
Abstract
Background:
Underrepresentation of various populations in clinical trials limits our understanding of efficacy and safety outcomes, as individuals with diverse backgrounds can respond differently to the same interventions. Inclusion of diverse groups in device clinical trials is essential for providing innovative, patient-centric solutions and achieving health equity.
Objective:
To evaluate how social determinants correlate with safety events in clinical trials with medical devices to identify safety signals, improve safety profiles, and develop risk-based safety and quality management strategies.
Design:
A single-center, retrospective study was conducted for clinical trials with medical devices at a safety-net hospital.
Methods:
Data obtained from 223 patients randomized into 19 device clinical trials were assessed retrospectively for correlation of social determinants of health variables and study protocol complexity with study protocol deviations, adverse events, and study dropout rates by using the Kruskal-Wallis and Spearman correlation tests.
Results:
Lower median income was associated with an increased number of adverse events (r = −0.18599, p = 0.0053) and protocol deviations (r = −0.27349, p < 0.0001). Higher study protocol complexity was associated with an increased number of adverse events (r = 0.55328, p < 0.0001) and a higher number of protocol deviations (r = 0.54079, p < 0.0001). Out of 701 adverse events reported, 472 were serious adverse events, and 3.8% of serious adverse events were related to the study device. Multiple comorbidities >3 were associated with an increased number of safety events and protocol deviations (r = 0.35929, p < 0.001; r = 0.17270, p = 0.0098, respectively).
Conclusion:
Examining the root causes of these outcomes, as well as patient-centric medical management, can improve data integrity and aid risk-based safety and quality management in device clinical trials.
Plain language summary
Why was this study done? Clinical trials often don’t include enough diverse groups of people, which makes it harder to understand how medical devices work for everyone. Including a wider range of people in these trials is important for creating better, more effective treatments for all and promoting fairness in healthcare.
What did the researchers do? The research team look at how factors like income and health conditions affect safety and problems in medical device trials. Researchers reviewed data from 223 patients in 19 clinical trials to see if things like income, health issues, and the complexity of the study were linked to safety problems, mistakes in the trial process, and people dropping out.
What did the researchers find? They found that people with lower incomes had more safety issues and more mistakes in following the study rules. They also found that the more complicated the trial, the more safety problems and mistakes occurred. People with multiple health conditions were more likely to have safety issues and mistakes. Most of the safety problems were serious, but only a small number were linked directly to the device being tested.
What do the findings mean? The study suggests that understanding these issues can help improve the safety of clinical trials and ensure that they better reflect the needs of all patients.
Keywords
Introduction
Diversity and inclusion in device clinical trials are necessary for developing patient-centric solutions to achieve health equity.1–3 Underrepresentation of various populations in clinical trials limits our understanding of efficacy and safety outcomes, as individuals with diverse backgrounds can respond differently to the same interventions.4,5 Inclusivity enables more comprehensive and tailored treatment approaches for all patient groups while addressing gaps in the safety and performance of new medical devices for those with a greater disease burden.1–3 The Food and Drug Administration (FDA) recommends including diverse populations with varying social determinants of health (SDoH) in clinical trials, as these factors influence patients’ health and trial outcomes.1–3 These guidelines reflect the current regulatory authority perspective, which conveys their current expectations to include in clinical trial protocols by design and development plans underrepresented racial and ethnic groups with different genders, ages, socioeconomic status, and comorbidities. 3
The Belmont Report was created to advocate for patients from diverse groups and classes, focusing on the ethics of clinical research. 4 The “Justice” principle from the Belmont Report highlights that clinical trials must never select or exploit specific populations due to “easy availability, their compromised position, or their manipulability.” 4 Rather, there should be an equitable balance of risks and benefits among all study subjects. 4 This principle is further reinforced by the World Health Organization’s Commission on Social Determinants of Health, and their report “Closing the Gap in a Generation.” 1 This report called on governments, organizations, and civilians worldwide to focus on improving justice and health equity among all people. 1
Safety in clinical trials involving medical devices is of paramount importance to protect trial participants and ensure that the device is safe and effective for its intended use. 6 Clinical trials for medical devices are highly regulated and are designed to assess both the risks and benefits of the device. Medical devices are categorized into three classes (I: low, II: moderate, and III: high risk) based on the level of risk they pose. 7 The FDA provides specific guidelines for ensuring safety in clinical trials involving medical devices.8,9 These guidelines are critical to ensuring that devices are safe and effective for their intended use before they are marketed to the public.8,9 Throughout the trial, investigators must closely monitor participants for any adverse events (AEs), which are any unintended medical occurrences during the trial. 6 AEs are recorded, evaluated, and reported to regulatory authorities as required. Serious adverse events (SAEs) must be reported to the FDA, investigators, and the sponsor immediately.10,11 If the device causes significant harm, the trial may be stopped or modified to mitigate the risk.10,11 AEs must be categorized and followed up with appropriate medical care if necessary. 10 The FDA requires manufacturers to include clear labeling and instructions for use (IFU) with the device to ensure that it is used correctly and safely. 12 The IFU should include:
Warnings and precautions.
Instructions for device operation.
Information on potential side effects or complications.
Any conditions under which the device should not be used (contraindications). 12
All clinical trial data, especially safety-related data, should be transparently reported to regulatory bodies, the public, and the scientific community.10,11 This ensures that the overall safety profile of the device is clearly understood and communicated. In clinical trials, AEs and protocol deviations (PDs) highlight areas where safety, efficacy, and quality can be improved.5,6 AEs emphasize where patients can be potentially exposed and are more at risk of harm due to the investigational device or study procedures, while PDs indicate noncompliance with the study protocol and procedures.5,6
Clinical trials are typically overseen by an Independent Data Monitoring Committee (DMC) or Data Safety Monitoring Board (DSMB). 11 This group reviews data periodically during the trial, especially safety data, to ensure that:
The device is not causing unexpected harm.
The risks of continuing the trial do not outweigh the potential benefits.
The trial can proceed or needs to be stopped early if safety concerns arise. 11
The DMC/DSMB has the authority to halt or modify the trial if safety concerns are identified. 11 Institutional Review Boards (IRBs) are independent committees that review and approve clinical trial protocols. The IRB ensures that the study complies with ethical standards and protects participants’ rights and welfare. The IRB evaluates whether the trial has adequate safety protections in place and whether risks are minimized to the extent possible. 11
Often in clinical trials with medical devices, the sponsor expects only SAEs related to investigational medical devices to be reported in an expedited fashion. However, local IRB policies and procedures may vary and impose more stringent requirements for safety reporting (e.g., any admission to the hospital with overnight stay for the subject enrollment in a clinical trial with medical device related or not to study participation or investigational device may be classified at SAE and subject to expedited reporting, if in a judgment of the principal investigator, this event is an unanticipated problem and poses increased risks to study participant or other subjects enrolled in this study). Risk management is an ongoing process in clinical trials. Sponsors/manufacturers and investigators must identify potential risks and implement measures to reduce those risks. 12 This includes:
Regular safety assessments and risk evaluations.
Developing safety protocols for managing risks, such as device failure or malfunction.
Clear instructions and training for clinical trial staff to handle potential safety issues.
The risk management process should be documented and continuously updated. 12
The objective of this project was to evaluate how social determinants correlate with safety events in clinical trials with medical devices to identify safety signals, improve safety profiles, and develop risk-based safety and quality management strategies.
Methods
Study design and settings
A single-center, retrospective study was conducted of 223 patients randomized into 19 clinical trials with medical devices at a safety-net hospital, which is a type of medical facility in the United States that by legal obligation and mission provides healthcare for patients regardless of their insurance status. Unlike many European countries, the United States does not have a universal health care policy or ability to pay.13,14 This legal obligation reinforces safety-net hospitals to be more inclusive in serving populations, which often are disadvantaged both socially and financially. These hospitals serve a proportionately higher number of uninsured, Medicaid, Medicare, Children’s Health Insurance Program (CHiP), low-income, and other vulnerable populations than their “non-safety net hospital” counterparts.14,15 The mission of these types of facilities is to provide the best possible care for those who are discouraged to interact with health care system and/or seek medical attention due to various circumstances such as concerns for financial aspects, lack of insurance coverage, potential to lose their job/missing work/lost wages, or health conditions high burden.13–15
Data sources and measurements
The de-identified, aggregate dataset was assembled based on data obtained from all 19 randomized clinical trials with medical devices, which were conducted in the department of surgery during a period of time from July 2007 to October 2024. Out of these 19 analyzed studies none of them were first in human or phase I trials; however, 10.5% were phase II, 15.8%—phase III, and 42.1%—phase IV post-marketing clinical trials. In terms of investigational device classification: 21.1% were Class III (high risk) implantable or intraoperative use investigational devices under Investigational Device Exemption regulatory designation; 78.9% were Class II (moderate risk) topical use devices for chronic wounds or surgical wound closure indications under 510k premarketing regulatory status.
All clinical trials were registered through a public trials registry by industry sponsors and received IRB approval prior to study initiation. Informed consent from each research subject was obtained prior to any research procedures being performed.
These clinical trials were similar in eligibility criteria, study design, and endpoints. In terms of investigational devices tested in these trials, the analyzed dataset consisted of indications in the fields of vascular, podiatry, and thoracic surgery, with study-specific eligibility criteria. The enrolled subjects were undergoing device studies targeting a wide range of conditions: lower extremity ulcers, hemodialysis, peripheral artery disease, critical limb-threatening ischemia, lymphedema, cardiovascular diseases, pulmonary embolism prevention, post-vascular surgery homeostasis control, airway obstruction management, etc. Measurement and/or selection bias was avoided as the statistician performing data analysis was blinded to study design or individual study participant characteristics, and/or treatment group assignment.
Variables and study size
Specifically, data obtained from 223 patients randomized into 19 device clinical trials were assessed retrospectively for correlation of SDoH variables (Table 1) and study protocol complexity, according to the complexity assessment model described by Malikova. 16 previously, with study PDs (e.g., visit out of window, visit missed, use of prohibited concomitant medication, test article handling, study procedure not performed/procedure performed late, eligibility criteria not met, or other), AEs (e.g., relatedness, seriousness, and expectedness), and study dropout rates (e.g., lost to follow-up, due to AE, withdrawn by investigator, or death).
The social determinants of health examined at baseline visit.
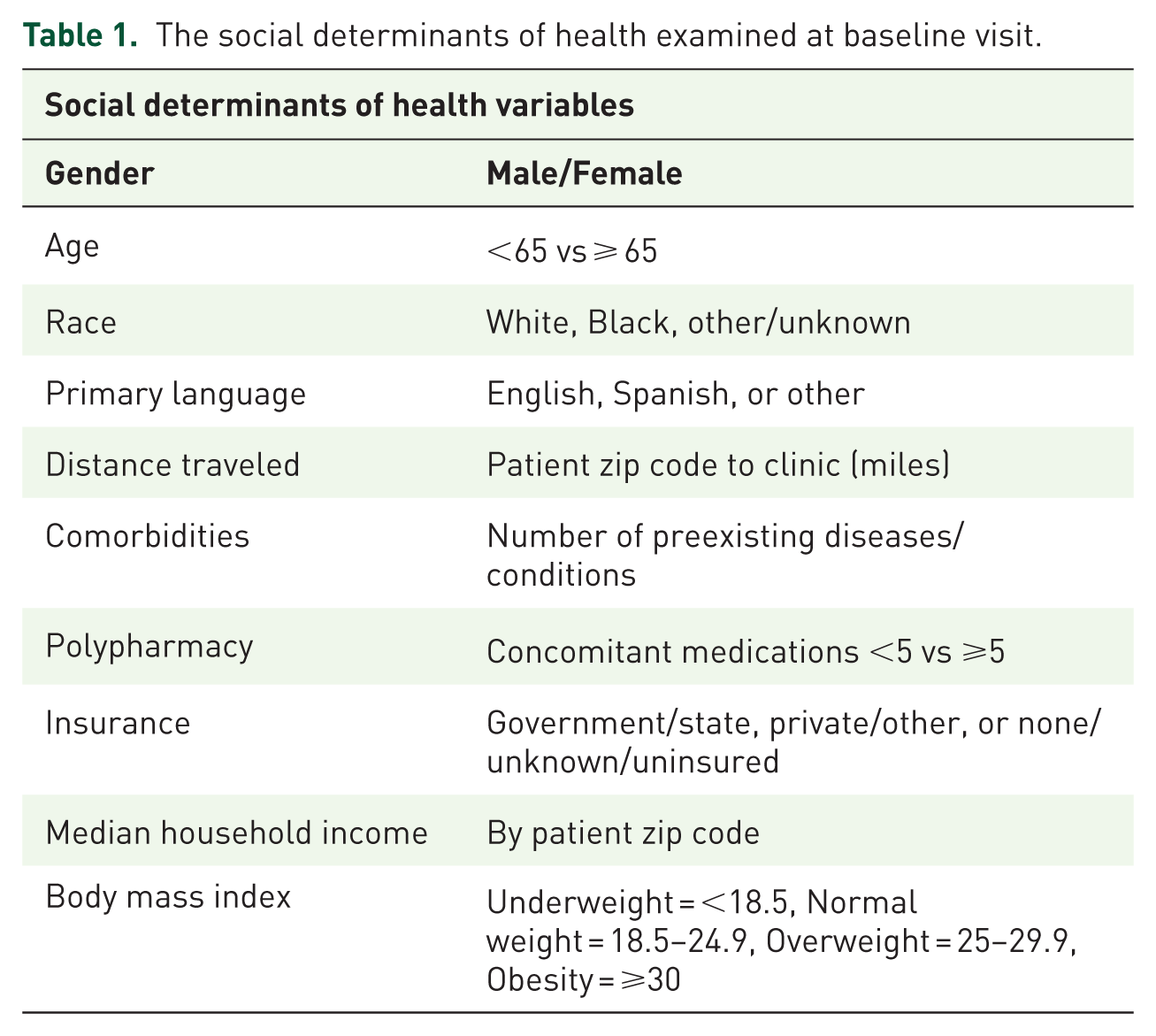
Patient demographics at the time of enrollment were evaluated, including gender (male/female), race (White, Black, or other), Hispanic ethnicity, weight (kg), height (meters), body mass index (BMI), 17 and age (years). Polypharmacy was defined as concomitant medication ⩾5 and included the following therapeutic indications: antiglycemics, antiplatelets, anticoagulants, antihypertensives, statins, nonopioid/opioid analgesics, antidepressants, antibiotics, etc. Comorbidities/preexisting conditions were recorded and categorized as shown in Table 2.
The comorbidities categorized by body system and recorded at baseline visit.
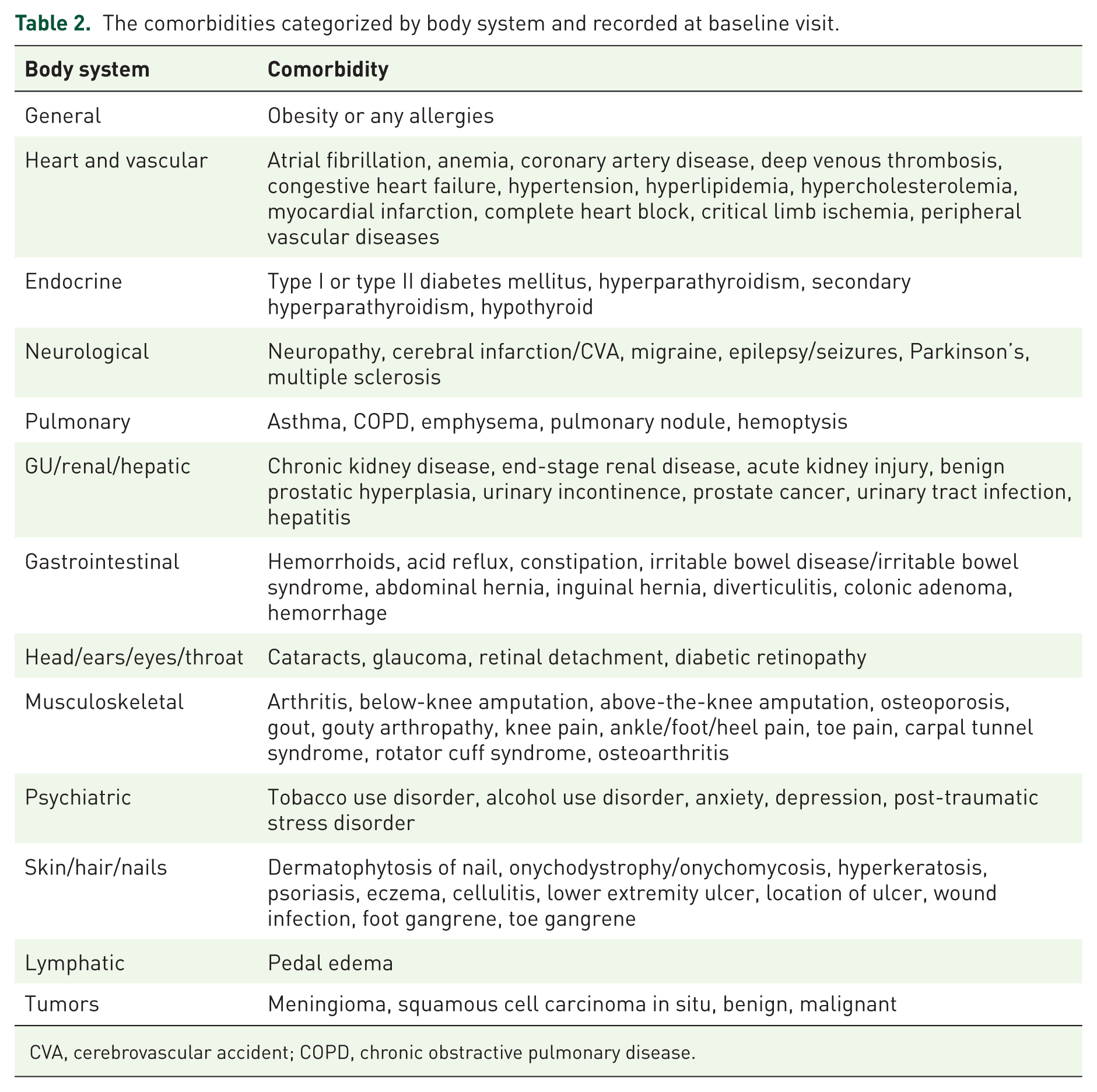
CVA, cerebrovascular accident; COPD, chronic obstractive pulmonary disease.
Socioeconomic variables were evaluated including primary language spoken (English, Spanish, or other), type of insurance (Medicare, Medicaid, private/other, or uninsured), median income of patient zip code, 18 distance traveled to visits (miles), and education level (tertiary/some or completed college, secondary/some or completed high school, primary/some or completed elementary school, or no formal schooling).
As part of standard clinical research operations at our site and as a method of maintaining metrics in real time, a database of SAEs and non-SAEs events was maintained, documenting event start and end date, and the date the event was reported to the sponsor and regulatory groups. Each safety event was assessed by categories of severity, expectedness, and relatedness to the study products (e.g., medical device) and/or study procedures. The aggregate dataset from 19 clinical trials was used in this retrospective analysis.
Each AE was classified and stratified by relationship to the investigational medical device and study procedures (e.g., relevant for implantable medical devices or procedures required by the study protocol only, not routine clinical care) to better understand the correlation between the type of event and exposure to the investigational device. An assessment of the expectedness of the event was performed based on the IFU or User Manual, which is in clinical trials with medical devices and is equivalent to the Investigators Brochure utilized in trials with drugs or biologics.
According to regulatory and study protocol definitions, an AE is any undesirable experience associated with the use of a medical product in a patient. 10 The event is SAE and should be reported to the FDA when the patient outcome is life-threatening, results in death, hospitalization (initial or prolonged), disability, congenital abnormality/birth defect, required intervention to prevent permanent impairment or damage (e.g., with medical devices); or other medically significant event that may jeopardize the patient and may require medical or surgical intervention (treatment) to prevent one of the other outcomes (e.g., significant blood abnormalities on laboratory tests, accidental finding on imaging tests, requiring immediate intervention, treatment in an emergency room, etc.).10,11
All other non-SAEs should be reported as part of continuing review per site organizational policies and procedures and captured and reported to the sponsor and IRBs in accordance with study protocol, data safety monitoring plan, DSMB if required, and site organizational policies and procedures. 11
Statistical methods
Using the Wilcoxon score distribution from the Kruskal-Wallis tests, the number of PDs, AEs, and dropout status were compared across the SDoH variables. The Spearman correlation test was used to examine associations between the continuous variables and the safety events. 19 The Spearman correlation coefficient ranges from −1 to +1, with a value of 0 indicating no correlation observed. The strength of the relationship between the two variables is categorized as weak (r = 0–0.3), moderate (r = 0.3–0.5), and strong (r > 0.5). 19 To analyze associations between two categorical variables, the Chi-Square test or Fisher’s test was used. A p value of less than 0.05 was considered statistically significant. In this project, SAS software, version 9.4 (SAS Institute Inc., Cary, NC, USA), was used for our statistical analysis. In preparation for this manuscript guidelines indicated on the STROBE checklist 20 were followed (attached as a Supplemental Material).
Results
A total of 223 patients were analyzed; out of which 60.54% were male (Figure 1(a)), and 93.99% were under 65 years old (Figure 1(b)); in terms of race: 47.53% were Black; 32.74% White, and 19.73% identified as others (Figure 1(c)). Most participants were English speakers (81.61%), 11.66% Spanish, and 6.73% spoke other languages (Figure 1(d)). In terms of BMI, 25.65% of the population was classified as underweight or normal weight, 31.30% as overweight, and 43.04% as obese (Figure 1(e)).

Study population demographic variables: (a) study participant gender; (b) study participant age; (c) study participant race group (White, Black, other); (d) study participant primary language (English, Spanish, other); and (e) study population BMI assessment (BMI < 25, 25–29.99, >30).
The Spearman correlation test demonstrated a statistically significant, albeit weak, positive correlation between the number of PDs and both subject age (r = 0.16929, p = 0.0113) and the number of comorbidities (r = 0.17270, p = 0.0098). In addition, a significant yet weak negative correlation was observed between the number of PDs and both distance traveled (r = −0.14277, p = 0.0331) and median income (r = −0.27349, p < 0.0001). The Kruskal-Wallis test similarly revealed a significant association between PDs and primary language spoken, with Spanish and “other” language speakers demonstrating higher rates of PDs compared to English speakers (p = 0.0451; Figure 2).

Wilcoxon score distributions for the number of protocol deviations based on primary language.
Conversely, the Spearman correlation test found no statistically significant relationship between the number of PDs and patient BMI (r = −0.06550, p = 0.3302). Similarly, the distribution of Wilcoxon scores showed no statistically significant associations between PDs and gender (p = 0.6966) or polypharmacy (group ⩾5 drugs vs <5 drugs were compared; p = 0.5471). The Kruskal-Wallis test confirmed the absence of significant relationships between PDs and gender (p = 0.6958), race (p = 0.1706), polypharmacy (p = 0.5460), and insurance type (p = 0.6296).
When analyzing AEs, the Spearman correlation test revealed several statistically significant relationships. Notably, a moderate positive correlation was identified between the number of AEs and the number of comorbidities (r = 0.35929, p < 0.001). In addition, a weak, but significant negative correlation was found between AEs and median income, suggesting that individuals with lower income experienced a higher number of AEs (r = −0.18599, p = 0.0053). In addition, a weak but statistically significant positive correlation was observed between AEs and subject age (r = 0.15842, p = 0.0179).
The Spearman test identified a weak negative correlation between distance traveled and AEs, though this was not statistically significant (r = −0.02425, p = 0.7188). Although there was a weak negative correlation between BMI and AEs, this association only approached significance (r = −0.12914, p = 0.0541). Furthermore, Kruskal-Wallis tests demonstrated no statistically significant differences in AEs based on the polypharmacy group (p = 0.0698) or gender (p = 0.1112; Figure 3(a) and (b)). Kruskal-Wallis tests also found no significant associations between AEs and race (p = 0.6424; Figure 3(c)), language spoken (p = 0.5648), or insurance type (p = 0.1830; Figure 4).

Wilcoxon score distributions for the number of adverse events based on (a) polypharmacy, (b) gender, and (c) race.

Wilcoxon score distributions for the rate of patient dropouts based on (a) age, (b) comorbidities, (c) protocol complexity, and (d) BMI.
Kruskal-Wallis test demonstrated a statistically significant association between age and those who dropped out (Median = 66), compared to those who remained in the study (Median = 59; p = 0.0002). In addition, there was a significant difference in the number of comorbidities between patients who dropped out (Median = 13) and those who remained (Median = 10; p = 0.0002). A significant difference in BMI was also observed, with patients who dropped out having a lower median BMI (Median = 25.90) compared to those who remained in the study (Median = 29.97), indicating that lower BMI was associated with a higher likelihood of dropout (p = 0.0005).
The Chi-Square test revealed a statistically significant association between gender and dropout status, with male subjects more likely to drop out (28.89%) compared to female subjects (15.91%; p = 0.0260). A significant association between polypharmacy and dropout status was also found, with polypharmacy patients dropping out at a higher rate (26.04%) compared to non-polypharmacy patients (9.68%; p = 0.0470). However, this result may be less reliable, as Fisher’s Exact test did not demonstrate statistical significance (p = 0.0665).
In contrast, the Kruskal-Wallis test found no significant difference in the distance traveled between patients who dropped out (Median = 5.20 miles) and those who remained (Median = 4.55 miles; p = 0.4978). In addition, there was no significant difference in median income between those who dropped out and those who remained in the study (p = 0.5293). Similarly, the Chi-Square test demonstrated no significant associations between dropout rates and race (White: 26.03%, Black: 24.53%, Hispanic/other/unknown: 18.18%; p = 0.6073), language spoken (English: 22.53%, Spanish: 23.08%, other: 40.00%; p = 0.3099), or insurance type (p = 0.4125).
Spearman correlation test showed a moderate positive correlation was observed between study protocol complexity and the number of comorbidities (r = 0.31200, p < 0.0001), alongside a strong positive correlation between protocol complexity and both the number of PDs (r = 0.54079, p < 0.0001) and total AEs (r = 0.55328, p < 0.0001). Furthermore, a significant difference in protocol complexity was found between subjects who dropped out of the study (Median = 12) and those who completed it (Median = 7), as indicated by both the Wilcoxon two-sample test (p < 0.0001) and Kruskal-Wallis test (p < 0.0001).
Alternatively, there was no statistically significant difference in protocol complexity between polypharmacy and non-polypharmacy patients, as shown by the Wilcoxon two-sample test (p = 0.0900) and the Kruskal-Wallis test (p = 0.0897). Similarly, the Spearman correlation test demonstrated no significant correlation between BMI and both median income (r = −0.00873, p = 0.8968) and the number of comorbidities (r = −0.11778, p = 0.0793). However, a weak, but statistically significant negative correlation was identified between median income and the number of comorbidities (r = −0.14481, p = 0.0306), suggesting that lower-income patients may present at baseline with a higher burden of comorbidities.
In 19 clinical trials conducted, a total of 701 AEs were observed. Of these, 229 events were classified as non-SAEs, while 472 events were categorized as SAEs (Figure 5(a)). This corresponds to an average of 1.03 nonserious and 2.12 serious events per subject (Figure 5(b)). In terms of device-related SAEs, 454 events (96.2%) were considered unrelated to the investigational device. In nonserious events, 200 events (87.3%) were unrelated (Figure 5(c)). Among the 472 SAEs, 290 events (61.4%) were expected based on the safety data available during study participation. The remaining 182 events (38.6%) were classified as unexpected, with the majority occurring across a range of comorbid conditions (Figure 5(d)).

Total number of nonserious and serious adverse events (a); incidence rate of nonserious and serious adverse events (b); relatedness of the total number of nonserious and serious adverse events to the investigational device (c); and expectedness of the total number of nonserious and serious adverse events to the investigational device (d).
Discussion
The research aim of this retrospective study was to evaluate how social determinants correlate with safety events in clinical trials with medical devices in order to identify safety signals, improve safety profiles, and develop risk-based safety and quality management strategies.
Our results indicated that a lower median income was associated with a higher number of AEs (r = –0.18599, p = 0.0053) and PDs (r = −0.27349, p < 0.0001). This income disparity was further exacerbated by the finding that a lower income at baseline was also associated with a higher number of comorbidities (r = −0.14481, p = 0.0306). Additionally, a higher number of comorbidities correlated with an increase in the number of AEs (r = 0.35929, p < 0.001); PDs (r = 0.17270, p = 0.0098), and dropouts from the study (p = 0.002). Noteworthy, most of the SAEs were unrelated to the investigational device in the analyzed trials. It further supports the findings that comorbidities led to greater amounts of AEs.
In comparison with publications from other authors, these findings reflect the well-documented trend that greater income and stronger social protections are associated with better health outcomes in populations.1,21 As a global example, the Commission on SDoH has reported that both sub-Saharan Africa and former Soviet Union countries failed to benefit from the economic gains of globalization in the latter half of the 20th century. 1 These nations have also experienced reversals in life expectancy, demonstrating how income disparities exacerbate health inequalities. 1 The persistence of these issues on both global and local scales underscores the urgent need for improved social protection.1,21
Social protections may enhance patient compliance and reduce PDs. For instance, increasing study stipends or providing affordable childcare may reduce missed visits. If patient compliance improves, it could be predicted that patients have a decrease in AEs or a decrease in comorbidities due to improved medical management. A comprehensive study examined self-perceived health and objective health (grip strength) in 50-year-old male and female patients across 20 European countries. 21 It found that higher social protection expenditures mitigated the socioeconomic inequalities that might normally lead to worse grip strength for both men and women. 21 The study did not find that social protections directly improved grip strength, but that social protections lessen the impact, demonstrating social protections may be one solution to income-related health disparities. 21
Complexity score for each study protocol was determined as described previously by Malikova. 16 We have observed that the greater the study protocol complexity, the more likely a subject enrolled in the device clinical trial would experience an AEs (r = 0.55328, p < 0.0001); have PDs (r = 0.54079, p < 0.0001); or drop out from the study before all study visits/procedures were completed (p < 0.0001).
Based on our findings, we recommend that adherence to study protocol and procedures should be monitored and patient-centric solutions provided to improve subjects’ retention in clinical trials. Assessment of the disease burden of individual subjects is also important for medical management of comorbidities to mitigate safety risks and prevent exacerbation of preexisting diseases/conditions, which can lead to hospitalizations, emergency visits, and affect adherence to study protocol/procedures.
Limitations
A limitation in our study can be attributed to the utilization of the United States Census Bureau search to find the median household income by each patient’s zip code during the year of consent, or the closest available year. 2 In future studies, it would be preferable to assess income at the individual level rather than relying on zip code medians to more accurately represent our patients. While we do not know whether patients fell above or below the median income of their zip code, this approach was the most streamlined method available for this analysis. Nevertheless, it remains a fair measurement as a patient’s social environment and lived experience can impact their health outcomes. There were no formal calculations of sample size performed, which could affect the power of the study. Instead, all randomized subjects were included in the retrospective aggregate analysis. Also, we have conducted an analysis of the safety and SDoH at a single clinical research site, affiliated with the safety-net hospital, with some characteristics and practices that can be specific to our routine clinical care and clinical research practices only. Therefore, large prospective studies at various sites in different settings (e.g., academic medical centers, rural clinics, private practices, etc.), with more diverse patient populations, and/or research practices are needed to further elucidate observed correlations and trends, increase the quality of research conducted and improve safety profiles in clinical trials with medical devices.
Conclusion
Lower median income was associated with an increased number of AEs and an increased number of PDs. Higher study protocol complexity and the presence of multiple comorbidities were associated with an increased number of AEs, PDs, and study dropouts. Examining the root causes of these outcomes, as well as patient-centric medical management, can improve data integrity and aid risk-based safety and quality management in device clinical trials with diverse study populations.
Supplemental Material
sj-docx-1-taw-10.1177_20420986251349005 – Supplemental material for Impact of social determinants of health on safety of patients in device clinical trials conducted at a safety-net hospital
Supplemental material, sj-docx-1-taw-10.1177_20420986251349005 for Impact of social determinants of health on safety of patients in device clinical trials conducted at a safety-net hospital by Anna C. Schneider, Katherine N. Cilley and Marina A. Malikova in Therapeutic Advances in Drug Safety
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.