
Abstract
The relatively new discipline of pharmacovigilance (PV) aims to monitor the safety of drugs throughout their evolution and is essential to discovering new drug risks. Due to their specific and complex physiology, children, pregnant women, and elderly adults are more prone to adverse drug reactions (ADRs). Additionally, the lack of clinical trial data exacerbates the challenges faced with pharmacotherapy in these populations. Elderly patients tend to have multiple comorbidities often requiring more extensive medication, which adds additional challenges for healthcare professionals (HCPs) in delivering safe and effective pharmacotherapy. Clinical trials often have inherent limitations, including insufficient sample size and limited duration of research; as some ADRs are attributed to long-term use of a drug, these may go undetected during the course of the trial. Therefore, the implementation of PV is key to insuring the safe and effective use of drugs in special populations. We conducted a thorough review of the scientific literature on PV systems across the European Union, the United States, and China. Our review focused on basic physiological characteristics, drug use, and PV for specific populations (children, pregnant women, and the elderly). This article aims to provide a reference for the development of follow-up policies and improvement of existing policies as well as provide insight into drug safety with respect to patients of special populations.
Plain language summary
Due to the particularity of physiological functions, the special population (children, pregnant women, and the elderly) are more susceptible to adverse drug reactions (ADRs) and have more drug safety problems. The implementation of PV is helpful for the detection of safety risks throughout the life cycle of drugs, so that healthcare professionals (HCPs) can take early measures to reduce the drug use risks of patients.
Many countries have implemented a PV system. However, PV policies and systems for the special population are not complete in various countries, or no independent PV system for the special population has been set up.
This article discusses the PV systems of the European Union, the United States, and China with special focus on basic physiological characteristics, use of drugs, and the implementation of PV with respect to children, pregnant women, and the elderly. Focus on these problems are of great importance for formulating a more complete drug management scheme in the special population and can provide a reference for the development of follow-up policies and improvement of existing policies.
Introduction
In 1968, the World Health Organization (WHO) established the global individual case safety report (ICSR) database VigiBase, which regularly receives data regarding adverse drug reactions (ADRs) from more than 140 countries. 1 In 1974, the concept of pharmacovigilance (PV) was first proposed in France. In 2002, WHO explicitly defined PV as ‘the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems’. 2 The trend in drug management has shifted from focusing on disease treatment to managing a drug’s development and clinical use. Compared with ADRs, PV has expanded with respect to its terms and definitions, range of coverage, and drug monitoring cycle. An ADR is defined as ‘an unintended harmful reaction of a drug product that occurs in the normal amount of use’. 3 In addition to covering adverse reactions and monitoring of adverse events, PV also covers drug safety issues such as substance abuse, drug quality, medication errors, drug interactions, and reactions to excipients. 4 PV involves not only conventional drugs but also blood products, biological products, and vaccines. PV monitors drug safety events during the whole drug ‘life cycle’ (pre-marketing, post-marketing, and any recall or withdrawal from market).
ADRs can cause serious physical harm to patients and also represent a significant economic burden. In one hospital in the United Kingdom, the cost of hospital admission due to ADRs was £466 million per year. 5 The cost of adverse reactions in the United States is as high as $30.1 billion per year. 6 These figures are striking, especially since in most cases, ADRs can be prevented or effectively treated. 7 PV is critical for discovering unexpected ADRs, and, if properly implemented, can warn of the potential for post-marketing ADRs and thereby reduce harm. Clinical trials are valuable when evaluating drug efficacy, but they are far from thorough in evaluating drug safety. Due to sample size limitation, ADRs or adverse drug events (ADEs) identified during clinical trials typically represent only the most common safety problems of the studied drug. The practice of PV is an effective strategy to systematically and comprehensively evaluate drug safety throughout the drug’s life cycle and to discover any rare ADRs.
There are many challenges facing the safe use of medication in children, pregnant women, and elderly adult populations. As physiologic systems in children are not fully developed, drugs may not behave in children the same way as they do in adults. Therefore, off-label use of drugs (the indication, dosage, course of treatment, administration method, or population of drug use are not within the scope of the medication package insert approved by the drug regulatory department) and the incidence of drug use errors (e.g. wrong dose, prescribing errors, dispensing errors, and administration errors) are more common.3,8 In addition, clinical trials in children often face challenges in subject recruitment, precluding the design of large-scale studies or impeding the use of adequate control groups for a given age range. 9 Clinical trials are also scarce with respect to the study of pregnant patients; therefore, the safety profiles of various drugs with respect to use in pregnancy remain unclear. Elderly patients are particularly prone to ADRs due to functional degradation, decreased immunity, memory decline, and the presence of multiple comorbidities. The implementation of PV systems in the European Union, the United States, and China and their related databases are discussed below, with special focus on the implementation of these systems with respect to special populations. Here, we aim to provide a resource for healthcare professionals (HCPs) with the goal of improving drug safety in children, pregnant women, and elderly adults.
Methods
We present a narrative review of the literature about PV in special populations. We searched in MEDLINE (PubMed) using the following Medical Subject Headings terms and their synonyms: ‘adverse effects’ OR ‘adverse drug effects’ OR ‘drug-related side effects’, ‘adverse reactions’ OR ‘adverse drug reactions’, ‘pharmacovigilance’, ‘child’ OR ‘children’, ‘pregnant women’ OR ‘gravida’, ‘elderly’, ‘physiological characteristics’ OR ‘absorption’ OR ‘distribution’ OR ‘metabolism’ OR ‘excretion’, ‘drug use’ OR ‘medication’ OR ‘medication use’. This search includes literature published as of December 2022. We conducted a preliminary screening of the identified records based on their titles and abstracts, and subsequently selected relevant studies for inclusion in this review based on a thorough review of the full text. Three researchers screened the articles, and any differences were resolved by consensus.
Articles included in the narrative review discussed the PV systems of the European Union, the United States, and China. Moreover, we evaluated the included literature related to the basic physiological characteristics, use of drugs, and the implementation of PV with respect to children, pregnant women, and the elderly.
PV implementation around the world
The discipline of PV has been introduced in the European Union, the United States, China, Japan, South Korea, and several other countries; these countries typically have differing institutional settings and ADR reporting systems. The three most famous ADR spontaneous reporting databases in the world are EudraVigilance, FDA Adverse Event Reporting System (FAERS), and VigiBase. The construction of these drug safety monitoring databases is a crucial aspect of PV implementation. Data mining is extensively utilized in medicine, particularly in ADE monitoring, to extract and assess potential rules from vast data sources. The disproportionality analysis is the frequently utilized approach to mine ADE signals. This method can efficiently identify the association between drugs and adverse effects. The disproportionality analysis is categorized into the frequency method and Bayesian method. It mainly compares the ratio of actual and expected ADE reports. A safety signal is indicated when the ratio surpasses a predetermined critical value. The frequency method, including reporting odds ratio (ROR) and proportional reporting ratio (PRR), utilizes statistical methods to examine relative risk ratios. Its logical principles are clear and easy to understand, with simple computations, high sensitivity, and specificity. However, it is prone to generating false positive signals.10,11 Bayesian methods, such as Bayesian confidence propagation neural network (BCPNN) and multiple item empirical Bayesian Gamma Poisson Shrinker (MGPS), are based on the Bayesian principle and are commonly used in data analysis. They provide stable calculation results and have no usage restrictions, making them suitable for analyzing large datasets. However, the use of these methods may lead to false negative signals, which should be taken into account. Despite these limitations, Bayesian methods continue to be an important tool for scientific research due to their reliability and accuracy.12,13 Both the frequency method and the Bayesian method are based on a four-cell table (Table 1). The specific equations and signal generation criteria of the four methods are shown in Table 2.
Four grid table for the disproportionality analysis.

a, number of reports containing both the target drug and target ADR; b, number of reports containing other ADR of the target drug; c, number of reports containing the target ADR of other drugs; d, number of reports containing other drugs and other ADR.
ADR, adverse drug reaction.
Four major algorithms used for signal detection.

Equation: a, number of reports containing both the target drug and target adverse drug reaction; b, number of reports containing other adverse drug reaction of the target drug; c, number of reports containing the target adverse drug reaction of other drugs; d, number of reports containing other drugs and other adverse drug reactions.
95% CI, 95% confidence interval; χ2, chi-squared; EBGM, empirical Bayesian geometric mean; EBGM05, the lower limit of 95% CI of EBGM; IC, information component; IC025, the lower limit of 95% CI of the IC.
The European Union and the United States, as the earliest countries to introduce PV, have more developed systems and policies based on larger bodies of data. Although introduced later, the review of PV systems in China may also be informative. Therefore, we will focus primarily on the implementation and management of PV systems in the European Union, the United States, and China.
PV in the European Union
The current laws and regulations of the European Union regarding PV were revised and improved in 2012 and comprise four parts: directive, regulation, non-legislative acts, and miscellaneous. 14 Member states manage their own individual PV systems within the framework of the larger European Union system. The European Medicines Agency (EMA) and the relative regulatory agencies of member states, respectively, approve and regulate drugs marketed through both centralized and non-centralized review procedures. The ADR reporting system in the European Union employs a combination of spontaneous and mandatory reporting. Among these, spontaneous reporting is the main reporting modality and is primarily used by medical institutions, monitoring agencies, and patients. Mandatory reporting is secondary, and typically used by drug manufacturing companies. Furthermore, PV laws and regulations stipulate that marketing authorization holders (MAHs) establish a ‘Qualified Person for Pharmacovigilance’ (QPPV) to oversee PV-related work and facilitate communication with EMA.
EMA has established seven scientific committees, one of which is the PV Risk Assessment Committee. This committee is responsible for assessing and monitoring drug safety issues in humans and consists of surveillance and management experts, patient representatives, and HCPs.
The main database used by EMA to monitor ADRs is EudraVigilance. Through data mining, potential drug risks are identified to provide the timeliest drug safety information possible to the public with the goal of controlling the impact of ADRs. Adverse reaction reports from the European Union are first sent to the individual member state PV contact or the MAH by way of an online or paper report form. The individual case safety report (ICSR) is only reported to EudraVigilance after it has been reviewed and confirmed as valid. This process is conducive to ensuring the accuracy and quality of data submitted to EudraVigilance and increasing the credibility of conclusions based on subsequent data mining of this database. For any high-risk drugs identified, EMA increases the frequency of surveillance and data analysis. European Union member states can freely access EudraVigilance, facilitating its use by medical and scientific professionals.
PV in the United States
The United States Food and Drug Administration (FDA) began collecting ADR reports in 1961. The agency largely responsible for PV in the United States is the Center for Drug Evaluation and Research (CDER), a subdivision of the FDA. There are no local PV institutions, and CDER is the only national PV center. The main responsibility of CDER is to monitor, identify, evaluate, and control risks throughout the life cycle of drugs. Also under the purview of this institution is the authenticity and integrity of drug-related data. Within CDER are the Office of New Drugs, the Office of Compliance, the Office of Generic Drugs, and the Office of Surveillance and Epidemiology (OSE). OSE is the main department responsible for drug safety work such as PV, drug epidemiology, and drug risk management. The adverse reaction reporting system of the FDA is also divided into two categories: the mandatory reporting system for drug manufacturers and the MedWatch voluntary reporting system for patients and medical professionals. The other two national voluntary reporting systems in the United States are the Institute for Safe Medication Practices Medication Errors Reporting Program (ISMP-MERP) and the United States Pharmacopoeia MEDMARX drug error-reporting system.15,16
The FDA’s active PV monitoring system was first introduced in 2008. The FDA collects and analyzes the electronic data of medical institutions and compiles and publishes drug monitoring information through its website. The FDA also has a passive detection system for safety signals called the FAERS, which contains data on all marketed drugs and is a helpful tool for use in the monitoring of drug safety. Professionals can identify high-risk drugs by reviewing data regarding adverse reactions in this database. From these data, active monitoring rules and any needed emergency measures can be established for the relevant drugs with the intent to reduce patient harm.
PV in China
In June 2017, the China National Medical Products Administration (NMPA) joined the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). In December 2019, China’s newly revised Drug Administration Law was officially implemented, in which Article 12 of Chapter I stipulated the establishment of a PV system. In May 2021, China’s first PV Quality Management Standard (No. 65, 2021) was issued; this standard took effect on 1 December 2021, representing China’s shift from monitoring of post-market ADRs to monitoring the safety and risks related to the whole life cycle of a drug. NMPA issued the Guiding Principles of PV Inspection on 15 April 2022 to act as a guide for regulatory authorities with respect to PV inspection and to further improve the overall implementation of PV.
The NMPA is the main organization for PV work in China and is responsible for the monitoring of ADRs and post-marketing safety evaluation. China’s ADR monitoring network is mainly divided into four levels: national, provincial, municipal, and county. Data from these levels combine to form a centralized spontaneous reporting system. ADRs are collected and recorded into the database according to a standardized reporting principle when events are suspected. However, this reporting method has several limitations including omission of information, non-standard filling of relevant forms, and incomplete reporting of events. To improve this system, realize the active monitoring of risk signals, and better promote the reporting and analysis of ADRs, China began to explore the establishment of the Chinese Hospital PV System in 2016. In 2020, the NMPA proposed a ‘one body and two wings’ adverse reaction monitoring system. The ‘one body’ refers to the ADR monitoring organization, which is the professional and technical organization within the system. The ‘two wings’ are the MAH and individual medical institutions, which mainly fulfill the relevant responsibilities set forth by law.
PV in China comprises two main modules. The NMPA is responsible for the supervision of policy implementation within regional drug administrations. The National Adverse Drug Reaction Monitoring Center, directly under the NMPA, is responsible for ADR monitoring and the collection and reporting of ADR information at a national level. Unlike WHO, the European Union, and the United States, China’s ADR database is not open to the public and external personnel have no access to its data.
Child patient
Basic physiological condition of children
In the context of medicine, children are defined those 0–14 years old. This group can be further divided into several subgroups, including neonates (0–28 days old), infants (0–3 years old), those in early childhood (1–7 years old), and juveniles (7–14 years old).17–21 For children, it is not sufficient to simply regard them as miniature adults [Figure 1(a)]. Drug pharmacokinetics (PK) and pharmacodynamics (PD) observed in children are different from those observed in adults.22,23 The underlying reason for this may be that compared to adults, children have higher ratio of water to lipids, lower total plasma protein, and lower intestinal activity of cytochrome P-450 1A1 (CYP1A1).3,24 In addition, children are more vulnerable to external factors as a result of immature organ function. For example, non-alcoholic fatty liver disease is present in about 10% of children 25 ; this impairment of liver function can complicate the safe use medication in children. Furthermore, children of different ages and weights can have varying PK and PD characteristics.24,26 Finally, the characteristics, course, and etiology of diseases in pediatric patients may also be different from those in adults. 22 These multiple factors all contribute to differences in the efficacy and safety of drugs seen in pediatric versus adult patients.

(a) Physiological characteristics of children, (b) Risk factors of ADRs in children, (c) Acts relevant to the safe use of drugs in children and (d) Common drugs causing ADRs in children.
Use of medication in the child patient
Currently, the issues surrounding drug use in children primarily revolve around challenging clinical trials, limited drug options specifically formulated for children, inadequate or unclear instructions on children’s medication usage in the package inserts, and insufficient production of specialized dosages and specifications tailored for children. A prospective cohort study showed up to 96.4% of newborns were exposed to off-label drugs. 27 The primary classifications associated with the off-label drug use include unapproved age group usage, off-label indications, and unapproved dose administration. 28 Additionally, for the treatment of pediatric patients, both the drug dosing strategy and the course of treatment must be thoroughly studied. The lack of dosages and specifications for children frequently necessitates fragmentation of tablets in clinical settings. This practice can disrupt the integrity of the medication’s dosage structure and lead to imprecise dosing. In clinical trials with children, determining the appropriate dosage is extremely important to ensure the safety and efficacy of drugs. Dosage for children is usually calculated based on body weight, body surface area, and clearance rate. 29 Previously utilized dose calculation rules include: Young’s age rule, Clark’s weight rule, Clark’s surface area rule, and Shirkey’s dosing recommendations.24,30 However, most of these rules are not considered accurate enough for dose calculation in children. A possible reason is that these rules were designed for use with small molecule drugs and did not universally apply well to new drugs. The allometric method and Salisbury Rule are able to predict the appropriate first dose for therapeutic proteins (i.e. monoclonal and polyclonal antibodies and non-antibody proteins) in clinical trials with children.22,31,32 However, it is clear that a single-drug administration strategy cannot be applied across all childhood age subgroups. Optimal dosage calculation rules may vary for different ages, body weights, types of drugs, and treatment courses.
Implementation of a PV system for children
Many lessons have been learned in the course of development of a pediatric PV strategy. In October and November of 1901, 22 children died from injection of diphtheria antitoxin contaminated with tetanus bacilli. 33 Subsequently, the Biological Agents Control Act was introduced on 1 July 1902. This act specifies that enterprises that want to produce and sell vaccines and antitoxins must obtain a license. In 1938, 107 children died from administration of oral sulfanilamide formulated with diethylene glycol. 34 Following this tragedy, the United States government enacted the Food, Drug, and Cosmetic Act. With the continuous development of the field of medicine, a large number of drugs intended for use in children are marketed every year. During January 1996 through December 2019, the European Union approved a total of 405 medicinal products and 322 active substances for use in pediatric populations. 9 Ensuring the quality, safety, and effectiveness of all drugs indicated for use in children after marketing is a major challenge that requires serious consideration by all parties involved in drug development, clinical use, and marketing.
In pediatric clinical drug trials, the mechanisms for assessing potential risks are inadequate, the criteria are unclear, and high-quality evidence is lacking. Therefore, there are more drug safety issues arising in pediatric patients than in adults [Figure 1(b)]. In order to inform evidence-based decision making with respect to the safety and long-term benefits of pharmacotherapy in children, many policies and regulations support the inclusion of children in relevant clinical studies [Figure 1(c)]. In 2001, the European Clinical Trials Directive allowed children to be included in clinical trials. 34 In 2002, the United States enacted the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act, implemented in 2003. 35 These bills promote drug manufacturers to conduct clinical drug trials in children. The EMA first published their pediatric PV guidance standard in 2006, which is now included in the Good PV guidance standard in the Population-Specific Considerations IV: Pediatric Population section. 36 The European Pediatric Regulation was officially implemented in 2007, and stipulates that except for certain exemptions, the marketing license applying to drugs must include research conducted in children. 37 Given that children do not have full consent rights, the FDA has established 21 Code of Federal Regulations 50, subpart D [2001/2013] to ensure the safety of children participating in clinical trials. 38 In addition, the FDA human subject protection regulations clearly stipulate that pediatric clinical trials can only be conducted when the benefits outweigh the risks, or there is sufficient evidence to prove that the risks are reasonable and provide the prospect of direct benefit to children. 35 Due to the paucity of evidence regarding drugs intended for use in children the many differences between children and adults, it is difficult to define this ‘direct benefit’. A workshop convened in 2019 by the FDA in collaboration with the Duke-Margolis Center for Health Policy produced expert advice on how to define this term. 39 The seminar mainly evaluated the prospect of direct benefit from five angles: biological plausibility, non-clinical data, clinical data, dosing justification, and trial duration. These efforts were made to ensure the maximum benefit of pediatric patients participating in clinical trials.35,39
With the vigorous development of PV systems and the cooperation and support of international agencies and institutions, more and more drug safety data are publicly obtainable [Figure 1(d)]. At present, the three major public ADR databases are EudraVigilance, FAERS, and VigiBase. Rasmussen et al. 40 found that 40% of children experienced drug-induced adverse reactions in cases related to immunoglobulin A (IgA) vasculitis in the French PV database and VigiBase. The drugs implicated included vaccines (measles, rubella, mumps, influenza, poliomyelitis, diphtheria, and tetanus), antibiotics, and immunomodulatory TNF-α blockers (e.g. adalimumab and infliximab). It is critical to analyze database cases like these to determine the mechanisms underlying ADRs and to take appropriate action in withdrawing the drug or providing symptomatic treatment. In addition, Haarman et al. 41 analyzed relevant data in the Netherlands PV Center Lareb and VigiBase and found that neuropsychiatric symptoms (e.g. nightmares, aggression, and depression) and headaches were related to the use of montelukast in children with asthma. Using these databases for risk factor analysis can help identify potential adverse reactions that go undetected in clinical trials due to small pediatric sample sizes. Aside from analyzing data from the database, the researchers also conducted prospective pharmacoepidemiology studies using patients from children’s hospitals. Yori et al. 42 analyzed 111 pediatric patients receiving intravenous immunoglobulin G and found that the incidence of adverse reactions was relatively low and could be treated effectively. A Canadian Pediatric Surveillance Program (CPSP) Study found that if a pediatric patient’s parents have acanthosis nigricans or type 2 diabetes (T2D), blood glucose should be closely monitored when using diabetogenic medication. 43 Another prospective observational study found that approximately 17.7% of treated pediatric patients experienced at least one ADR, and the incidence of adverse reactions in children who had received general anesthesia was more than six times that of non-anesthetized children. 44 It is important to recognize that any neglected adverse reaction may result in long-term consequences that can affect the whole life of a pediatric patient. Therefore, whether in the process of treatment, research, or exploration, HCPs and researchers should approach drug-related issues with a meticulous and rigorous attitude.
Despite many difficulties, there are many professionals working toward improving PV in pediatric populations. The conect4children expert group white paper introduced various considerations regarding pediatric PV, including protocol development, risk management plans, Pediatric Investigation Plans, and collection and analysis of safety data. 34 The EMA also requires a benefit-risk assessment if clinical trial research is to include children. 45 Good PV in pediatrics can help optimize treatment, reduce harm, help prescribing physicians understand the potential long-term effects of drugs, and assist physicians in designing optimal treatment strategies. We believe that over time, the management of drug safety and efficacy in pediatric patients will improve greatly.
Pregnant and lactating patients
Basic physiological condition of pregnant women
The processes of maternal absorption, distribution, metabolism, and excretion in different stages of pregnancy experience various changes [Figure 2(a)]. Therefore, in order to ensure the safety and efficacy of drugs intended for use during pregnancy, it is necessary to adjust the dose and frequency of administration according to these physiological changes. Early pregnancy is prone to physiological reactions that often result in nausea and vomiting, which can in turn cause the reduced absorption and bioavailability of orally administered drugs. In addition, gastric acidity decreases and gastric emptying and peristalsis slowdown, which can also lead to changes in drug absorption. 46 Drug distribution can also vary with the increase in plasma volume and the associated relative decrease in plasma albumin. 47 Changes in hepatic enzyme activity, renal blood flow, and glomerular filtration rate have also been shown to affect drug metabolism and excretion. 46 Besides physiological pregnancy changes, the chance of developing maternal anxiety and antenatal or postpartum depression increases. 48 Furthermore, despite the placental barrier between the mother and fetus, most drugs can still reach the fetus, potentially resulting in fetal drug exposure, and subsequent adverse outcomes. During breastfeeding, drugs can reach the newborn through the milk. However, the extent of the risk to the fetus and newborn for most drugs is still unclear.
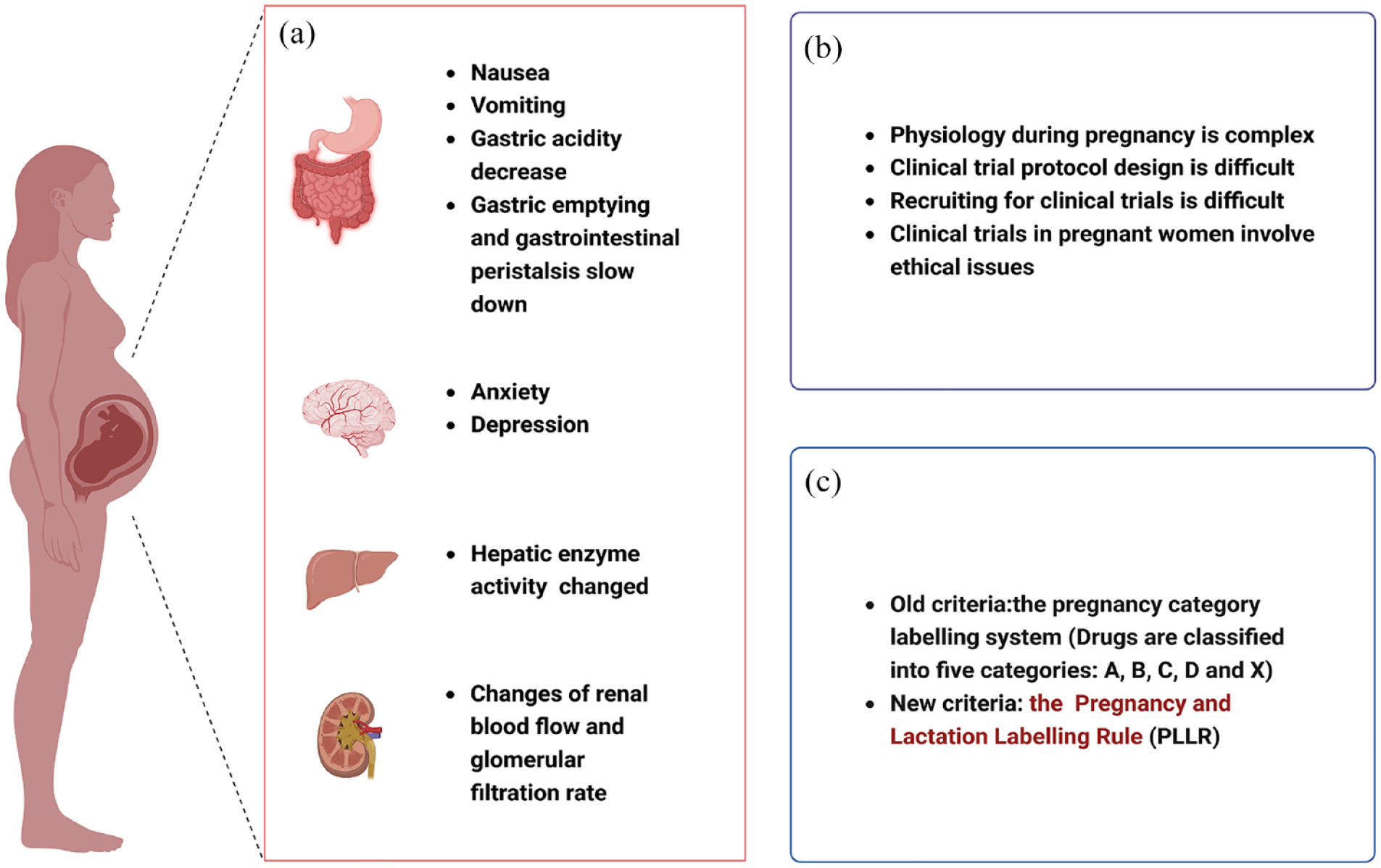
(a) Physiological characteristics of pregnant women, (b) Risk factors of ADRs in pregnant women and (c) Evaluation criteria for drug toxicity during pregnancy and lactation.
Medication during pregnancy and lactation
Over 90% of pregnant women will use at least one prescription or over-the-counter medication, with 80% being in the first trimester. Nonetheless, information on PK and drug safety and efficacy in pregnancy is extremely lacking.49,50 There is also little available clinical safety information to guide the rational use of drugs in pregnant and lactating women [Figure 2(b)]. Approximately 97.7% of approved drugs do not have safety information relevant to use during pregnancy and lactation. 51 During the COVID-19 pandemic, the majority of clinical trials to treat this disease excluded pregnant women, and both pregnant and lactating women were excluded from COVID-19 vaccine studies.52,53 Population pharmacokinetics (popPKs) modeling and physiologically based pharmacokinetic (PBPK) modeling can be used to predict PK during pregnancy and lactation, providing reference for the formulation of drug dosage and frequency in clinical trials. 53 It is worth noting that these pharmacometric tools, while promising, are not yet fully developed.
The WHO recommends exclusive breastfeeding for the first 6 months of fetal life and breastfeeding until the age of two if conditions permit.54,55 However, more than 50% of women take at least one medication in the postpartum period, and most of these medications have unclear effects with respect to infant health.56,57 As of now, discontinuation of breastfeeding is the safest practice in the event of uncertainty about the safety of a given drug. However, breastfeeding positively affects immune system development in early infancy, promoting subsequent healthy growth of the child. Inadequate information about medicines can lead to premature or inappropriate termination of breastfeeding. Adequate knowledge of drug safety among HCPs is critical to maintain the longest possible duration within the recommended infant age window of breastfeeding.
To better manage medication use in pregnant patients, studies on the safety and effectiveness of prophylactic and therapeutic drugs indicated for use in this population are increasing. According to data published by WHO, 10% of pregnant women and 13% of postpartum women will experience mental disorders. 58 A cross-sectional study in 12 European countries found that approximately 4.3–7.6% of pregnant and postpartum women had moderate to severe depressive symptoms. 59 Another study showed that women over 40 years of age and living in impoverished areas were more likely to use antidepressants. 60 Anxiety and depression in pregnancy are strongly associated with adverse pregnancy outcomes (e.g. miscarriage, preterm birth) and impaired infant development.61–63 Aside from depression and anxiety, primary headache is also common during pregnancy and the postpartum period. One retrospective study found that drugs used to treat primary headache such as antipsychotics, antiepileptics, tricyclic antidepressants, benzodiazepines, β-blockers, acetaminophen, indomethacin, and oral or intravenous magnesium may be associated with adverse fetal reactions. 64 In response to the urgent need to address the treatment of disease in pregnant patients, relevant studies are also underway to provide data for the safe and effective use of certain drugs during pregnancy. Preeclampsia is one of the leading causes of maternal death, and one clinical study (ClinicalTrials.gov NCT01717586) found that prophylactic use of pravastatin was associated with better pregnancy outcomes in women at high risk. 65 Gestational diabetes causes 5−13% of complications seen during pregnancy. 66 Metformin is commonly used in the treatment of this condition, but the clearance rate of metformin during pregnancy is high; therefore, to achieve the ideal effective concentration, the dose of must be increased. 66 However, Faure et al. 67 showed that metformin exposure in utero may reduce the fertility of male offspring in adulthood. Indomethacin is used to treat preterm labor, but studies have found that as pregnancy progresses, maternal drug exposure decreases while fetal exposure increases. 68 Therefore, the dose of indomethacin should be adjusted as pregnancy progresses to ensure the efficacy of the drug and reduce unnecessary fetal exposure. Tacrolimus, a commonly used immunosuppressive drug in kidney transplant patients, is almost undetectable in breast milk 3 weeks after delivery; as such, it is likely safe to use during lactation.69,70
Maternal physiology during pregnancy is complex, clinical trial protocol design is difficult, and trials involving pregnant women carry many ethical issues. Only 6% of clinical trials registered between 2007 and 2012 included pregnant women, with only 11% of these reporting outcomes. 71 Due to the lack of clinical trials during pregnancy and lactation, most marketed drugs have little available safety and efficacy information with respect to use in these patients. Furthermore, drug effects on human embryos are not fully clear. For these reasons, more attention must be paid to the monitoring and prevention of ADRs during conception and pregnancy. However, it is far from adequate to rely solely on clinical trials and spontaneous reporting adverse drug effects to fully evaluate the safety of drugs indicated for use in pregnant patients. Proper treatment of disease during pregnancy is essential for the health of the mother and fetus; however, medication safety in pregnant patients also impacts both mother and fetus, so the risk assessment at all points of the life cycle of a drug is essential.
Implementation of a PV system for pregnant patients
In addition to considering the therapeutic effect of drugs on the mother, the effect of drugs on the growth and development of the fetus also must be considered [Figure 2(c)]. Inappropriate medication regimens can cause serious and even life-long adverse consequences to both mother and fetus. Diethylstilbestrol was used to treat pregnancy complications from the 1940s to the 1960s. It was subsequently found that women with in utero exposure to diethylstilbestrol had a significantly increased risk of infertility, adverse pregnancy outcomes (e.g. spontaneous abortion, preterm delivery, loss of second-trimester pregnancy, ectopic pregnancy, and stillbirth), vaginal and cervical clear cell adenocarcinoma, and breast cancer.72,73 In 1960, the thalidomide tragedy shocked the world, resulting in the birth of tens of thousands of infants with seal deformities.74,75 This event reminds of concerns regarding drug-induced diseases and represents an important inciting factor for the formation of PV systems.
In 1979, the FDA created the pregnancy category labeling system based on reproductive toxicity of drugs. 76 In this system, drugs are classified into five categories: A, B, C, D, and X. 76 Class A and B drugs have no toxicity to the fetus, Class C drugs have uncertain toxicity to the fetus, and Class D and X drugs have been proven to be toxic to the fetus. The first Teratogen Information Service in Brazil has been providing free teratogen information to HCPs and the general public since 1990. This information is especially important for pregnant women and women planning to become pregnant. 77 This organization participated in the discovery of two teratogens, misoprostol and Zika virus. 77 In 1993, the National Institutes of Health recommended that pregnant women be included in clinical studies to obtain more clinical and PK data. 78 In 1994, the FDA established the Office of Women’s Health (OWH) in order to better protect the safety of pregnant and lactating patients with respect to certain drugs and to promote related studies. 79 With advances in medical care and increasingly individualized treatment, the FDA also implemented a new Pregnancy and Lactation Labeling Rule in 2015, which replaced the letter-based categories. 80 The new guidelines require data from both human and animal studies, including information on the risks of drug use during pregnancy and lactation and the effects of drugs on reproductive function. 81 In addition, the United States Congress established the Task Force on Research Specific to Pregnant Women and Lactating Women (PRGLAC) in 2017. This organization aims is to investigate the safety and effectiveness of drugs in pregnant and lactating women and provide guidance to the Secretary of Health and Human Services (HHS). 82 Additionally, the EMA highlights PV with respect to drugs used in pregnant and lactating women in their Guidelines on Good PV Practices (GVP), Chapter P.III. 4 In addition to using EudraVigilance, FAERS, and VigiBase for large-scale data analysis, there are many other channels for obtaining relevant drug safety information. For example, Klein et al. 83 collected the data regarding the use of ß-blockers during pregnancy via Twitter and analyzed associated pregnancy outcomes as supplementary data to evaluate the safety of these drugs.
Information about the safety of drugs during pregnancy and lactation is complex and often inconsistent, making the determination of drug safety difficult for many HCPs. Currently, no separate PV system for pregnant patients has been established in any country. However, in view of the characteristics of the use of drugs in this population and the particularity of drug safety during pregnancy, it is necessary to establish pregnancy PV systems. Governments should issue relevant laws and regulations to encourage and guide the establishment of these systems and promote public platforms for the release of drug safety information so medical institutions, pharmaceutical companies, patients, and others can report and understand the necessary drug information.
Elderly patients
Basic physiology of the elderly
Aging is an increasing trend in population change globally. In 2018, the number of people over 65 years old exceeded the number of people under 5 years old for the first time. 84 It is estimated that the proportion of people aged greater than 60 years will reach 20% by 2050. 85 There are differences in the definition of ‘elderly’ as set forth by various guidelines. For example, JNC 8 defines the elderly as the population older than 60 years, but China defines this group as those older than 65 years. 86 Age and the influence of the surrounding environment combine to affect changes in gene expression, resulting in cellular and metabolic dysfunction. 87 Compared to that of the young, liver volume in elderly adults is reduced by 20–40%, hepatocytes are more susceptible to stress, and the risk of chronic liver disease is greater. 88 With increasing age, the physiological structure of the kidney also changes with the number of functional glomeruli decreasing, leading to the decline of renal function. 89 Once an elderly person develops chronic kidney disease, the probability of acute kidney injury increases. 89 Medication compliance can also be problematic in elderly patients due to hypomnesis and polypharmacy. Most elderly patients with multiple drug prescriptions have been shown to have liver and kidney dysfunction. 90 For elderly patients with multiple diseases and polypharmacy, it is often difficult to determine whether ADRs are caused by a single drug, multiple drugs, or disease itself. Therefore, there are many challenges to the safe, correct, and proper use of drugs in elderly patients. The basic physiological characteristics of the elderly are summarized in Figure 3(a).

(a) Risk factors of ADRs in elderly, (b) Common drugs causing ADRs in elderly and (c) Common tools for identifying PIMs.
Use of medication in elderly patients
Among elderly hospitalized patients, approximately 16–25% will experience at least one ADR.91,92 Drug-related factors are the most important predictors of ADR [Figure 3(b)]. Common drugs leading to ADRs in elderly patients include diuretics, non-steroidal anti-inflammatory drugs (NSAIDs), antibiotics, anticoagulants, benzodiazepines, antithrombotic drugs, analgesics, and digoxin.92–95 Multiple comorbidities present in elderly patients often lead to the use of multiple drugs. As alluded to above, polypharmacy is one of the major risk factors leading to ADRs. Polypharmacy is generally defined as taking five or more drugs per day. 96 An Australian study showed that polypharmacy was present in 36% of people over 70 years old and 44–46% of patients between 80 and 89 years old. 97 In China, the proportion of polypharmacy in elderly patients is 48% and up to 73% in hospitalized elderly patients. 98 Notably, polypharmacy was associated with hospitalization due to any cause, with the likelihood of hospitalization associated with five to nine medications being 34% and with 10 or more medications being up to 98%. 99
Due to the complex physiology of the elderly, ADRs are an important cause of high morbidity, mortality, and hospitalization rates. A United States study found that among the cases of death due to ADRs in 1999–2006, patients older than 75 years had the highest risk of death. 100 Furthermore, the rate of fatal ADRs reported in elderly patients is three times that of younger patients. 101 The proportion of elderly patients hospitalized due to ADRs is 6–35%.93,94,102
Implementation of PV systems for elderly patients
Polypharmacy is common in elderly patients, and drug–drug interaction is one of the important causes of ADRs. Most ADRs in elderly patients are caused by common prescription drugs. 103 One study showed that 53.97% of ADRs caused by drug interactions could be prevented. 104 Another study showed that greater than 75% of hospitalizations of elderly patients due to ADRs could be avoided. 94 The study of the predictors of adverse reactions in elderly patients is key to formulating preventive strategies. Cabré et al. 94 analyzed 3292 elderly inpatients and found that the risk factors leading to ADRs included personal factors (e.g. female sex, renal insufficiency) and external factors (e.g. the use of inappropriate medications, multiple medications, or sedatives). The implementation of PV can help to detect risks associated with a given drug, can assist HCPs in selecting drugs with the best therapeutic effect and the smallest chance of ADR, and can help to reduce economic burden.
The complexity of medication use in elderly patients increases with the incidence of potentially inappropriate medications (PIMs) (the potential risks of drugs exceed the potential benefits, and there are safer alternatives). PIMs are closely related to falls, ADRs/ADEs, higher treatment costs, and decline of bodily function in elderly patients. 105 Research shows that PIMs can increase the all-cause hospitalization rate of elderly patients by 27%. 99 Common tools for identifying PIMs include the Medication Appropriateness Index (MAI), HEDIS DAE, Beer’s criteria, STOPP/START criteria and EU-7-PIM list [Figure3(c)].106–109 Wang et al. 110 used Beer’s criteria, STOPP/START criteria, and the EU-7-PIM list to evaluate the use of drugs in 560 elderly inpatients and found that Beer’s criteria best predicted avoidable ADRs. However, for the elderly in Africa (Nigeria and South Africa), PIMs identified using Beer’s criteria did not correlate to ADRs in hospitalized patients. 111 The adjusted Beer’s criteria developed by a consensus of local experts is more applicable to the healthcare environment in Nigeria and South Africa, and may be used as a guide for the prescription of drugs to the elderly in these regions. 112 In Europe, STOPP/START criteria are used to identify PIMs in elderly patients. 113 In a multi-center prospective study in Spain, the STOPP/START criteria were used to evaluate PIMs, and the most common drugs identified were proton pump inhibitors and benzodiazepines. 114 Additionally, Chen and Zhang. 115 used these criteria to evaluate PIMs in elderly outpatients and found that benzodiazepines and hypnotic Z-drugs (zolpidem) were the main drugs identified. In central Portugal, the EU-7-PIM list was used to assess the use of drugs in elderly patients, with 83.7% of these patients found to be at potential risk of inappropriate drug use. 116 There are differences in pharmacotherapy in countries in different levels of development. For example, Yadesa et al. 107 applied the Prediction of ADR in Old Patients (PADROI) model as a risk assessment tool for elderly inpatients (⩾60 years old) in low-income countries.
The resistance of elderly cancer patients is decreased, and the possibility of comorbidity, polypharmacy and PIMs is increased. 117 Because of specific characteristics of cancer treatment, drugs considered as ‘possibly inappropriate’ in the general elderly population may be necessary for the treatment of elderly cancer patients. Therefore, the suitability of the above PIMs assessment tools for elderly cancer patients still needs to be evaluated. As of now, there is no definitive tool for the evaluation of PIMs in this population. Miller et al. 117 suggest that combining Beer’s criteria with MAI may be an effective method to identify PIMs in elderly cancer patients. Alternatively, Whitman et al. 109 suggest that three assessment tools, STOPP/START, the Beer’s criteria, and the MAI be used simultaneously to identify PIMs in this population.
Using real-world data of PV systems for analysis is conducive to identifying drug safety risks and taking countermeasures in a timely fashion. Generally, older patients are more likely to experience ADRs than younger patients. Age is one of the main risk factors for hypertension, and orthostatic hypotension is more likely to be induced by antihypertensive therapy in elderly patients. 86 Salem et al. 118 found that adverse cardiovascular drug reactions caused by ibrutinib mainly occurred in male patients older than 70 years, as identified by analysis of data in VigiBase. Mikami et al. 119 analyzed the FAERS database and found that elderly patients may be more prone to fatal neurologic adverse events when using immune checkpoint inhibitors. However, the incidence of ADRs in elderly patients is not always higher than that in non-elderly adults. Endrikat et al. 120 found that the risk of hypersensitivity reactions in patients older than 65 years after iopromide administration was lower than that in non-elderly adults as identified by analysis of the data in four observational studies and the PV database. Gouverneur et al. 121 found no more or more severe ADRs with the use of drugs targeting metastatic colorectal cancer (mCRC) in elderly patients in their analysis of ICSRs in VigiBase.
Gomes et al. 102 used the Portuguese PV system to identify PIMs and found that cardiovascular and nervous system drugs are common culprits. As such, supervision should be increased when using these drugs. Montastruc et al. 95 used the data in the French Midi-Pyrénées PV Center to analyze the PIMs in patients older than 75 years and found that the risks of benzodiazepines, imipramine antidepressants, and atropine-related drugs were the highest. Dubrall et al. 101 analyzed the ADR database of the German Federal Institute for Drugs and Medical Devices (BfArM) and found that anticoagulants were the most common drugs implicated in ADRs observed in the elderly. In addition to using databases, the analysis of articles published on the Internet is also a good way to identify high-risk drugs in elderly patients. Motter et al. 122 systematically evaluated the articles in PubMed, AgeLine, Academic Search, Academic Search Premier, and CINAHL from January 1991 to April 2017 and found that benzodiazepines and NSAIDs were the most reported PIMs in elderly patients. Aguiar et al. 123 conducted a meta-analysis of articles published on PubMed, MEDLINE, and Google Scholar from 1991 to September 2017 and found that the top three PIMs were tricyclic antidepressants, centrally acting antiadrenergic agents, and NSAIDs.
The WHO and relevant member states jointly formulated the Decade of Health Aging 2020–2030 program with the goal of promoting the maintenance of normal function and obtaining happiness in the elderly. 124 However, there are still many difficulties facing drug management methods, the formulation of relevant policies, and the research and development of helpful tools for elderly patients. In the face of an aging society, it is necessary for people from all walks of life to formulate strategies together in order to achieve the purpose of fine-tuned management of the use of drugs in elderly patients.
Problems and suggestions
Due to their specific physiology, special populations are more prone to ADRs. The implementation of PV can improve the speed and increase the quantity and quality of ADR reporting. Pre-marketing clinical research is more conducive to identifying common ADRs, while post-marketing monitoring is indispensable for finding rare or long-term ADRs. To fully understand the safety of a drug requires the study of large numbers of patients and long-term monitoring data. PV is a useful tool for evaluation of drugs. Real-world data based on PV can aid in discovering potential drug risks and bridging gaps in drug safety information. The most up-to-date data will provide HCPs more insight into drug risks and aid in the close monitoring of patients, with the ultimate goal of reducing patient harm and societal financial burden.
The following problems still exist with respect to the implementation of PV:
Most medical staff have not received PV training, the training provided by professional courses and talents related to PV is not perfect, and the number of PV professionals in practice is insufficient.
MAHs are ultimately responsible for drug safety. However, MAHs are faced with the challenge of ensuring drug quality without increasing costs. Moreover, with the increase of users, the workload of PV is also increasing.
The sufficient quantity and quality of data is the basis of relevant research and necessary conditions for establishing and improving PV systems.
Public databases for specific populations are deficient in providing important information pertaining to drug combinations, comorbidities, and laboratory indicators for relevant tests. As a result, establishing a causal relationship between drugs and adverse events can prove to be challenging.
Suggestions on how to better develop PV in special populations are as follows:
Focus on drug safety risk monitoring for special populations, set up corresponding system segments based on these populations, and establish and improve management norms and institutional systems. Different special populations should be equipped with corresponding pharmacists to review prescriptions with the goal of effectively intercepting inappropriate prescriptions and achieving early detection and resolution of any issues.
Strengthen cooperation between countries with the help of information systems to create an efficient global pattern of PV cooperation.
In addition to mining drug safety data, PV professionals should also provide timely information to patients, the public, and HCPs to promote the safe and effective use of drugs.
To improve the accuracy and analyzability of reported medical data, it is highly recommended that comprehensively report complete medical records, excluding any identifying patient information. As an incentive to increase the enthusiasm for reporting, a certain reward will be offered to individuals who submit complete medical records. This will also help address the ongoing issue of missing or incomplete reports, ultimately improving the overall quality of medical research.
By establishing a collaborative drug safety legislation, drug manufacturers are compelled to proactively shoulder accountability, and companies that show exemplary PV practices for particular demographics are incentivized to extend drug patent protection duration. This approach fosters greater responsibility and diligence in the pharmaceutical industry, while also prioritizing the well-being of patients.
Conclusion
Pregnant women and children are considered ‘treatment orphans’, and the deterioration of physiological functions in elderly patients leads to an increase in drug-related risks. Therefore, more evidence and careful consideration are needed to ensure safe and effective use of drugs in these special populations. In the face of contradictory evidence, doctors must rely on personal experience and more unified treatment standards. PV can monitor the whole life cycle of a drug and provide reference for HCPs to use drugs safely in special populations. Complete safety data are a prerequisite for risk minimization, and a scientific and comprehensive assessment of these data is the basis for the development of appropriate management strategies. However, there are differences in PV systems, disease diagnostic criteria, and treatment protocols in different countries, leading to the challenge of multi-country safety data analysis. To establish a global PV system for special populations and develop unified standards for diagnosis, treatment and risk management require the participation of global academic, medical, pharmaceutical, and information experts.