an inhibitor of mTORC1.102,106 However, it should also be noted that the role of mTORC1 in ketamine’s antidepressant action may not be as clear-cut as originally surmised. In mice, ketamine administration was not noted to affect mTOR phosphorylation in hippocampal or cortical tissue44,96 and rapamycin did not block ketamine-induced antidepressant effects. 96
There is significant support for the role of BDNF pathways in ketamine’s antidepressant mechanism. Use of genetic mutant mice lacking BDNF prevented the behavioral antidepressant responses of ketamine. 96 The authors also proposed that ketamine-mediated antagonism of NMDA receptors deactivates eukaryotic elongation factor 2 (eEF2) kinase, resulting in de-suppressing BDNF translation. Mice with a Val66Met single-nucleotide polymorphism in the BDNF gene exhibit impairments in BDNF release and mRNA trafficking; ketamine administration was demonstrated to have reduced synaptogenesis in the PFC and impaired antidepressant behavioral effects in these animals. 107
In rats vulnerable to chronic mild stress, ketamine was noted to result in reversal of anhedonic behavior, partial attenuation of hippocampal impairments in presynaptic release of glutamate and GABA, along with complete restoration of dendritic atrophy and dendritic BDNF mRNA trafficking. 108 In a mouse model of social defeat stress, ketamine was noted to attenuate reductions in BDNF, dendritic spine density, GluA1 and PSD-95 (both markers of synaptogenesis) in the PFC, dentate gyrus and CA3 region of the hippocampus at 8 days following drug administration. 109 In an investigation employing two-photon imaging in the PFC of living mice exposed to chronic stress, ketamine rescued elimination of postsynaptic dendritic spines and reversed the loss of coordinated activity of multicellular ensembles in projection neurons. 110 Of note, the authors observed that ketamine’s rescue of dendritic spine formation occurred prior to its acute behavioral effects but was later correlated with behavioral effects 2–7 days after treatment. In addition, optogenetic ablation of newly formed dendritic spines disrupted the maintenance of ketamine’s behavioral effects. Taken together, this may suggest that ketamine’s effect on synaptogenesis may be related to longer-term maintenance of antidepressant activity. Furthermore, in a mouse social defeat model of depression, ketamine restored deficits in markers of neuronal and astroglial metabolic activity in the PFC to normal levels. 111 This led the authors to suggest that ketamine may improve neurotransmitter cycling.
Ketamine’s neurochemical effects were also investigated, to a lesser extent, in depressed patients. Ketamine responders with TRD demonstrated rapid elevations in plasma BDNF levels112,113; however, another study did not support this finding. 114 In the aforementioned investigations, higher levels of BDNF were correlated with lower severity of depressive symptoms on rating scales. Interestingly, patients with a Val66Met single-nucleotide polymorphism associated with impairments in BDNF release and mRNA trafficking were also found to have reduced responses to ketamine.115,116 In a study of three depressed patients responding to ketamine, the authors observed an increase in expression of plasma mTOR and eEF2 phosphorylation. 117 While the increase in mTOR is supported by animal studies, it was surprising to note an increase in eEF2 phosphorylation that was previously shown to be reduced in animal models. 96 A recent RCT of 20 patients demonstrated the surprising finding that pre-treatment with rapamycin, an mTORC1 inhibitor, actually tripled the response rate at 2 weeks after treatment. 118 The authors suggested that rapamycin may have augmented ketamine’s effects by targeting neuroinflammation via its immunosuppressant actions or by promoting homeostasis of synaptic density. However, it has also been noted that it is unknown whether low-dose rapamycin would reach appropriate levels to inhibit mTOR in the brain and that it may exert its augmenting effects through dampening inflammation in the periphery. 41
The possible interactions between D-serine, a potent co-agonist at the NMDA receptor that has been implicated as a possible therapeutic agent and/or biomarker in both depression and schizophrenia, are also of interest and warrant further investigation. Several animal studies and clinical investigations suggest that D-serine levels may be abnormal in depression and that D-serine has antidepressant properties.119–123 In this regard, it is interesting that ketamine inhibits transport of D-serine, 124 ketamine metabolites decrease intracellular concentrations of D-serine in PC-12 cells, 125 and that plasma D-serine levels predict a response to the antidepressant effects of ketamine.126,127
Other possible cellular targets of ketamine include binding to opioid (mu, delta and kappa) receptors, monoaminergic receptors and transporters, and muscarinic and nicotinic cholinergic receptors.46,47,128 It has been proposed that ketamine’s anti-suicidal and antidepressant effects may depend on activation of the opioid system, since pre-treatment with naltrexone (an opioid receptor antagonist) attenuated these effects in depressed patients.128,129 However, other studies have disputed the effect of naltrexone on ketamine’s mechanism of action.130,131 Agonists of the opioid receptors, such as buprenorphine and methadone, do not seem to affect ketamine’s antidepressant properties. 131 Based on the results of studies on laboratory animals, Zhang and Hashimoto 132 suggested that the opioid system may not play a role in ketamine’s antidepressant effects. Overall, it appears that the role of the opioid system in ketamine’s mechanism of action is still relatively unclear and controversial.
Other potential biomarkers for predicting response to ketamine
Sleep
It is natural to review the potential interactions of ketamine, sleep and MDD, given the well-known use of higher doses of ketamine as a general anesthetic as well as the myriad of clinical and neurophysiological interactions of sleep and MDD.
Ketamine has well-described effects on increasing total sleep and slow-wave sleep/slow-wave activity (SWS/SWA), 133 and its antidepressant response has been linked to this effect. 113 Improving SWS/SWA, especially early in the night, is thought to be a critical factor in ketamine’s mechanism of rapid antidepressant action in MDD, and similar results have been seen with repetitive transcranial magnetic stimulation (rTMS). 134 This increase in SWA correlates strongly with increases in synaptic plasticity and plasma BDNF preclinically, 135 and in ketamine-responsive MDD patients.113,133 BDNF is a well-known potential marker of antidepressant response, 136 and the magnitude of increase has been seen to predict acute mood response to ketamine. 112 Interestingly, this improvement in SWS/SWA may be unique to patients with unipolar depression. Ketamine responders with bipolar depression were found to experience the opposite effect – that is, a reduction of SWA. This may be due to known phenotypic differences of sleep (more hypersomnolence) associated with bipolar depression 137 or the effect of mood-stabilizing medication. 133 Low baseline delta sleep ratio, defined as decreased SWA earlier in the night compared with later in the night, has also been suggested to predict acute antidepressant effects of ketamine in MDD. 138
Overall sleep improvement, especially reduction in objective electroencephalogram early-night awakening, may be a mechanism by which ketamine exerts its anti-suicidal effects. 139 This should be an interesting area of future inquiry as non-antidepressant mechanisms need to be elucidated to completely explain the anti-suicidal effects of ketamine. 140
Ketamine also appears to have significant effects on circadian rhythm systems, and its effects on glutamate likely underlie part of this. Synchronization of light/dark and the internal clock is partially mediated by glutamate in the retinothalamic tract. Small studies have shown ketamine responders exhibit more phase advance and a stronger amplitude increase in 24 h motor activity, indicating a more robust circadian rhythm.141,142 Baseline higher amplitude and a delayed 24 h motor activity pattern in the circadian rhythm were associated with nonresponse. Low and blunted amplitude 24 h activity patterns were also seen to associate with rapid relapse and brief response to ketamine, respectively. 142
Some of these circadian rhythm changes are very similar to the effects of sleep deprivation and light therapy in MDD, 143 and it may be these changes to the circadian clock that underlie some of ketamine’s rapid antidepressant effects. It is well known that circadian rhythm disruption is a key biological feature of MDD and it often appears to return to normal as symptoms remit. 144 It has been postulated that people with disrupted circadian rhythms may be a subtype of mood disorders, but clinically they could be quite responsive to ketamine or other antidepressant treatments with rapid effects on the body clock.
Clock genes are known to control circadian rhythms, and ketamine has been noted to induce their rapid expression, 145 suggesting that clock genes may play a role in ketamine’s rapid antidepressant effects.146,147 Newer preclinical data have shown an overlap between clock gene expression in both sleep deprivation and ketamine, both fast-acting treatments for depression. 148 However, slower-acting treatments such as escitalopram 149 and lithium 150 have also demonstrated this effect. As such, clock gene expression could be more a long-term mechanism of ketamine, with the increase in sleep, SWS/SWA (which has been described as a proxy for sleep homeostasis) and more acute circadian rhythm changes being more linked to the rapid effects. 141 The interaction is likely critical to sustain any antidepressant response overall. 151
To date, data regarding ketamine’s effects on sleep are very limited, typically restricted to being short term with intravenous infusions. No reported data could be found on sleep effects of other less bioavailable forms of ketamine. The authors’ combined clinical experience is that drowsiness is seen in about 15–20%. Effects of ketamine on subjective sleep complaints in clinical trials is also not well reported. A major meta-analysis indicated no difference between ketamine and placebo groups on patient reports of ‘tired/fatigued’ and ‘vivid dreams’. 55 However, when reviewing the major esketamine trials, significantly increased rates of somnolence versus placebo have been reported, especially in relapse-prevention studies,74,152 but this has not been seen consistently. 153 While ketamine-specific data are limited, evidence from other MDD treatments indicates that both normalization of sleep homeostasis and circadian rhythm stabilization could be response predictors for ketamine. Associated rapid expression of clock genes could also be a marker for sustained improvement with ketamine in MDD. A prospective trial dosing ketamine at different points of the circadian rhythm is currently underway to shed more light on this question both clinically and biologically. 154
Cognition
Cognition and cognitive symptoms (CCS) have a key role in recovery and functional outcome in MDD. 155 Hence, looking at the effects of ketamine on CCS is critical to both understanding the mechanism and potentially predicting ketamine-responsive patient subtypes in MDD. There are emerging lines of evidence to support the theory that pro-cognitive effects of lower-dose ketamine are a foundational component in its putative efficacy. 156
It can be confusing to examine the ketamine literature with respect to CCS, as it has traditionally been considered to have significant negative cognitive effects and has even been used as a model of schizophrenia, psychosis and cognitive dysfunction. 157 Animal models show a differing, dose-dependent effect of ketamine on cognition, depression and anxiety, with a major postulated mechanism being altered BDNF levels. Sub-anesthetic doses of ketamine were seen to have positive effects on BDNF levels in the hippocampus, while the opposite is seen with anesthetic doses. 158 A single infusion of low-dose ketamine (0.5 mg/kg) also increased hippocampal volume (often a proxy for increased BDNF) in a small group of unmedicated MDD patients as well. 159 Infusion of much higher analgesic doses (8–20 mg/h) in healthy volunteers was also shown to produce significant deficits in cognition, 160 indicating a potential dose-dependent effect with acute ketamine on CCS. Yet, there appears to be a distinct lack of long-term side effects of any sort with anesthetic doses, 161 and sub-anesthetic doses appear to carry a very low risk in clinical trials. 162 Animal models have also demonstrated impairment in episodic memory with a single ketamine infusion, 163 but this may be an acute finding and unrelated to long-term cognitive dysfunction. Human results of acute ketamine use on memory are mixed, 164 and a recent study on intranasal esketamine administration in healthy volunteers showed significant cognitive dysfunction at 40 min, but not at 2, 4, and 6 h post-dose. 165
Ketamine treatment in actual MDD treatment protocols also shows promising results in CCS. Three groups of patients – with treatment-resistant unipolar and bipolar depression, 166 TRD 167 and anxious/non-anxious depression 168 – were given six ketamine infusions over 12 days, with similar cognitive testing in a 2 week follow-up period. No deterioration in cognitive function was seen in any of the studies. Processing speed and verbal learning improved, but this was significantly correlated with improvement in depressive symptoms.166,167 Only the group of anxious depressed patients demonstrated a similar change in the third study. 168 Improvement in many domains of memory in another study of repeated infusions over 12 days with a 4 week follow-up period was seen, but was not significant when controlling for depressive symptom improvement. 169 A single infusion of 0.5 mg/kg in TRD patients was also seen to be slightly beneficial in attention and response control as well. 170 In terms of predictive cognitive variables of response to ketamine, greater baseline visual learning predicted degree of MDD response to ketamine treatment in two of the above studies.166,168 Low attention 169 and processing speed have also been seen to be predictive. 171
There has long been concern that chronic ketamine use could lead to cognitive deficits. Heavy and chronic ketamine users have been seen to have a variety of cognitive function deficits across multiple domains. These include word reading and memory, 172 verbal/visual memory, motor speed and executive function, 173 as well as verbal fluency, processing speed and verbal learning specific to frontal and medial temporal cognition. 174 A small group of chronic ketamine users have also demonstrated spatial memory disturbances and altered hippocampal activity. 175
First, these cognitive deficits may be reversible. A large group recovered substantial cognitive function in domains of executive function, verbal and visual memory after stopping heavy ketamine use for 12 weeks. 176 The ex-ketamine users who had been abstinent for a mean of 189 days in another study showed no cognitive deficits, even though their prior use had been just as significant as the chronic usage group. 173
Second, the negative effects reported above could be related to the fact that abusers were likely using very high doses for long periods, creating an effect similar to ongoing anesthetic doses. Patients in a study demonstrating similar cognitive deficits of ketamine psychosis to schizophrenia had an average ketamine consumption of 3.8 g per day over 7 years. 157 Even taking into account reduced bioavailability and purity, this would be an exponential order of magnitude greater than what is used in even regular TRD treatment. Cognitive deficits were also only seen in very frequent users (over four times weekly) in a study of chronic self-administered ketamine 177 and the frequent user group in a small group of regional pain disorders 178 versus people who used ketamine less often. Interestingly, the type of diagnosis may interact with ketamine in its potential to create cognitive deficits. Nonpsychotic ketamine abuse patients were seen to have significantly fewer cognitive deficits than either schizophrenic or ketamine-abusing psychotic patients. 157 Clinically, this could indicate that more caution may be needed when treating patients who have a history of ketamine-induced psychotic disorders, psychotic illnesses and perhaps even psychotic depressions.
As mentioned previously, there are a number of potential mechanisms of the pro-cognitive effect of ketamine in depression. Some include de-emphasizing the link between cognition and emotion pathways in the brain, creating less interference of cognitive processing by negative emotional content. 179 Glutamatergic modulation of numerous neural circuits (especially PFC networks) involved in cognition through mechanisms such as synaptogenesis, synapse stabilization, increased BDNF and mTORC1 production may also be involved. 156 The specific anti-suicidal effect of ketamine may also be related to pro-cognitive effects. Better executive function and control could reduce the irrationality sometimes seen with the impulsivity of acute suicide attempts. 180 This is partially supported by the finding of a single infusion of ketamine reducing explicit suicidal cognition and a performance-based index of implicit suicidal cognition compared with another anesthetic, although it was mediated by depressive symptom improvement. 181 Another consideration supporting cognition as a key driver of ketamine response could be the potential decreased efficacy of ketamine clinically when benzodiazepines (which are known to impair cognitive processes) are co-administered.182–184
In summary, doses of ketamine used in TRD appear to have overall pro-cognitive effects that may mechanistically underlie their rapid effectiveness. The negative cognitive side effects of ketamine are present likely transiently in acute dosing of ketamine but only consistently in long-term heavy ketamine users, and appear to be reversible. These potential dose-dependent opposing actions of ketamine in CCS could be analogous to amphetamines, where low doses can greatly help cognitive measurements in disorders such as MDD179–185 and attention deficit hyperactivity disorder (ADHD),180–186 whereas much higher doses or different formulations of abuse can be detrimental cognitively or even cause psychosis in those predisposed.181–187 Indeed, there are significant overlaps in the cognitive dysfunction pattern between chronic ketamine and methamphetamine users.182–188 Cognitive impairment is not likely an issue in the doses given in MDD trials, but longer-term data are needed and, given the mechanistic underpinnings, consideration could be given to avoiding use in patients with psychosis.
Inflammation and metabolism
Metabolic syndrome, a constellation of symptoms including hypertension, hypercholesterolemia, hyperglycemia and increased waist circumference, is common in patients with mood disorders. A recent study reported a 38% prevalence of metabolic syndrome in patients with TRD, and it has been estimated that approximately one-third of depressed patients have elevated inflammatory markers. 189 A significant yet complex relationship exists between mood disorders and metabolic syndrome, and this link appears to involve inflammation. Metabolic syndrome in patients with TRD is three times more common in patients with elevated C-reactive protein (CRP), an inflammatory marker. 190 A systematic review looking at predictors of response in TRD suggested that the inflammatory markers interleukin-6 (IL-6), CRP and high-sensitivity CRP (hsCRP) may predict response to antidepressant medications with anti- inflammatory properties, including ketamine. 191 While there is some suggestion from animal studies that racemic ketamine may have anti-inflammatory effects, 192 human studies remain contradictory. Some authors have reported that decreases in inflammatory mediators IL-6 and interleukin-1 alpha (IL-1 alpha) following ketamine treatment have been only transient in the form of hours and have not correlated with antidepressant response,193,194 but changes in fibroblast growth factor (FG-2) were correlated with antidepressant response. 193 Another study reported that a rapid decrease in the pro-inflammatory mediator tumor necrosis factor-alpha (TNF-α) was correlated with rapid antidepressant effects of ketamine, suggesting that changes in inflammatory cytokines may play a direct role. 195 Of note, peripheral cytokine levels may not reflect central levels, and to date, studies have consisted of small sample sizes. Of particular interest, racemic ketamine has been suggested to protect against inflammation-induced vulnerability to stress behaviors in mouse models of depression. 192
Mechanisms of the relationships between ketamine, depression, metabolism and inflammation remain unclear, but are likely mediated by multiple factors. Elevated body mass index (BMI) has been found to be a predictor of response to ketamine, 196 but in another study 197 elevated BMI was not correlated with ketamine response, and presence of metabolic syndrome was negatively correlated with ketamine response. It has been shown that fatty acid metabolism differs between depressed subjects and non-depressed controls,198–200 and it has been suggested that differences between patients who respond to ketamine and those who do not are due to alterations in the mitochondrial β-oxidation of fatty acids. 201 Adipokines such as adiponectin, resistin and leptin regulate inflammatory and neuroplasticity pathways, as well as influence insulin sensitivity. 202 It has been suggested that low levels of adiponectin, which typically acts as an anti-inflammatory and improves insulin sensitivity, may be predictive of ketamine response. Resistin is a pro-inflammatory molecule, and its levels have been noted to decrease with positive response to ketamine, suggesting that it may play a role in ketamine’s anti-inflammatory antidepressant action. 202 Fat cells also release monocyte chemoattractant protein-1 (MCP-1), which leads to macrophage infiltration and more inflammation. 203 It has been previously described that inflammation leads to glutamate excitotoxicity and synaptic destruction in depression. 189 As noted previously in this review, ketamine’s antidepressant properties are thought to be at least in part due to its elevation of BDNF, which supports synaptic repair and regeneration.
Neuroimaging
There is a growing body of literature on neuroanatomical biomarkers of response to ketamine treatment. MDD has been shown to affect, among other areas, the PFC, the hippocampus and the anterior cingulate cortex (ACC), and there is evidence that ketamine affects these areas preferentially.204–206
Results from Lehmann et al. 207 have implicated the ACC by utilizing task-related functional magnetic resonance imaging (fMRI) to investigate the effect of a single dose of intravenous ketamine versus placebo in a sample of healthy subjects. They found greater blood-oxygen-level-dependent (BOLD) reactivity in patients with high levels of rumination on negative experiences and a potentially larger effect at the pregenual ACC. A study using a magnetoencephalographic (MEG) task-related technique also found evidence for dysregulation in the ACC being implicated in a more favorable response to a single infusion of ketamine. 208 This may correlate with findings of anhedonia being a possible clinical biomarker of response. 17
Ketamine’s mechanism of action has also been associated with the glutamatergic system, especially in the PFC, as shown in neuroimaging studies. Using resting state fMRI, researchers have shown reduced PFC global connectivity to be implicated in MDD.209,210 Abdallah et al. 211 demonstrated that 24 h after a single infusion of ketamine, PFC global connectivity could be normalized in responders and the extent of the PFC global connectivity increase was associated with response. Interestingly, lanicemine, another NMDA receptor antagonist, did not produce this.
Structural MRI has been performed on human subjects undergoing single-infusion ketamine, and evidence has been provided showing increased hippocampal volumes and decreased nucleus accumbens volumes 24 h post-infusion, which was correlated to treatment response. 212
However, it should be mentioned that neuroimaging data overall are limited by small sample sizes and the low number of investigations to date.
Future directions
There has been increasing interest surrounding ketamine in recent years, largely owing to its rapid antidepressant and anti-suicidal properties in patients with TRD and its unique mechanism of action. The preclinical and clinical studies to date have led to further understanding of its use in psychiatry, and this will continue to be an active area of research as more investigations are conducted to determine optimum conditions for ketamine treatment in patients (see the work of Phillips et al.60, 213 for examples of very recent studies on comparisons of single, repeated and maintenance ketamine infusions on TRD and suicidal ideation in TRD). There will need to be further exploration of individual differences in response between patients (including sex differences214,215) and determination of appropriate regimens for maintenance therapy and discontinuation, given ketamine’s transient antidepressant effects. There is also a need to investigate further reliable biomarkers for prediction of ketamine response and adverse effects. As indicated in this review paper, studies on ketamine’s roles in sleep, cognition and inflammation have resulted in some interesting findings worthy of further research. Additional exploration of ketamine’s possible role in treatment of bipolar depression is also warranted.
It should be remembered that there are also many publications in the literature that urge caution in the use of ketamine as an antidepressant,76,216–222 and clinicians planning to use this drug would be well advised to be familiar with the extensive literature available on it. A 2017 consensus statement on ketamine use encourages consideration of the current data limitations and potential risks associated with the drug. 222 Nonetheless, ketamine remains a promising option for those suffering from TRD, and it is exciting to surmise that understanding ketamine’s neurochemical mechanisms and related biomarkers will lead to the development of other, much needed, next-generation antidepressants.
Footnotes
Conflict of interest statement
JS has been paid speaking honoraria by Otsuka and Lundbeck and has served on advisory boards for Otsuka, Lundbeck, and Janssen. RKT and SMD have also served on advisory boards for Janssen. AK has been paid honoraria for speaking from, and has served on advisory boards for Pfizer, Takeda, Lundbeck, Janssen-Ortho, Purdue, Sunovion, Allergan, Otsuka and Merck, and has received speaking honoraria from Bausch Health; he has also received research funding from AstraZeneca, Sanofi-Aventis, Pfizer and Merck. None of the companies mentioned above has had a role in the preparation of this review paper.
Funding
The applicants are grateful to the Department of Psychiatry and the Faculty of Medicine & Dentistry, University of Alberta, for funding.