
Abstract
Background:
Gilteritinib and midostaurin are FLT3 inhibitors that have made significant progress in the treatment of acute myeloid leukemia. However, their real-world safety profile in a large sample population is incomplete.
Objectives:
We aimed to provide a pharmacovigilance study of the adverse events (AEs) associated with gilteritinib and midostaurin through the Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) database.
Design:
A retrospective analysis of the FAERS database was conducted by disproportionality analyses.
Methods:
We conducted disproportionality analyses to identify drug-AE associations, including the reporting odds ratio and the Bayesian confidence propagation neural network. A signal was detected if both methods achieved statistical significance.
Results:
There were 1887 and 2091 case reports for gilteritinib and midostaurin, respectively. We have separately retained significant disproportionality AEs across two algorithms, with a total of 53 AEs for gilteritinib and 46 for midostaurin. The common AEs observed with gilteritinib included febrile neutropenia, pyrexia, anemia, and thrombocytopenia. Similarly, the prevalent AEs associated with midostaurin were nausea, vomiting, diarrhea, pyrexia, and febrile neutropenia. The common AEs of both drugs are consistent with previous clinical trials. Notably, we also revealed unexpected significant AEs for both drugs. For gilteritinib, 29 positive signals for AEs not mentioned in its instructions were identified, such as cerebral hemorrhage, tumor lysis syndrome, and interstitial lung disease. Midostaurin exhibited 24 positive signals for AEs not listed in its instructions, including neutropenic colitis, neutropenic sepsis, and septic shock.
Conclusion:
This study highlights the need for continued monitoring and evaluation of these drugs in clinical practice, as it first reveals their AEs in a large real-world sample population. Some AEs are generally consistent with the instructions and previous studies, but some unexpected AEs are detected for each drug. Due to the limitations of the spontaneous report database, such as including potential underreporting, overreporting, lack of causal relationship, unable to calculate incidence, and other confounding factors, more pharmacoepidemiology studies are needed to validate our findings.
Plain language summary
Introduction:
To monitor and evaluate the safety of drugs, the U.S. Food and Drug Administration (FDA) created the FDA Adverse Event Reporting System (FAERS) database to collect post-marketing adverse event (AE) reports of drugs. This study aims to explore the signals of gilteritinib and midostaurin using the FAERS database.
Methods:
We retrieved gilteritinib and midostaurin-related reports submitted between the year of initial FDA approval and March 31, 2023, from the FAERS database. We not only counted information about patients’ gender, age, reporting country, and outcome, but also analyzed the system organ classes (SOCs) of AEs, and concomitant medication and frequency of gilteritinib and midostaurin.
Results:
We collected 1,887 and 2,091 case reports for gilteritinib and midostaurin, respectively. We have detected that the common AEs of gilteritinib and midostaurin are consistent with clinical trials. Gilteritinib and midostaurin also produce unexpected AEs that were not mentioned on the label, including interstitial lung disease, cerebral hemorrhage, and tumor lysis syndrome for gilteritinib, septic shock, neutropenic sepsis, and neutropenic colitis for midostaurin.
Conclusion:
Through the FAERS database, we identified more AEs associated with gilteritinib and midostaurin than those indicated in the instructions. However, our findings cannot conclude a clear causal relationship between the two drugs and AEs. Additional studies are required to assess unexpected AEs.
Introduction
Acute myeloid leukemia (AML) is the most common form of leukemia in adults and the second common in children. 1 Due to its rapid development and high recurrence rate, the prognosis of AML remains unsatisfactory with a 5-year survival rate of 60%–75% for children and only 5%–15% for elderly patients.2,3
Approximately 25%–35% of AML patients have FMS-like tyrosine kinase 3 (FLT3) gene mutations.4,5 FLT3 is a transmembrane receptor tyrosine kinase protein predominantly expressed by hematopoietic stem cells. It plays a crucial role in cell development by regulating the growth and differentiation of hematopoietic stem cells. FLT3 is overactive and stimulates the growth of a large number of leukemia cells in FLT3-mutant patients, leading to a significantly increased risk of recurrence and reduced survival rates. 4 Therefore, AML patients with FLT3 mutations have a worse prognosis than wild-type FLT3 patients. 6
FLT3 inhibitors impeded the progression of AML by blocking the FLT3 signaling pathway and inhibiting leukemia cell proliferation.4,7 Midostaurin was approved on April 28, 2017, for the treatment of FLT3-mutant AML patients. 8 Clinical trials have demonstrated midostaurin plus standard first-line chemotherapy can prolong overall survival (OS) and event-free survival, and reduce the cumulative incidence of relapse in FLT3-mutant AML patients.4,9 Gilteritinib was approved on November 28, 2018, for the treatment of adult patients with FLT3-mutant relapsed or refractory (R/R) AML. 10 Compared with salvage chemotherapy, gilteritinib can significantly extend the OS and improve the survival rate of FLT3-mutant R/R AML patients.11 –13
Both drugs have shown promising results in AML patients with FLT3 mutations. Nevertheless, adverse events (AEs) are discovered gradually during the clinical use of the drug, causing concern among practitioners. Common AEs observed in midostaurin were thrombocytopenia, neutropenia, anemia, febrile neutropenia, infection, nausea, and vomiting.9,14 In addition, a retrospective study found that midostaurin was associated with an increased risk of cardiovascular AEs. 15 The common AEs of gilteritinib were diarrhea, neutropenia, fatigue, QTc interval prolongation, creatine kinase, and liver transaminase elevation. 14 Posterior reversible encephalopathy syndrome occurred in rare cases. 14 Due to AEs, gilteritinib interrupted treatment in 29% of patients, discontinued treatment permanently in 7%, and died in 2%. 10 AEs are one of the main causes of drug discontinuation and treatment failure.
Although some AEs have been reported in phase II and III clinical trials, there have been no studies on the safety profiles of midostaurin and gilteritinib in a large real-world sample population. This study aims to investigate the occurrence of AEs since the approval of two drugs for marketing using the Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) database, the largest publicly pharmacovigilance database in the United States.
Methods
Study design and data source
We conducted a retrospective, observational pharmacovigilance study based on the FAERS database, a spontaneous passive surveillance system. FAERS is a publicly available database that since 1968, allows healthcare professionals, consumers, and drug manufacturers worldwide to submit AE reports, medication error reports, and product quality complaints. Each report in the FAERS includes patient demographics and administrative information, medical history, drug/biologic information, the AEs, patient outcomes, report sources, therapy information, and indications of drugs. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 26.0, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) and grouped by system organ class (SOC). However, a characteristic of MedDRA is multiaxiality that a preferred term (PT) may correspond to one or more SOC. To avoid double-counting, we only analyzed the primary SOC related to PT.
The exposure time was set from the year of initial FDA approval for each drug to March 31, 2023: midostaurin (April 28, 2017), gilteritinib (November 28, 2018). We queried the FAERS database using any one of the following both generic and brand names: “midostaurin,” “rydapt,” “PKC412,” “CGP41251”; “gilteritinib,” “XOSPATA,” “ASP2215.” According to the recommendations of the FDA, we excluded duplicate reports by the following steps: when CASE ID are identical, we select the record with the latest FDA_DT; and in instances where both CASE ID and FDA_DT coincide, we choose the record with the higher PRIMARYID. 16 To focus our results on the drug most likely to cause the AE, the role code “primary suspect (PS)” was used to limit our analysis on reports that consider the drug as the primary suspect. In addition, we collected and organized the top 30 drugs with a high frequency of concomitant medication of gilteritinib and midostaurin to analyze the influence of concomitant medication. In this study, combination medication specifically refers to instances where gilteritinib or midostaurin serves as the primary suspected drug, accompanied by drugs identified with a role code of “concomitant,” distinct from the conventional understanding of “combination therapy,” which denotes the simultaneous initiation and continued administration of multiple drugs. Furthermore, to ensure the accuracy and completeness of data extraction and analysis, two researchers independently completed the two tasks, followed by cross-checking. In case of discrepancies, a consensus was sought through discussion; otherwise, the third researcher was referred to for adjudication.
Statistical analysis
Descriptive analyses were used to summarize the clinical features of AE reports associated with gilteritinib and midostaurin in the FAERS. For signal detection, we performed two disproportionality analysis methods to increase the consistency and robustness of results, including the reporting odds ratio (ROR) and the Bayesian confidence propagation neural network (BCPNN). The relative calculation formula and criteria are shown in Table 1. For ROR, a statistically significant signal was identified if the lower limit value of the ROR 95% confidence interval (CI) exceeded one, with at least three cases. For BCPNN, we employed the traditional threshold used by the Uppsala Monitoring Center, where the lower limit of the 95% CI (IC025) for the information component (IC) greater than zero indicates statistical significance. 17 Only when both methods achieved statistical significance, a signal was considered. Besides the signal detection at the PT level, we also carried out signal analysis at the SOC level only based on the ROR.
Summary of calculation formula and criteria used for signal detection.

a, number of reports with target AE caused by the target drug; AE, adverse event; b, number of reports with other AEs caused by the target drug; BCPNN, Bayesian confidence propagation neural network; c, number of reports with target AE caused by other drugs; CI, confidence interval; d, number of reports with other AEs caused by other drugs; IC, information component; IC025, the lower limit of the 95% CI; ROR, reporting odds ratio.
Sensitivity analysis
To evaluate the robustness of the findings, sensitivity analysis was performed using the following methods: (1) We conducted a time scan diagram for several significant AEs with clinical interest or life-threatening and calculated the IC values and 95% CI, to reflect the change tendency of the target drug-target AE association in the database as the number of reports increasing over time. The calculation method is shown in Table 1. When the lower 95% CI limit of the IC from a negative value to a positive value, a signal appears. In addition, a stable upward trend in the IC curve with a narrowed 95% CI indicates a robust and stable correlation. 18 (2) To improve the specificity and accuracy of safety signal assessment of gilteritinib and midostaurin, we excluded AE reports where either drug was used in combination with other medications, and indications other than AML. We undertook the following procedures to filter the extracted reports: Initially, we excluded AE reports with other drugs designated as secondary suspects, concomitants, or interacting drugs. Subsequently, AE reports with indications that were not relevant or did not correspond to our research theme of AML were also eliminated from the analysis.
Results
Clinical characteristics
We identified 1887 and 2091 case reports with gilteritinib and midostaurin in the FAERS, respectively. The characteristics of AE reports are displayed in Table 2. Male patients had a higher reporting percentage than female patients (gilteritinib: 50.34% vs 42.82%; midostaurin: 44.76% vs 41.46%). The age group with the majority of patients was 45–64 years for gilteritinib and midostaurin (24.59% and 23.86%, respectively). The number of case reports peaked in 2021 (gilteritinib: 545, 28.88%) and 2019 (midostaurin: 463, 22.14%). Physicians accounted for the highest proportion of reports for gilteritinib (1027, 54.43%) and midostaurin (891, 42.61%). Of note, 52.62% of gilteritinib case reports came from Japan, and the United States was the main reporting country for midostaurin (35.01%). AEs leading to death occurred more than 20% in gilteritinib (22.95%) and midostaurin (21.04%).
Characteristics of AE reports associated with gilteritinib and midostaurin in the FAERS.

AE, adverse event; FAERS, FDA Adverse Event Reporting System.
In addition, we compiled the top 30 drugs with the high frequently used in combination with gilteritinib and midostaurin, respectively, as shown in Figure 1. Cytarabine (85), azacitidine (77), and fluconazole (72) were the top three frequently used drugs in combination with gilteritinib, and the top three drugs with the high frequency of combination with midostaurin were cytarabine (201), daunorubicin (132), acyclovir (35).

Top 30 ranked concomitant medication reported with gilteritinib and midostaurin. (a) concomitant medication of gilteritinib; (b) concomitant medication of midostaurin.
Analysis at the SOC level
As shown in Figure 2, we found that midostaurin-induced AEs involved nine SOC with statistically significant ROR, and gilteritinib involved seven SOCs. The top three SOCs with high signal values were observed for midostaurin: blood and lymphatic system disorders, cardiac disorders, and hepatobiliary disorders. While gilteritinib presented the top three signals in blood and lymphatic system disorders, hepatobiliary disorders, and investigations.
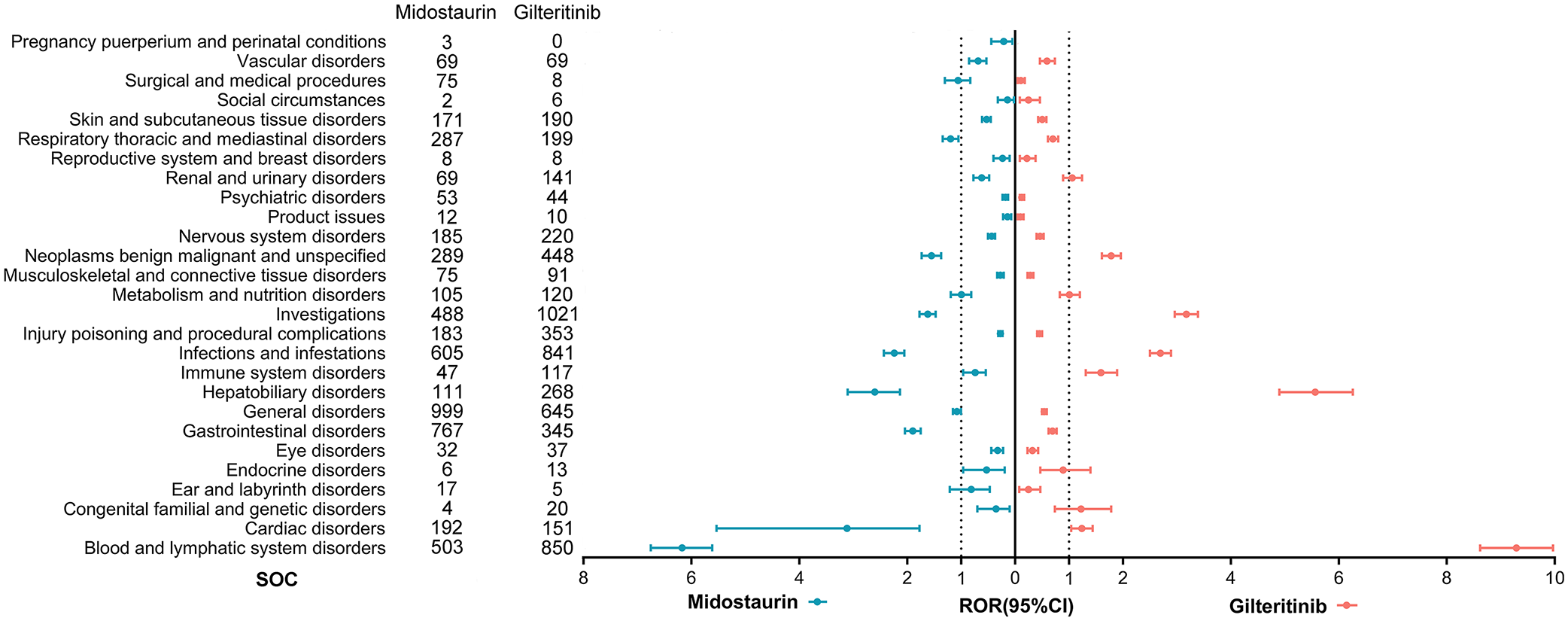
ROR of reports for gilteritinib and midostaurin at the SOC level.
Analysis at the PT level
Table 3 provided the results of disproportionality analysis at the PT level, grouped by SOC. We identified 46 positive signals for midostaurin. Besides the AEs mentioned in the instruction (nausea, vomiting, diarrhea, pyrexia, febrile neutropenia, pneumonia, electrocardiogram QT prolonged, etc.), there remained 24 unexpected AEs for midostaurin that showed signals, such as neutropenic colitis, septic shock, neutropenic sepsis, and hepatotoxicity. For gilteritinib, a total of 53 significant signals were found, 29 of which were not listed in the instruction, including septic shock, neutropenic sepsis, interstitial lung disease, subdural hematoma, and cerebral hemorrhage. Similarly, we also detected common AEs of gilteritinib that are mentioned in the instructions, such as anemia, differentiation syndrome, QT interval prolongation, and pneumonia.
Overview of statistically significant ROR and IC (PT level, organized by SOC) for gilteritinib and midostaurin in FAERS.

AEs are mentioned in the instruction.
AE, adverse event; FAERS, FDA Adverse Event Reporting System; IC, information component; PT, preferred term; ROR, reporting odds ratio; SOC, system organ class.
In the unexpected AEs, we found that some PTs, which may be indicative of treatment response or disease status, were detected as positive signals. For example, minimal residual disease (MRD), bone marrow failure, pancytopenia, white blood cell count decreased, and hemoglobin decreased were detected as positive signals in the case of midostaurin. For gilteritinib, bone marrow failure, pancytopenia, hemoglobin decreased, blast cell count increased, and white blood cell count increased were detected as positive signals.
Sensitivity analysis
We plotted a time scan of clinical concerns or life-threatening AEs to observe the change tendency of signal over time. The results are shown in Figure 3, of which Figure 3(a) and (b) show the time scan of differentiation syndrome and electrocardiogram QT prolonged related to gilteritinib, Figure 3(c) and (d) for electrocardiogram QT prolonged and sepsis associated with midostaurin. Figure 3(a) and (b) show that with the increasing number of reports of differentiation syndrome and electrocardiogram QT prolongation related to gilteritinib, the IC values significantly increased in 2019, with a 95% CI lower limit above zero, indicating the emergence of the two signals was first detected in 2019. From 2019 to 2022, the IC values gradually increased, and the 95% CI gradually narrowed and showed an upward trend, suggesting that the two signals were stable and the correlation between gilteritinib and the two AEs was strong. Similarly, a time scan of electrocardiogram QT prolongation and sepsis associated with midostaurin was conducted. As shown in Figure 3(c) and (d), the signals of electrocardiogram QT prolongation and sepsis for midostaurin were first exhibited in 2017. Over the subsequent 5 years, the IC and 95% CI values gradually stabilized, indicating the presence of a consistent and stable safety signal for these two AEs. These observed trends preliminarily suggest the four signals are unlikely to be false positives.

Time scans of IC for gilteritinib- and midostaurin-related adverse events: (a) gilteritinib-related differentiation syndrome; (b) gilteritinib-related electrocardiogram QT prolonged; (c) midostaurin-related electrocardiogram QT prolonged; (d) midostaurin-related sepsis. When the lower 95% CI limit of the IC from a negative value to a positive value, a signal appears. In addition, a stable upward trend in the IC curve with a narrowed 95% CI indicates a robust and stable correlation.
To more precisely assess the safety signals of gilteritinib and midostaurin, we excluded all potential confounding factors from concomitant medications and AE reports unrelated to our target indications. We definitively identified 1167 reports associated with gilteritinib and 1230 reports associated with midostaurin, with both exhibiting a certain degree of reduction in case numbers. The subsequent disproportionality analysis revealed that gilteritinib exhibited 24 positive signals, all consistently present prior to confounding factors exclusion (Supplemental Table S1). In contrast, for midostaurin, 19 positive signals were observed, also preexisting before exclusion, with the emergence of “Malaise” as a novel significant association (Supplemental Table S2). After excluding confounding factors, gilteritinib still exhibited a strong correlation with the following unexpected AEs: blast cell count increased, blood bilirubin increased, blood lactate dehydrogenase increased, bone marrow failure, cytopenia, infection, liver disorder, myelosuppression, pancytopenia, and white blood cell count increased (Supplemental Table S1). Midostaurin showed a significant correlation with unexpected AEs such as bone marrow failure, cytopenia, dysphagia, malaise, neutropenic colitis, pancytopenia, and septic shock (Supplemental Table S2).
Discussion
In this study, the statistical methods of ROR and BCPNN were used to evaluate the safety profile of midostaurin and gilteritinib in large real-world samples from the FAERS database.
We included 2091 and 1887 AE reports associated with midostaurin and gilteritinib. AE reports of gilteritinib were mainly from Japan, while AE reports of midostaurin were mainly from the United States. It seems likely that there is a higher incidence of AEs with gilteritinib in Japan and midostaurin in the United States. However, due to coding and spontaneous reporting, most of these are considered to be man-made rather than biological. 19 The reasons for the variation in AE reporting rates may be likely multifactorial: (a) Gilteritinib was first launched in Japan, while midostaurin was launched in the United States, which allowed clinicians and patients in the first countries to have earlier exposure to the drug and increased experience with its use and case reports.8,20 (b) “Stimulated reporting,” “the Weber Effect,” and media attention seem to affect reporting rates. “Stimulated reporting” refers to the concept that public disclosure of a safety issue by the issuance regarding the drug and/or the specific AE mentioned in such an alert, 21 for example, an FDA alert will result in substantially increased AE reporting rates. “The Weber Effect” is often generalized as a phenomenon where, after regulatory approval, AE reporting peaks at the end of the second year and then rapidly diminishes over time. 22 (c) Institutions engaged in risk evaluation and mitigation strategies, regulatory authorities, and the Internet might influence the reporting rate. 23 In addition, compared to midostaurin, we detected more AE-positive signals in gilteritinib, which may be attributed to various reasons. First, patients with FLT3-mutated R/R AML who received gilteritinib monotherapy usually experienced treatment failure, resulting in a more compromised physical condition, immune function, and tolerance compared to newly diagnosed patients. 24 Second, patients with relapsed/refractory conditions often require higher doses or longer durations of treatment to achieve more pronounced therapeutic effects. However, this may also increase the risk of AEs, as the cumulative impact of the drug within the body and its sustained challenge to the body may exceed the patient’s threshold of tolerance. Nevertheless, gilteritinib still provides a novel therapeutic option for patients with R/R AML. 25 In the future, it is crucial to investigate the impact of varying doses, treatment durations, and combination therapy regimens on AEs and efficacy. Additionally, we will pay attention to individualized differences among patients, including genotype and past treatment history, to devise more precise treatment strategies.
Our study found that midostaurin and gilteritinib are often used in combination with chemotherapy and antifungal agents during the treatment of AML. This combination therapy aims to improve treatment outcomes, but it may also increase the risk of AEs. Gilteritinib is often combined with traditional chemotherapy drugs such as cytarabine and azacitidine, and midostaurin is often combined with standard induction chemotherapy such as 7 + 3 regimens (daunorubicin and cytarabine).9,26 These chemotherapy drugs can cause adverse reactions, including bone marrow suppression, nausea, vomiting, hair loss, and abnormal liver function. In addition, mold-active primary antifungal prophylaxis is widely recommended in neutropenic patients with newly diagnosed AML. 27 Therefore, gilteritinib and midostaurin are often used in combination with antifungal drugs. The common adverse reactions of antifungal drugs include nausea, vomiting, abnormal liver function, and abnormal renal function. The adverse reactions commonly associated with chemotherapeutic agents and antifungal drugs exhibit some overlap with those observed in patients treated with gilteritinib and midostaurin. Consequently, when administering midostaurin and gilteritinib in clinical settings, once adverse reactions occur, clinicians should carefully discern the etiology of these reactions. Specifically, they must determine whether the adverse reactions are primarily caused by midostaurin or gilteritinib alone, whether they are the result of concomitant medications, or whether they arise from the synergistic effects of both drugs.
Our results indicated that blood and lymphatic system disorders were one of the most common SOCs in midostaurin and gilteritinib. AEs were reported in 83.1% of patients treated with gilteritinib, and the most common AEs with grade ⩾3 were hematological toxicities (e.g., anemia, febrile neutropenia, and thrombocytopenia). 25 A clinical trial of midostaurin plus chemotherapy treated FLT3-mutant AML patients (NCT00651261) revealed that up to 97% of participants experienced grade ⩾3 hematological toxicities. 9 In response to such hematologic toxicities, therapeutic interventions including thrombopoietin receptor agonists, granulocyte colony-stimulating factors, and erythropoietin can be administered, with platelet transfusions and packed red blood cell transfusions provided when necessary. The plasma protein binding rate of gilteritinib is about 90%–94%, midostaurin is more than 99%, and CYP3A4 is their main metabolic enzyme.25,28 –30 Therefore, to prevent the exacerbation of AEs, CYP3A4 inhibitors should be avoided when using midostaurin and gilteritinib, such as itraconazole, ketoconazole, and grapefruit juice. The common (⩾10%) non-hematological AEs of midostaurin and gilteritinib included: nausea, vomiting, diarrhea, edema, abdominal pain, fatigue, constipation, fever, headache, and dyspnea. A phase II, randomized, open-label trial of midostaurin (NCT01883362) mentioned that the most common AEs leading to drug dose adjustment were vomiting (27%), nausea (20%), and elevated of aspartic aminotransferase (10%). 31 In a phase III, randomized, controlled clinical trial, 11.3% of patients discontinued gilteritinib because of AEs. 32 The most common AEs were elevated aspartate aminotransferase level, elevated alanine aminotransferase level, and pneumonia. 32 One study mentioned that the elevated liver enzyme levels (i.e., alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase), blood creatine phosphokinase level, and diarrhea increased with gilteritinib in a dose-dependent manner. 33 If the above common AEs are not controlled in time, they may lead to drug reduction, interruption, or even permanent discontinuation. Ultimately, they promote the progression of the patient’s disease and endanger their lives. In addition, in our study, pneumonia and septic shock were detected as positive signals in midostaurin and gilteritinib. Pneumonia and septic shock are common AEs that cause death in patients treated with gilteritinib. 32 Furthermore, the instruction of midostaurin recommends discontinuing the drug if patients have signs or symptoms of pulmonary toxicity. Prolonged QT interval may result in arrhythmias such as Torsades de Pointes and ventricular fibrillation, which can cause sudden cardiac death in patients.34,35 One study found that 10.3% of patients treated with gilteritinib experienced QT interval prolongation. 36 Another retrospective analysis showed that midostaurin and/or FLT3 inhibitors were independent risk factors for cardiovascular AEs. 15 This is consistent with the results of our study, where electrocardiogram QT prolonged was detected as a positive signal in midostaurin and gilteritinib, and the signal stabilized from 2019 to 2022. Regarding the detection and management of QT interval prolongation during treatment with midostaurin, existing research suggests that if the QTc interval is >500 ms, midostaurin should be immediately discontinued. If the QTc interval is between 470 and 500 ms, the midostaurin dose should be reduced to 50 mg qod. If the QTc interval at the beginning of the next cycle is ⩽470 ms, midostaurin can be restarted at the original dose. 9 Similarly, according to the FDA’s recommendations, patients need to take an electrocardiogram before starting the treatment of gilteritinib, including the 8th and 15th day of the first cycle, and before the start of each subsequent treatment cycle. If an increase in QTc interval >30 ms is detected on the eighth day of the first cycle, it must be confirmed on the ninth day. If confirmed, a dose reduction to 80 mg qod should be considered. In particular, patients with QTc > 500 ms must discontinue gilteritinib. 37 It should be used with caution in patients with relevant cardiac diseases. Additionally, hypomagnesemia and hypokalemia may increase the risk of QT interval prolongation, therefore, electrolyte abnormalities should be corrected before and during treatment. Furthermore, other medications with a risk of prolonged QT interval should be avoided during the treatment with midostaurin and gilteritinib. 30 Differentiation syndrome is a severe and specific AE of gilteritinib. Approximately 3% of patients may develop differentiation syndrome, which can be life-threatening if not promptly diagnosed and managed. 10 The clinical manifestations of differentiation syndrome are mainly pulmonary edema, pericardial effusion, renal dysfunction, hypotension, rapid weight gain, dyspnea, unexplained fever, and rash. 10 In addition, it has been reported that some patients with differentiation syndrome have skin diseases characterized by neutrophilic dermatoses.38,39 If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution. If severe symptoms and/or signs persist for more than 48 h after initiation of corticosteroids, gilteritinib should be discontinued until the severity of symptoms and signs decreases. Once symptoms resolve, corticosteroids can be tapered and should be administered for at least 3 days. 37 Patients with high-risk factors of differentiation syndrome, such as elevated white blood cell count, creatinine, lactate dehydrogenase, and peripheral blood blast count, can be given corticosteroids for prevention. 40
In addition to the QT interval prolongation, sepsis, and pneumonia mentioned in the instruction of midostaurin, our study also found serious unexpected AEs, such as neutropenic colitis (N = 20), neutropenic sepsis (N = 11), and septic shock (N = 52). The typical symptoms of AML do not include these unexpected AEs related to midostaurin. In contrast, the potential association of midostaurin with these AEs has been revealed in existing study reports. Neutropenic colitis is a necrotizing infection of the intestine, often accompanied by fever, diarrhea, abdominal pain, nausea, and vomiting. In the current study, only Dotson et al. 41 reported three cases of neutropenic colitis after treatment with midostaurin. A total of 20 cases of neutropenic colitis were reported in our study, and the association was strong (ROR: 100.26; IC: 4.12). The exact pathogenesis of neutropenic colitis is still unclear. Intestinal mucosal injury, neutropenia, and weakened immune systems may be the main causes of the disease. 42 Midostaurin is usually combined with chemotherapy drugs in the treatment of AML patients. Chemotherapy drugs can cause direct damage to the gastrointestinal mucosa and immune dysfunction, thereby increasing the risk of such diseases. 43 It has been reported that neutropenic colitis can cause fatal septic shock, with estimated mortality rates ranging from 0% to 50%.41,44 Neutropenic colitis usually progresses rapidly, and clinicians need to make an accurate differential diagnosis on time for early treatment. In clinical management, patients without gastrointestinal bleeding, peritonitis, or intestinal perforation usually receive medical treatment, including granulocyte colony-stimulating factors and antibiotics. Patients with severe conditions may require surgical intervention, such as bowel resection and stoma. 45 Additionally, the positive signals of neutropenic sepsis (ROR: 18.52; IC: 2.91) and septic shock (ROR: 14.86; IC: 3.54) caused by midostaurin were reported for the first time. Neutropenic sepsis can rapidly progress to septic shock and multiorgan failure, thereby increasing the risk of death.46,47 Prevention and appropriate treatment of neutropenic sepsis is important to maintain dose intensity and continue treatment as planned. Therefore, guidelines recommend that patients with neutropenic sepsis should immediately receive empirical antibiotic therapy with broad-spectrum antibiotics against Pseudomonas aeruginosa. As the initial treatment recommendation, piperacillin-tazobactam or carbapenems are usually chosen. 48 In addition, dexamethasone, anticoagulants, and blood products may be necessary for management. It is also important to monitor the patient’s blood glucose level, bicarbonate level, and other relevant indicators. 49
We also found serious unexpected AEs of gilteritinib, such as cerebral hemorrhage (N = 21), subdural hematoma (N = 11), tumor lysis syndrome (TLS; N = 21), and interstitial lung disease (N = 20). Perrone et al. 50 reported the first case series on five intracranial hemorrhages (including cerebral hemorrhage) in patients exposed to gilteritinib and highlighted that this side effect may be underreported. In our study, a total of 33 cases related to cerebral hemorrhage and subdural hematoma were detected. The mortality rate of cerebral hemorrhage is as high as 50%, and the main risk factors include advanced age, coagulopathy, platelet dysfunction, hypertension, diabetes mellitus, and vascular lesions. 51 Gilteritinib inhibits glutamine uptake and utilization by interfering with glutamine transporters, which leads to cellular senescence and death. Glutamine inhibition leads to a block in glutathione synthesis, further increasing the stimulation of brain tissue by oxidative free radicals. 52 In addition, gilteritinib can also cause coagulation disorders by reducing fibrin polymerization and impairing clot stability. 53 Currently, no research has proposed an effective therapeutic drug for this AE, so timely discontinuation of the drug and active symptomatic treatment may be a better choice. An open-label phase I study revealed that one of the 24 patients treated with gilteritinib developed grade 3 TLS. 33 However, we detected a total of 21 cases of TLS, and the signal intensity of TLS ranked prominently (ROR: 22.04; IC: 3.49). The mechanism of TLS is extensive tumor cell breakdown, leading to the rapid release of intracellular contents into the systemic circulation, exceeding homeostatic mechanisms. 54 This process can lead to hypocalcemia, hyperphosphatemia, hyperkalemia, hyperuricemia, and the accumulation of xanthine. These electrolyte and metabolic imbalances pose significant risks of acute kidney injury, arrhythmia, seizures, multiorgan failure, and even death. TLS usually occurs between 3 days before and 7 days after treatment initiation. 54 The risk factors for TLS in AML patients include tumor burden, cellular proliferation rate, and sensitivity to therapy, as well as patient-related factors such as hyperuricemia, dehydration, and preexisting renal disease. For high-risk patients with TLS in AML, prevention is crucial. Before and during treatment, patients’ electrolyte levels and renal function indicators should be closely monitored, and preventive measures such as the use of allopurinol and intravenous hydration can be taken to reduce the risk. Interstitial lung disease (ROR: 4.12; IC: 1.84) was the positive signal detected in our study. A post-market surveillance study in Japan targeting FLT3-mutated relapsed/refractory AML patients showed interstitial lung disease was associated with gilteritinib, a specific AE leading to discontinuation in multiple patients, with an incidence of 4.7%. 36 Interstitial lung disease is characterized by inflammation and/or fibrosis of lung parenchyma, which is associated with progressive dyspnea and can progress to respiratory failure. The pathogenesis of interstitial lung disease caused by gilteritinib has not been elucidated yet. The current mechanisms of drug-induced interstitial lung disease may be related to direct damage to alveolar epithelial or capillary endothelial cells, as well as immune system dysregulation, systemic cytokine release, cell-mediated lung injury, and free radical production caused by oxidative damage. 55 Drug withdrawal is the main treatment for interstitial lung disease. For patients with progressive disease after drug withdrawal, glucocorticoid therapy is usually used, but there is no reliable data on the efficacy of glucocorticoid therapy for interstitial lung disease. 56 In conclusion, although not mentioned in the instructions, healthcare professionals should remain vigilant and monitor patients treated with gilteritinib or midostaurin for potential severe AEs.
Among the unexpected AEs we detected, we noticed that some AEs may be associated with treatment response or disease status. For example, MRD serves as a crucial indicator of treatment response for midostaurin. The presence of MRD typically indicates a high risk of AML recurrence, compared to patients without MRD. 57 The decrease in white blood cell count and hemoglobin may not only be a manifestation of drug toxicity but also a signal of disease progression. This is particularly noteworthy when these changes are accompanied by other adverse prognostic factors, such as advanced age, genetic mutations including TP53 mutations, and high expression of internal tandem duplication of FLT3, which should alert clinicians.58 –60 Bone marrow failure and pancytopenia may be caused by various factors, including drug toxicity, the disease itself, or the patient’s underlying health status. However, during midostaurin treatment, they are more likely to be symptoms or serious complications of the disease itself rather than simply drug toxicity, which requires careful clinical judgment on the part of the physician. The symptoms caused by drugs are usually related to the dosage and duration of drugs and may be relieved after discontinuation. Conversely, symptoms caused by AML are often more complex, potentially accompanied by the emergence or exacerbation of other complications, such as infections, hemorrhage, anemia, etc. Especially in the treatment of midostaurin, if these symptoms persist and worsen, accompanied by the deterioration of disease indicators, such as a sustained increase in MRD, additional gene mutations, and an elevated proportion of blasts, it is more likely to reflect a worsening of the disease state rather than AEs of midostaurin. Similarly, in the case of gilteritinib, bone marrow failure, and pancytopenia may primarily reflect the disease state, especially when the disease progresses or the treatment response is suboptimal. In addition to possibly being a marker of disease progression, hemoglobin decreased may also be associated with the myelosuppression effects of gilteritinib, which requires careful monitoring and evaluation in clinical practice. Clinicians can comprehensively assess the causes of hemoglobin reduction through regular blood routine tests, bone marrow aspiration examinations, and consideration of the patient’s clinical manifestations, such as fatigue, pallor, and malnutrition. During gilteritinib treatment, an increase in blast cell count and white blood cell count may signify a treatment response, particularly when accompanied by improvements in other disease indicators, such as the decrease in the proportion of bone marrow blasts and the negative conversion of MRD. Nevertheless, an elevated white blood cell count may also correlate with infection or other inflammatory processes, for example, viral or bacterial infections, autoimmune diseases (rheumatoid arthritis, systemic lupus erythematosus). Therefore, in order to accurately discern the causes of changes in white blood cell count, a comprehensive assessment of the patient’s clinical status is required, including a detailed inquiry into their medical history, necessary laboratory tests for virus and bacterial detection, as well as imaging examinations.
In this study, the ROR value serves as a measure of the statistical association strength between a drug and an AE. High ROR values may indicate a strong association between the drug and the reported AE but may also result from inherent limitations of spontaneous reporting systems (reporting bias, such as the notoriety effect). Therefore, severe or novel AEs with high ROR values should be interpreted with caution. Firstly, we can use it in combination with CIs, where a narrow CI means a more precise estimation of ROR values, while a wide CI indicates greater uncertainty in the estimation. Secondly, we can conduct a comprehensive assessment of external factors that may have contributed to a relative increase in specific AE reports, conduct a review and evaluation of individual case reports, or even undertake large-scale epidemiological studies to confirm or refute these associations. Additionally, the ROR value is an indicator that dynamically changes over time as more data accumulates. Consequently, it is essential to continuously monitor and evaluate the ROR value. ROR is only a statistical association indicator and not direct evidence of causality. Therefore, the ROR value can be used as a tool for initial assessment of drug safety, but it should not be solely relied upon as the basis for clinical decision-making. When making clinical decisions, clinicians need to comprehensively consider factors such as other research evidence, the specific circumstances of the patient, treatment goals, the safety of alternative treatment options, and clinical guidelines. Especially when serious or novel AEs with high ROR values occur, a more cautious assessment is required to avoid blindly altering the treatment plan. Nevertheless, the ROR value still has significant implications for clinical practice. It serves as a reminder to pay attention to potential AEs induced by drugs, strengthen pharmacovigilance efforts, conduct closer monitoring of patients, and adjust treatment plans as necessary to ensure patient safety.
Limitations
However, given a retrospective study of FAERS, there is an inherent limitation. For example, as a spontaneous reporting system, the FAERS database is inevitably impacted by reporting biases and underreporting, and varies quality of reports. 61 Reporters tend to prioritize reporting more severe or novel AEs, which may not comprehensively reflect the full spectrum of AEs associated with a drug in the real world. 62 This inherent reporting bias within the FAERS database can lead to an overemphasis and overestimation of severe or novel AEs, while common, less prominent AEs may be underestimated. There are also duplicate reports where the same report was submitted by a consumer and by a sponsor, which may result in an inflation of certain AE reports and consequently impact their reporting rates and ROR values. 63 The FAERS does not require a proof of causal relationship between the drug and the AE and lacks information about the number of patients treated with a drug and the detailed severity of AEs. Therefore, we could not obtain a causal relationship from the observed associations, nor could we calculate the incidence and the severity of related AEs in midostaurin and gilteritinib.
In addition, the severity of AML, a highly heterogeneous disease, can significantly impact patient outcomes and treatment strategies. This heterogeneity likely extends to the occurrence and reporting of AEs associated with AML therapies. More severe cases of AML may require more aggressive treatment regimens, which could increase the likelihood of AEs. Additionally, patients with severe AML may be more closely monitored during treatment, leading to a higher rate of AE detection and reporting. Conversely, patients with less severe AML may experience fewer AEs or have their AEs underreported due to less intensive monitoring. However, due to the limitations of spontaneous reporting systems, we were unable to acquire more comprehensive information regarding the reports, including the severity of AML. Therefore, we can’t fully exclude the possibility that AML severity may affect AE signal detection.
Moreover, the AE reports in the FAERS database may have seasonal or temporal biases. Firstly, the prevalence of seasonal diseases often prompts a surge in the use of related therapeutic drugs, which in turn indirectly leads to fluctuations in the number of AE reports. Secondly, the reporting behavior of healthcare professionals and consumers for AEs can be influenced by seasonal factors such as holiday schedules and workload changes, further exacerbating the extent of seasonal biases. Reporting frequency might be influenced by media attention and publicity, such that a well-publicized safety concern increases reporting rates, thereby the ROR values could be inflated, a phenomenon referred to as the “notoriety effect.” 64 Furthermore, the inherent time lag in the submission and entry process of AE reports implies that we may fail to promptly capture all relevant AE information when assessing the safety of new drugs or novel drug uses. Such seasonal and temporal biases have the potential to underestimate or overestimate the AE risks associated with certain drugs.
To mitigate the impact of these biases on research findings, continuous and systematic monitoring of drugs is of paramount importance. For example, the time scan for key signals of interest to observe their variations over time. Additionally, some key signals can be further investigated in real-world studies with robust designs. Despite these limitations, the FAERS remains an important tool in identifying safety concerns for drugs, and also communicating concerns to physicians and patients.
Conclusion
This is the first study highlighting the comprehensive post-marketing safety between midostaurin and gilteritinib based on real-world data from the FAERS database. Common AEs mentioned in the instructions were detected, such as anemia, nausea, and vomiting for midostaurin; febrile neutropenia, and pneumonia for gilteritinib. We also detected some serious AEs that were not included in the instructions, such as septic shock, neutropenic sepsis, and neutropenic colitis for midostaurin; interstitial lung disease, cerebral hemorrhage, and TLS for gilteritinib. Due to the limitations of FAERS, including potential underreporting, overreporting, lack of causal relationship, and unable to calculate incidence, it is crucial to acknowledge that our findings may be biased and need to be interpreted with caution. We recommend conducting prospective, observational studies or randomized controlled trials to validate our results and better understand these AEs, especially the incidence, severity, and risk factors associated with the unexpected AEs. In addition, pharmacovigilance programs should be strengthened to continuously monitor the safety of midostaurin and gilteritinib, with a particular focus on newly identified serious AEs.
Supplemental Material
sj-docx-1-taw-10.1177_20420986241308089 – Supplemental material for Unveiling unexpected adverse events: post-marketing safety surveillance of gilteritinib and midostaurin from the FDA Adverse Event Reporting database
Supplemental material, sj-docx-1-taw-10.1177_20420986241308089 for Unveiling unexpected adverse events: post-marketing safety surveillance of gilteritinib and midostaurin from the FDA Adverse Event Reporting database by Tingting Jiang, Yanping Li, Ni Zhang, Lanlan Gan, Hui Su, Guiyuan Xiang, Yuanlin Wu and Yao Liu in Therapeutic Advances in Drug Safety
Supplemental Material
sj-docx-2-taw-10.1177_20420986241308089 – Supplemental material for Unveiling unexpected adverse events: post-marketing safety surveillance of gilteritinib and midostaurin from the FDA Adverse Event Reporting database
Supplemental material, sj-docx-2-taw-10.1177_20420986241308089 for Unveiling unexpected adverse events: post-marketing safety surveillance of gilteritinib and midostaurin from the FDA Adverse Event Reporting database by Tingting Jiang, Yanping Li, Ni Zhang, Lanlan Gan, Hui Su, Guiyuan Xiang, Yuanlin Wu and Yao Liu in Therapeutic Advances in Drug Safety
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.