
Abstract
Introduction:
Tuberculosis is a major respiratory disease globally with a higher prevalence in Asian and African countries than rest of the world. With a larger population of tuberculosis patients anticipated to be co-infected with COVID-19 infection, an ongoing pandemic, identifying, preventing and managing drug–drug interactions is inevitable for maximizing patient benefits for the current repurposed COVID-19 and antitubercular drugs.
Methods:
We assessed the potential drug–drug interactions between repurposed COVID-19 drugs and antitubercular drugs using the drug interaction checker of IBM Micromedex®. Extensive computational studies were performed at a molecular level to validate and understand the drug–drug interactions found from the Micromedex drug interaction checker database at a molecular level. The integrated knowledge derived from Micromedex and computational data was collated and curated for predicting potential drug–drug interactions between repurposed COVID-19 and antitubercular drugs.
Results:
A total of 91 potential drug–drug interactions along with their severity and level of documentation were identified from Micromedex between repurposed COVID-19 drugs and antitubercular drugs. We identified 47 pharmacodynamic, 42 pharmacokinetic and 2 unknown DDIs. The majority of our molecular modelling results were in line with drug–drug interaction data obtained from the drug information software. QT prolongation was identified as the most common type of pharmacodynamic drug–drug interaction, whereas drug–drug interactions associated with cytochrome P450 3A4 (CYP3A4) and P-glycoprotein (P-gp) inhibition and induction were identified as the frequent pharmacokinetic drug–drug interactions. The results suggest antitubercular drugs, particularly rifampin and second-line agents, warrant high alert and monitoring while prescribing with the repurposed COVID-19 drugs.
Conclusion:
Predicting these potential drug–drug interactions, particularly related to CYP3A4, P-gp and the human Ether-à-go-go-Related Gene proteins, could be used in clinical settings for screening and management of drug–drug interactions for delivering safer chemotherapeutic tuberculosis and COVID-19 care. The current study provides an initial propulsion for further well-designed pharmacokinetic-pharmacodynamic-based drug–drug interaction studies.
Plain Language Summary
Introduction:
Tuberculosis is a major respiratory disease globally with a higher prevalence in Asian and African countries than rest of the world. With a larger population of tuberculosis patients predicted to be infected with COVID-19 during this period, there is a higher risk for the occurrence of medication interactions between the medicines used for COVID-19 and tuberculosis. Hence, identifying and managing these interactions is vital to ensure the safety of patients undergoing COVID-19 and tuberculosis treatment simultaneously.
Methods:
We studied the major medication interactions that could likely happen between the various medicines that are currently given for COVID-19 and tuberculosis treatment using the medication interaction checker of a drug information software (Micromedex®). In addition, thorough molecular modelling was done to confirm and understand the interactions found from the medication interaction checker database using specific docking software. Molecular docking is a method that predicts the preferred orientation of one medicine molecule to a second molecule, when bound to each other to form a stable complex. Knowledge of the preferred orientation may be used to determine the strength of association or binding affinity between two medicines using scoring functions to determine the extent of the interactions between medicines. The combined knowledge from Micromedex and molecular modelling data was used to properly predict the potential medicine interactions between currently used COVID-19 and antitubercular medicines.
Results:
We found a total of 91 medication interactions from Micromedex. Majority of our molecular modelling findings matched with the interaction information obtained from the drug information software. QT prolongation, an abnormal heartbeat, was identified as one of the most common interactions. Our findings suggest that antitubercular medicines, mainly rifampin and second-line agents, suggest high alert and scrutiny while prescribing with the repurposed COVID-19 medicines.
Conclusion:
Our current study highlights the need for further well-designed studies confirming the current information for recommending safe prescribing in patients with both infections.
Keywords
Introduction
Tuberculosis (TB) is a major respiratory disease occurring globally, particularly in the WHO (World Health Organization) regions of South-East Asia (44%), Africa (25%) and Western Pacific (18%).1,2 The countries with high incidences of TB include India (26%), Indonesia (8.5%), China (8.4%), Philippines (6.0%), Pakistan (5.7%), Nigeria (4.4%), Bangladesh (3.6%) and South Africa (3.6%), which account for two-thirds of the total global TB incidences in 2019. 2 The global impact of Coronavirus disease-19 (COVID-19) on TB services has shown a reduction in the detection and diagnosis of TB worldwide. Moreover, there is a patient delay before the first presentation to TB care and decreased patient treatment initiation rates compared with pre-pandemic levels. These could lead to increased deaths due to TB in settings with a high TB burden. 3 A modelling analysis developed by Stop TB Partnership in collaboration with Imperial College, Avenir Health, Johns Hopkins University and United States Agency for International Development (USAID) has projected that the COVID-19 pandemic could increase by 6.3 million TB cases and 1.4 million TB deaths globally between 2020 and 2025. 4 COVID-19 pandemic is projected to cause severe adverse impacts on poverty levels in developing countries. As poverty is a significant risk factor for the development of TB, there is an expected anticipation for a higher incidence of TB during this COVID-19 pandemic era.5–7 Currently, there is a paucity of data on the prevalence of TB among COVID-19 patients. TB infection has been found to increase the susceptibility, severity and mortality related to COVID-19.8,9 COVID-19 could occur before, simultaneously, or after the diagnosis of TB. A study with 49 COVID-19 patients from 26 centres of 8 countries reported that 26 (53.0%) had TB before COVID-19, 14 (28.5%) had COVID-19 first and 9 (18.3%) had both diseases diagnosed within the same week (4 on the same day). 10
Drug–drug interactions (DDIs) represent an austere and ubiquitous problem in patient safety, especially with geriatrics and patients on polypharmacy. Indeed, DDIs are culpable for a significant proportion of adverse drug reactions, hospital admissions, healthcare expenditures, morbidities and mortalities.11–15 Repurposed COVID-19 drugs have been reported to interact with drugs used in the management of other diseases.16,17 DDIs screening software has emerged to be an important tool for identifying and managing DDIs for clinicians, pharmacists and other healthcare providers. Micromedex is a widely used software in clinical practice for the identification of DDIs. Micromedex provides information regarding severity (contraindicated, major, moderate, minor and unknown), level of evidence (excellent, good, fair, unknown), clinical consequence, underlying mechanism, clinical management and onset of the adverse outcome (either rapid or delayed) of the DDIs.18,19
DDIs are identified by various approaches such as in silico, in vitro and in vivo experiments. In vitro and in vivo methods are laborious and expensive. However, in silico modelling approaches have become robust means for examining hypotheses and understanding mechanisms related to DDIs in health research. The optimality and cost-effectiveness give an extra edge to in silico modelling.20,21 Hence, combining the data derived from DDI checker software and in silico computational methods are expected to provide a more comprehensive picture of the prediction of potential DDIs associated with antitubercular drugs and repurposed COVID-19 drugs.
TB treatment regimens are characterized by multidrug combinations and a longer duration of use. 22 Several drugs have been reported as substrates, inhibitors and inducers for CYP3A4 and P-gp.23,24 CYP3A4 and P-gp are reckoned to be key molecular targets related to DDIs occurring with TB treatment.25,26 Hence, there is an increased potential for CYP3A4 and P-gp-based DDIs between antitubercular drugs and repurposed COVID-19 drugs. QT prolongation is also a key feature of many second-line antitubercular drugs and repurposed COVID-19 drugs.27–30 The hERG is a gene that encodes for the pore-forming subunit of a delayed rectifier voltage gated K + (VGK) channel, which is variously referred to as Kv11.1, IKr or as hERG. This ion channel plays an important role in the repolarization phase of the cardiac action potential by mediating the rapid component of cardiac delayed rectifier K+ current.31,32 The most common mechanism of drug-induced QT prolongation is due to hERG inhibition.33,34 Several antitubercular agents and repurposed COVID-19 drugs have been reported to inhibit hERG and result in QT interval prolongation.35–38 Hence, there is an increased potential for DDIs due to QT prolongation. This could be expected when antitubercular drugs and repurposed COVID-19 drugs with a potential for inhibition of hERG are used concomitantly. Hence, predicting these potential DDIs related to induction or inhibition of CYP3A4, P-gp and hERG in TB patients co-infected with COVID-19 could help deliver safe chemotherapeutic TB and COVID-19 care for better clinical outcomes.
With a larger population of TB patients anticipated to be co-infected with COVID-19, it may be imperative to assess the potential DDIs occurring with the antitubercular drugs with the repurposed COVID-19 drugs. To our knowledge, no studies have reported a comprehensive drug information database and molecular docking integrated data on DDIs between antitubercular drugs and repurposed COVID-19 drugs till the date. In this scenario, we assessed the potential DDIs between the repurposed COVID-19 drugs and antitubercular drugs using the drug interaction checker of IBM Micromedex®. In addition, relevant literature was extracted from PubMed and Google Scholar for gathering information regarding the mechanism of DDIs found from the drug interaction checker of Micromedex. We excluded non-scientific commentaries and only English literature were included in the study. Extensive computational studies were performed to validate and understand the DDIs found from the Micromedex drug interaction checker database at a molecular level. The information derived from this integrated data is collated and curated for providing an easy and clear picture to clinicians, nurses and pharmacists for improved identification, detection and management of DDIs of antitubercular drugs with repurposed COVID-19 drugs.
Methodology
Step 1: identifying repurposed COVID-19 drugs and antitubercular drugs for DDI assessment
The repurposed COVID-19 drugs were identified from the medications list of American Society of Health-System Pharmacists (ASHP; https://www.ashp.org/-/media/8CA43C674C6D4335B6A19852843C4052.ashx). 39 We have included repurposed drugs used as antiviral, supportive and other agents for the management of COVID-19. The final consensus list of drugs included for the final search for DDIs in the Micromedex® software was decided by a multidisciplinary and multi-institutional team comprising infectious diseases/COVID-19-treating physician, TB physician specialist, clinical pharmacist, clinical researcher, basic scientist, medicinal chemists, and medical and pharmacy academicians. We excluded COVID-19 convalescent plasma therapy from our list. The repurposed COVID-19 drugs, favipiravir, umifenovir and etesevimab, were not found in the search list of drug interaction checker of Micromedex. The potential DDIs of the repurposed COVID-19 drugs were assessed with the following antitubercular drugs: isoniazid, rifampin, pyrazinamide, ethambutol, streptomycin, amikacin, kanamycin, capreomycin, clarithromycin, levofloxacin, moxifloxacin, ofloxacin, cycloserine, amoxicillin-clavulanate, ethionamide, imipenem-cilastatin, linezolid, clofazimine, delamanid, bedaquiline and pretomanid.
Step 2: searching for DDIs in Micromedex® software
We searched to identify potential DDIs between antitubercular drugs and repurposed COVID-19 drugs by inputting all the generic names of the drugs into the drug interaction checker of IBM Micromedex®. The severity of the potential DDIs from Micromedex was classified into five groups: contraindicated, major, moderate, minor and none. All potential DDIs were collected from the Micromedex® till 24 March 2021.
Step 3: induced-fit docking (IFD) and binding free energy calculation
The protein crystal structure was downloaded from the Protein Data Bank (PDB). 40 In the crystal structures of human CYP3A4 (PDB ID: 6MA7), the active site cavities are much larger. They are located near haem iron, allowing these enzymes to accommodate substrates or inhibitors of diverse size. 41 Since the human protein structure is not available for P-gp protein, the amino acid protein sequence for human P-gp protein was obtained from the FASTA sequences from the Uniport database. For this, id P08183 and references protein (PDB ID: 3G60) in complex with the inhibitor was selected for generating a homology model. hERG protein crystal structure was downloaded from the well-established homology database of Schrodinger. 42 The crystal structure of the protein was optimized using the Protein Preparation Wizard module of Schrodinger 2021-1. Pre-processing of the protein was achieved by assigning bond order, hydrogen addition and disulphide treatment. All the water molecules beyond 5 Å from the hetero groups were removed. Hydrogen bonds were assigned, and the orientations of the remaining water molecules were rectified. Finally, the energy of the protein structure was optimized to root mean square deviation (RMSD) of 0.30 Å by using OPLS4 force field at pH 7.4. The co-crystallized ligand was retained using default parameters in the prepared protein and was further used for grid construction.
As there was no bound ligand in the case of hERG protein and P-gp protein, site map analysis was performed using the site map analysis module to identify top-ranked receptor binding sites of the enzymes. Site maps with a site score of more than 1.15 were taken for grid generation for docking study. 43 After extra precision docking protocol, compounds with better docking score and pose were selected for induced fit docking with extended sampling protocol (generates up to 80 poses using automatic docking protocol). This IFD output pose was taken further for the analyses of binding energy using MM-GBSA. The purpose of this MM-GBSA is to facilitate the calculation of binding free energy using ligand binding energies and ligand strain energies.
The binding energy is calculated according to the equation:
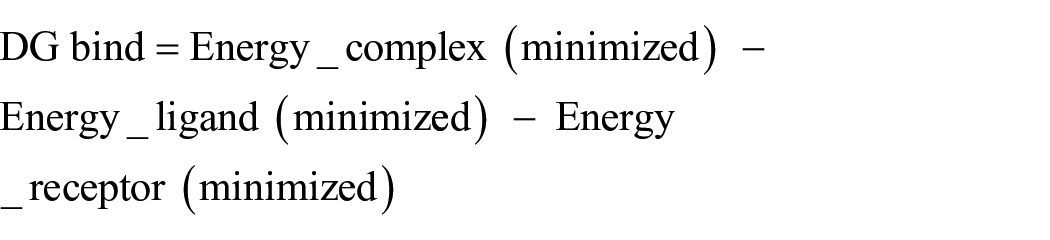
Energy properties are calculated for the complex (E_complex), the ligand (E_ligand) and the receptor (E_receptor), and strain energies can be calculated for the ligand and the receptor.
We propose that most of the interactions occurring between repurposed COVID-19 drugs and antitubercular drugs are due to synergistic inhibition of hERG protein (pharmacodynamic interaction resulting in QT prolongation) or due to inhibition of CYP3A4 and P-gp (pharmacokinetic interaction leading to alteration of drug levels). Information about each drug and its role in inhibiting CYP is taken from literature and extensive computational studies are performed to validate and understand the interactions at a molecular level. The computational studies also assessed the possibility of two drugs interacting together with hERG’s binding pocket.
Extensive induced-fit docking was performed for each drug on each of the target proteins viz. CYP3A4, hERG and P-gp. Binding free energy was calculated for each of the generated poses in IFD for each drug. A scoring method is then applied to calculate the net dG bind (N.S) where:
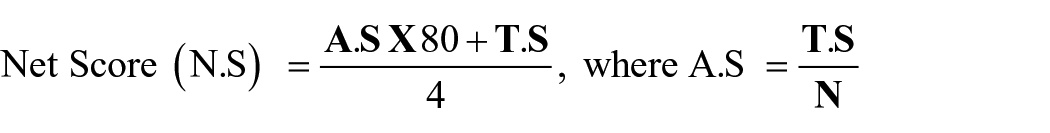
A.S = average score; T.S (total score) = sum of dG bind (from all poses) and N = total number of poses.
For simultaneous docking studies of two molecules inside the hERG binding pocket, a single molecule was initially docked and a sitemap was run to generate the binding pocket with the first molecule and the second molecule then docked on the generated site (Table 2). Again, this step was repeated with the second molecule from step one being docked first and the first molecule docked after that (on the generated site).
Step 4: categorizing the drugs based on QT prolongation drug risk assessment
For an individual QT prolongation drug risk assessment, we have categorized the QT-prolonging drugs into four categories as per the Arizona Center for Education and Research on Therapeutics (AZCERT) classification. 44 AZCERT now maintains the Web-based lists of drugs that have a risk of QT prolongation and Torsades de Pointes (TdP). The lists are now available on the CredibleMeds Website. 45 As per the website, the drugs were categorized into:
Known Risk of TdP – These drugs prolong the QT interval and are clearly associated with a known risk of TdP, even when taken as recommended.
Possible Risk of TdP – These drugs can cause QT prolongation but currently lack evidence for a risk of TdP when taken as recommended.
Conditional Risk of TdP – These drugs are associated with TdP but only under certain conditions of their use (e.g. excessive dose, in patients with conditions such as hypokalemia, or when taken with interacting drugs) OR by creating conditions that facilitate or induce TdP (e.g. by inhibiting the metabolism of a QT-prolonging drug or by causing an electrolyte disturbance that induces TdP).
Drugs to Avoid in Congenital Long QT Syndrome (cLQTS) – These drugs pose a high risk of TdP for patients with cLQTS. They include all those in the above three categories PLUS additional drugs that do not prolong the QT interval per se but have a special risk (SR) because of their other actions.
Results
A total of 91 potential DDIs along with their severity and the level of documentation were identified from Micromedex between repurposed COVID-19 drugs and antitubercular drugs, as shown in Figure 1. There were 47 pharmacodynamic DDIs, 42 pharmacokinetic DDIs and 2 unknown DDIs. We identified 4 contraindicated, 65 major, 21 moderate and 1 minor DDI based on the severity of interaction. Based on the level of documentation, there were 21 DDIs with excellent documentation, whereas 25 and 45 DDIs had good and fair level of documentation, respectively.

DDIs, along with the severity and level of documentation between repurposed COVID-19 drugs and with antitubercular drugs.
The net binding energy of individual repurposed COVID-19 drugs and antitubercular drugs with hERG, CYP3A4 and P-gp are shown in Table 1.
Net binding energy of individual repurposed COVID-19 drugs and antitubercular drugs with hERG, CYP3A4 and P-gp.

ND = Not determined.
Pharmacodynamic interactions
Among the 47 pharmacodynamic DDIs between the repurposed COVID-19 drugs and antitubercular drugs identified from Micromedex, 31 interactions resulted in QT prolongation effects. These interactions were integrated with the free binding energy of the repurposed COVID-19 drugs and antitubercular drugs and their AZCERT classification in Table 2.
Pharmacodynamic DDIs resulting in QT prolongation, identified from Micromedex with the free binding energy and combination binding energy to hERG protein along with their AZCERT classification.
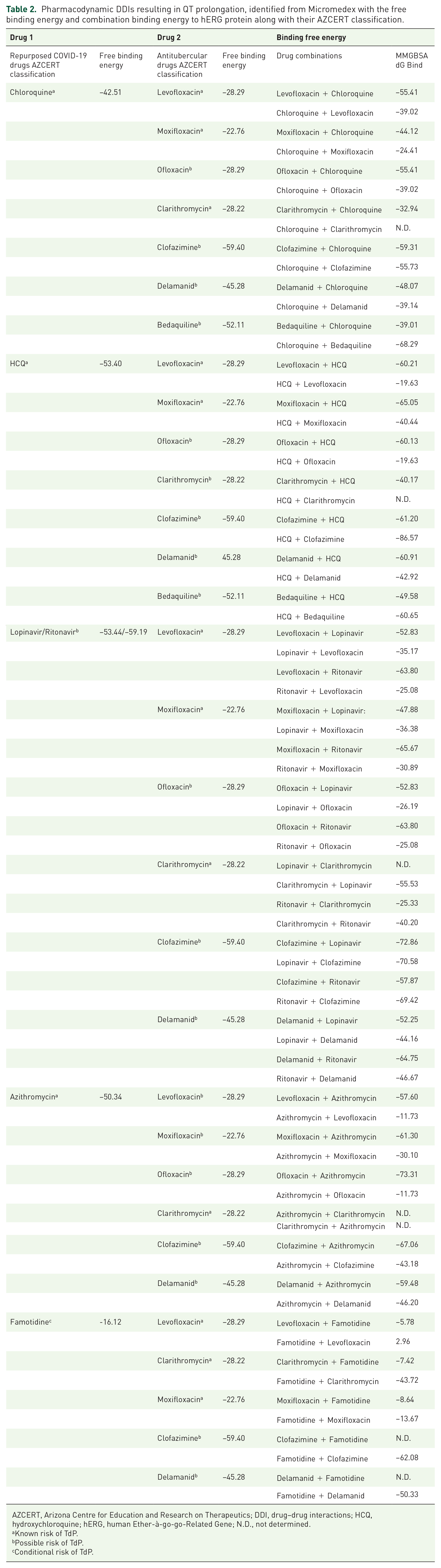
AZCERT, Arizona Centre for Education and Research on Therapeutics; DDI, drug–drug interactions; HCQ, hydroxychloroquine; hERG, human Ether-à-go-go-Related Gene; N.D., not determined.
Known risk of TdP.
Possible risk of TdP.
Conditional risk of TdP.
Chloroquine and hydroxychloroquine (HCQ) are known for their QT prolongation effect. The potency of these drugs towards inhibition of hERG was also established from the N.S of −1662.35 and −1803.59 for chloroquine and HCQ, respectively, by molecular modelling. However, ritonavir (N.S for hERG = −1298.24) and lopinavir (N.S for hERG = −1451.95) had a relatively lower binding with hERG (Table 1). The docking poses for chloroquine, HCQ, ritonavir and lopinavir in the hERG pocket are shown in Figure 2(a)–(d).

Docked pose of repurposed COVID-19 drugs: (a) chloroquine, (b) HCQ, (c) lopinavir and (d) ritonavir at hERG binding pocket.
It has been reported previously that few fluoroquinolones are usually either substrates or inhibitors for P-gp.46–48 The N.S against P-gp for levofloxacin, moxifloxacin and ofloxacin are −1653.33, −1470.86 and −1658.35, respectively (Table 1). When these fluoroquinolones are co-administered with chloroquine, HCQ, ritonavir and lopinavir, which have a higher affinity for P-gp (Table 1), the elevated concentration of the later drugs could result in synergistic QT prolongation effects. The induced-fit docking poses of levofloxacin, moxifloxacin and ofloxacin with P-gp pocket are displayed in Figure 3(a)–(c).

Docked pose of second-line antitubercular drugs, fluoroquinolones: (a) levofloxacin, (b) moxifloxacin and (c) ofloxacin at the P-gp binding pocket.
Clofazimine is known to show a potent inhibitory effect on CYP3A4 in vitro. 25 The N.S of −2608.84 against CYP3A4 validates the claim. It is also a potent inhibitor of hERG. 49 The N.S of clofazimine against hERG and P-gp were found to be −2698.49 and −2943.46, respectively (Table 1). The CYP3A4 and P-gp inhibitory effect of this drug would result in high levels of chloroquine, HCQ, ritonavir and lopanivir, thereby increasing the later drug’s hERG inhibitory effect when co-administered. The fact that clofazimine itself binds to hERG strongly may also result in synergism with the other drugs. Besides these, we strongly suggest the possibility of simultaneous drug interactions in the hERG pocket. Our computational predictions assert that the hERG-drug complexes are more stable with clofazimine when combined with another drug (chloroquine/HCQ/lopinavir/ritonavir/azithromycin/famotidine) than this or the other individual drugs being present in the pocket individually. The simultaneous docking poses are shown in Figure 4(a)–(f).

Simultaneous docking of clofazamine with (a) chloroquine, (b) HCQ, (c) lopinavir, (d) ritonavir, (e) azithromycin and (f) famotidine in hERG’s binding pocket.
The DDI profile of delamanid is similar to that of clofazimine. Experimental data reports that it does not inhibit or induce CYP isoforms. 50 Therefore, inhibition of P-gp by delamanid (N.S = −2505.75) may be a strong reason that maybe attributed to the increased QT prolongation effect observed when it is co-administered with chloroquine, HCQ, ritonavir and lopinvir (Table 2). From the simultaneous docking study on the hREG pocket, we propose that delamanid may also interact very strongly (better than individual affinity) inside the hERG binding region with co-administered drugs like chloroquine, HCQ, ritonavir, lopinavir, azithromycin and famotidine (Table 2). If proven experimentally, the presence of two molecules inside the pocket and their high stability may establish new mechanisms in DDIs for QT prolongation. The simultaneous docking poses are shown in Figure 5(a)–(f).

Simultaneous docking of delamanid with (a) chloroquine, (b) HCQ, (c) lopinavir, (d) ritonavir, (e) azithromycin and (f) famotidine in hERG’s binding pocket.
Bedaquiline itself is a potent hERG inhibitor (N.S = −1717.55). Hence, it is expected to have a synergistic DDI with other QT prolonging drugs such as ritonavir, lopinavir, chloroquine and HCQ (Table 2). Our computational studies have shown a good binding score with CYP3A4 (N.S = −1597.47) and P-gp (N.S = −2141.56), which may be another possible reason for its synergistic effect on QT prolongation with the above drugs. Despite extensive IFD studies, we could not generate sufficient pose for azithromycin with hERG, CYP3A4 and P-gp pockets after docking. In the case of clarithromycin, molecular modelling data could not be generated due to the complexity of the mechanisms it exhibits. Famotidine also had a very low interaction with hERG protein (Table 1). Molecular modelling results have revealed that remdesivir (N.S = −1636.25) and streptomycin (N.S = −2137.80) had higher binding affinities towards hERG protein. Further clinical data is required to validate these modelling results.
We also identified 16 synergistic pharmacodynamic DDIs. Of these, 12 DDIs were between corticosteroids (dexamethasone, hydrocortisone, methylprednisolone and budesonide) and fluoroquinolones (levofloxacin, moxifloxacin and ofloxacin) that could increase the risk of development of tendinopathy. The remaining 4 DDIs between non-steroidal anti-inflammatory drugs (NSAIDs; ibuprofen and indomethacin) with fluoroquinolones (levofloxacin and ofloxacin) could increase the risk of central nervous system stimulation and convulsive seizures.
Pharmacokinetic interactions
We identified a total of 42 pharmacokinetic DDIs between repurposed COVID-19 drugs and antitubercular drugs. The majority of these pharmacokinetic DDIs were explained by the ability of the antitubercular drugs and repurposed COVID-19 drugs to inhibit or induce CYP3A4 and P-gp. This was further evidenced by computational studies as several drugs were found to be strong substrates of CYP3A4 and P-gp as shown in Table 1.
The first-line antitubercular drug rifampin was shown to interact with several drugs that were substrates of CYP3A4 and P-gp such as lopinavir/ritonavir, dexamethasone, hydrocortisone, methylprednisolone, ruxolitinib, losartan, apixaban, dabigatran, rivaroxaban, edoxaban and simvastatin. Clarithromycin also was found to pharmacokinetically interact with substrates of CYP3A4 and P-gp such as colchicine, dexamethasone, methylprednisolone, budesonide, ruxolitinib, calcifediol, losartan, warfarin, dabigatran, rivaroxaban, edoxaban, atorvastatin, lovastatin and simvastatin.
Computation studies have shown that hydrocortisone (N.S = −2377.34), methylprednisolone (N.S = −2832.10), colchicine (N.S = −2537.80), ruxolitinib (N.S = −2339.74), calcifediol (N.S = −3982.01), losartan (NS = −2452.40), apixaban (N.S = −3299.81), dabigatran (N.S = −2483.90), rivaroxaban (N.S = −3246.04) and edoxaban (N.S = −2836.16) are strong substrates of CYP3A4. The induced-fit docking poses of lopinavir, ritonavir and apixaban with CYP3A4 pocket are displayed in Figure 6(a)–(c).
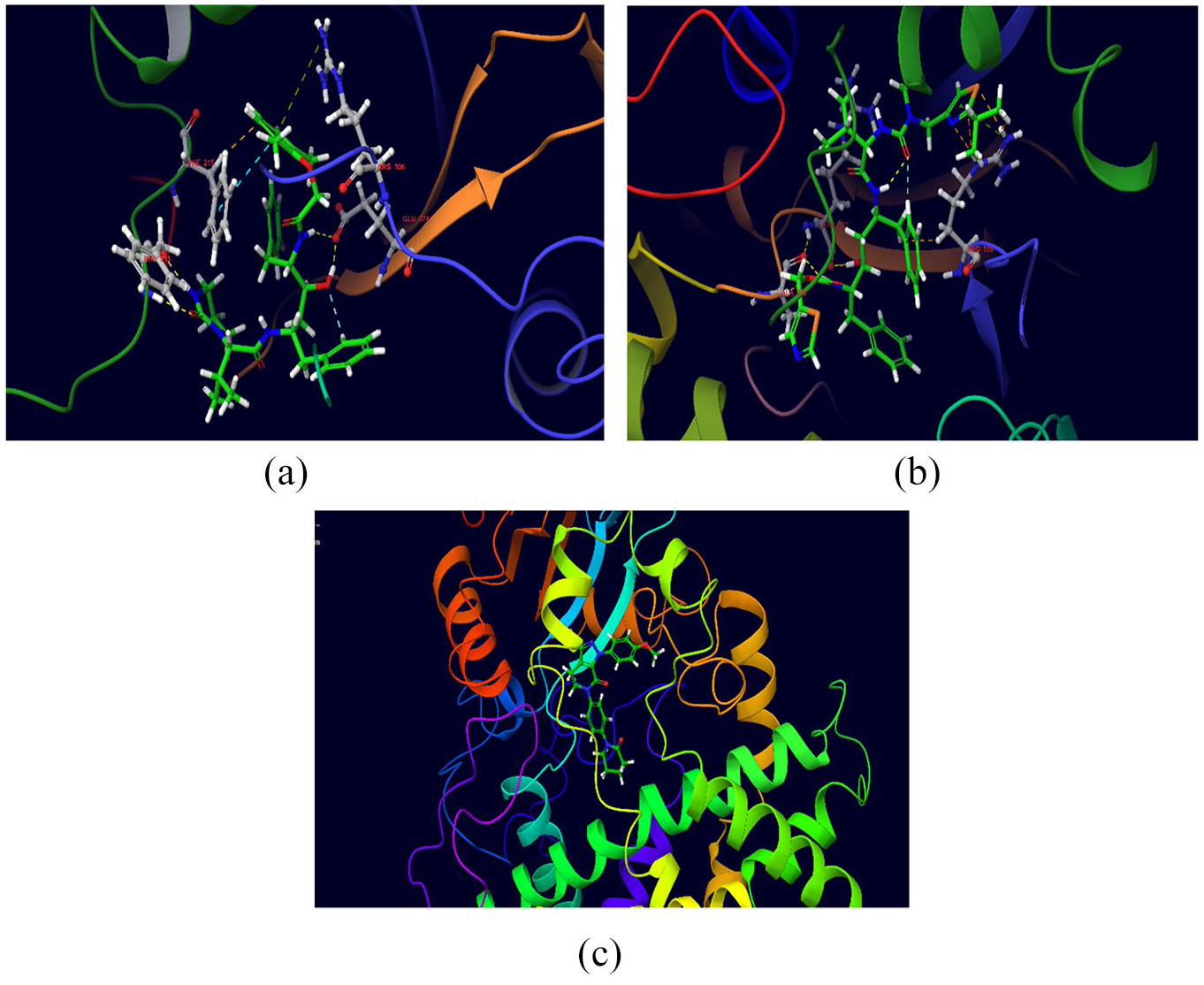
Docking pose of (a) lopinavir, (b) ritonavir and (c) apixaban at the CYP3A4 binding pocket.
Ritonavir (N.S = −2095.04), hydrocortisone (N.S = −2846.07), methylprednisolone (N.S = −2683.53), calcifediol (N.S = −3379.04), losartan (N.S = −2296.83), apixaban (N.S = −2589.28), dabigatran (N.S = −2656.35), rivaroxaban (N.S = −3171.51) and edoxaban (N.S = −2943.56) were found to be strong substrates of P-gp. The computational studies also indicate that remdesivir is an excellent substrate of P-gp (N.S = −2687.21) as shown in Figure 7(c). The induced-fit docking poses of ritonavir, hydrocortisone and remdesivir with P-gp pocket are displayed in Figure 7(a)–(c).

Docking pose of (a) ritonavir, (b) hydrocortisone and (c) remdesivir at the P-gp binding pocket.
First-line antitubercular drugs like ethambutol and pyrazinamide as well as the second-line drugs capreomycin, cycloserine, ethionamide, imipenem-cilastatin and pretomanid did not show any interactions with the repurposed COVID-19 drugs (Figure 1). These drugs had only weak-to-moderate binding with CYP3A4 and P-gp (Table 1).
Discussion
A large proportion of patients who develop active TB infection are immunocompromised. 51 COVID-19 infection also possesses a perilous impact on the immune system. 52 A synergy of these two infections could result in severe menacing consequences. 53 COVID-19 co-infection with TB has been reported to worsen the prognosis and increase morbidity and mortality in patients affected with this dual infection.54–56 There is a sparsity of data regarding the number of patients who receive antitubercular as well as repurposed COVID-19 drugs simultaneously. Stochino and colleagues reported that 20 cases of COVID-19 infection were observed among 24 inpatients diagnosed with TB in a hospital setting in northern Italy. Among these 20 patients co-infected with COVID-19 and TB, drug sensitivity test results showed that 3 patients were isoniazid resistant and 2 patients were multidrug-resistant. The standard first-line antitubercular treatment regimen was given in 14 cases, whereas a modified antitubercular regimen was given to 6 patients based on clinical characteristics and drug sensitivity test results. HCQ (200 mg twice a day) was administered to all TB patients co-infected with COVID-19 infection. 57 However, with the arrival of second and third wave of COVID-19 infection, there is a high potential for a significant population of TB patients to be infected with COVID-19 and vice versa, particularly in countries with a high TB burden. In this scenario, withholding the administration of antitubercular drugs is not a rational option as it has been reported that interruption of TB treatment is significantly associated with poor treatment outcomes and is one of the major hurdles associated with TB management, particularly of multidrug-resistant tuberculosis (MDR-TB).58–60 Hence, it is anticipated that in the coming days, there would be a significant proportion of TB patients who are likely to be co-infected with COVID-19 and could receive antitubercular as well as repurposed COVID-19 drugs simultaneously. Molecular docking is a widely used in silico method that aids in predicting interactions between molecules and biological targets.61,62 This is usually performed by first predicting the molecular orientation of a ligand within a receptor and then developing a scoring function based on IFD poses and energy calculations to estimate their complementarity. 61 IFD incorporates the principle that ligand binding causes changes in the residue side chain conformations within the specified pocket, resulting in flexibility in the binding pocket of the receptor. 63 These tools are fundamentally based on molecular docking approach that has been demonstrated to be a potential tool in the study of drug interactions with hERG, CYP3A4 and P-gp.64–66 CYP3A4 has a broad ligand specificity and is involved in the metabolism of many drugs. Hence, inhibition/induction of CYP3A4 metabolic activity by one substrate can have a significant impact on the metabolism of other substrates. 67 Drugs that are inhibitors or inducers of P-gp could alter the pharmacokinetic profiles of co-administered drugs that are P-gp substrates, resulting in DDIs.68,69 The structure-assisted docking model has been reported to predict P-gp inhibition quite effectively. 70 There is a substantial overlap in drugs that interfere with CYP3A4 and P-gp. Dual P-gp and CYP3A4 inhibitors, on the contrary, may not always have the same inhibitory potency against P-gp and CYP3A4.71,72 Hence, by studying the extent of binding of drugs with CYP3A4 and P-gp through docking studies could provide critical preliminary information about hERG, CYP3A4 and P-gp-mediated DDIs. We have explored the possibility of two drugs interacting in the same pocket, thereby causing better inhibitory effect on hERG than a single drug itself. Thereby, we propose a direct DDI with respect to hERG inhibition in many instances where the interactions are occurring independent of the metabolic enzymes. The DDIs reported in the current study through drug interaction checker and molecular modelling study are intended to aid clinicians in better management of COVID-19 and TB infection and should not impede the delivery of regular treatments for both the infections.
High costs involved in the new drug development process and concerns pertaining to production and distribution of COVID-19 vaccines have paved the way for instant ubiquity for repurposing the available drugs to manage COVID-19.73,74 TB patients develop poor medication adherence due to multiple factors such as poverty, lack of social support and food security, adverse drug effects and pill burden. 75 There is an anticipation of a higher incidence of COVID-19 infection among TB patients and vice versa, particularly in countries with a high TB burden. Therefore, a significant population would be expected to undergo repurposed COVID-19 drug therapy along with antitubercular therapy. Hence, it is very important to foresee and prevent or manage DDI in these patients to allay the development of poor TB and COVID-19 treatment outcomes. Changes in the expression and activity of transporters and drug metabolizing enzymes induced by the inflammatory response in COVID-19 infection could also possibly play a crucial role in developing DDIs. 76 Studies from Spain have reported a high frequency of DDIs identified between repurposed COVID-19 drugs and concomitant medications.77,78 Addition of corticosteroids in COVID-19 management resulted in a dramatic increase in the development of potential DDIs of class B (moderate DDIs: drug combinations requiring dose adjustment and drug concentration monitoring). 79 Recent studies have reported on the potential risk of developing QT prolongation, CYP450 and P-gp-based DDIs between repurposed COVID-19 drugs and various drugs used for management of lung cancer, neuropsychiatric diseases, stroke, asthma and arrhythmia.16,17,80–82 We have identified several pharmacodynamic DDIs resulting in a potential risk of QT prolongation as well as CYP450 and P-gp-based pharmacokinetic DDIs between repurposed COVID-19 drugs and antitubercular drugs through Micromedex drug interaction checker. Most of the pharmacodynamic interactions were supported by the results of the molecular modelling data.
Pharmacodynamic interactions: Jain and colleagues reported that QT prolongation was prevalent among 19.65% (n = 103/524) of the COVID-19 patients with electrocardiogram reports. Among these patients, 95.1% (n = 98) of patients were on COVID-19-related QT-prolonging medications. 83 Chloroquine, HCQ, lopinavir, ritonavir, azithromycin and famotidine have been documented to cause QT prolongation.84–91 Concomitant use of azithromycin with HCQ caused a greater change in the QTc prolongation than HCQ alone. 86 Approximately 5% of patients who were on remdesivir therapy for COVID-19 had developed QT prolongation. 83 Several second-line antitubercular drugs such as clarithromycin, clofazimine, delamanid, levofloxacin, moxifloxacin, ofloxacin and bedaquiline are associated with QT prolongation.36,92–99 The hERG channel has been reported to interact with numerous drugs of widely varying structure, possibly due to the unusual shape of the ligand-binding site and its hydrophobic character. Several in silico approaches for predicting the blockade of hERG channel have been attempted. 100 Our molecular modelling study also found that the aminoglycosides given as second-line antitubercular drugs had higher affinity for binding with hERG pockets (Table 1). However, we did not find any DDIs related to the same in Micromedex. Therefore, it can be said that the induced fit docking study was not sufficient enough to explain these findings. We expect that analysis with more advanced studies like molecular dynamics simulation and further clinical studies investigating the QT prolongation potential of aminoglycosides are required to give conclusive evidence. Our findings show that several combinations of repurposed COVID-19 drugs with antitubercular drugs had a higher combination binding energy towards hERG. Hence, concomittant administration of the repurposed COVID-19 drugs and antitubercular drugs can result in synergistic QT prolonging effects, which require vigilant monitoring. The majority of the identified DDIs from Micromedex, resulting in QT prolongation, were concordant with our molecular modelling results (Table 2). Further clinical studies are required to validate these data from patients who are co-infected with COVID-19 and TB. Necessary cautions should be taken to avoid QT prolongation arising as a DDI between these repurposed COVID-19 drugs and antitubercular drugs. Developing various early identification and management tools such as that of the Situation Background Assessment Recommendation (SBAR) and electronic registries for arrhythmias are some of the potential strategies that could be employed for screening and management of QT prolongation, especially in this co-infected situation.29,83 Concomitant use of the second-line antitubercular drugs such as fluoroquinolones (levofloxacin, moxifloxacin and ofloxacin) with corticosteroids such as dexamethasone, hydrocortisone, methylprednisolone can cause synergistic tendinopathy/tendon rupture, with a higher risk among females, patients aged above 60 years and long-term use of fluoroquinolones.101–104 Hence, corticosteroid therapy should be used with caution among MDR-TB patients taking fluoroquinolones. The concurrent administration of fluoroquinolones (levofloxacin and ofloxacin) with NSAIDs (ibuprofen and indomethacin) may increase the risk of central nervous system stimulation and convulsive seizures.105–107 Alternative therapy should be considered, especially in patients who are predisposed to seizure activity.
Pharmacokinetic interactions: The first-line antitubercular drug rifampin reduces the plasma concentration of several drugs such as lopinavir/ritonavir, dexamethasone, hydrocortisone, methylprednisolone, ruxolitinib, losartan, apixaban, dabigatran, rivaroxaban, edoxaban and simvastatin via indirect induction of CYP3A4 and P-gp. Rifampin binds to the pregnane X receptor (PXR) and constitutive androstane receptor (CAR) in the cytoplasm. It translocates into the nucleus to form a heterodimer with retinoic acid receptor RXR. Subsequently, the heterodimer binds to the promoter region of the target genes CYP3A4 and P-gp and activates the transcription of its open reading frame, leading to increased CYP3A4 and P-gp expression.26,108 As CYP3A4 and P-gp are major pharmacokinetics determinants of lopinavir/ritonavir, dexamethasone, hydrocortisone, methylprednisolone, ruxolitinib, losartan, apixaban, dabigatran, rivaroxaban, edoxaban and simvastatin, CYP3A4 and P-gp induction could increase the metabolism of these drugs and consequently reduces its plasma concentration.109–123 Valsartan is a substrate of efflux transporters P-gp (N.S = −1861.07) and MRP (multidrug resistance-associated protein) and uptake transporter OATP (organic anion transporter polypeptide). Therefore, co-administration with rifampin, an inducer of all the above transporters, may increase valsartan exposure. 124 Concurrent use of atorvastatin and rifampin may result in decreased atorvastatin concentration when administered separately after rifampin or increased atorvastatin exposure when administered simultaneously with rifampin which is due to induction of CYP3A4 metabolism of atorvastatin by rifampin and inhibition of organic anion-transporting polypeptide (OATP1B1)-mediated atorvastatin hepatic reuptake by rifampin, respectively.125,126 Concurrent use of fluvastatin and rifampin may result in decreased fluvastatin effectiveness. Fluvastatin is a substrate for BCRP (intestinal), OATP1B1, OAT1B3, OAT2B1, CYP2C9 and CYP3A4. Fluvastatin interactions are predominantly related to complete inhibition of CYP2C9, and rifampin is a CYP2C9 inducer.127,128 Besides CYP3A4 and CYP2C9, rifampin is also an inducer of CYP1A2, CYP2C19 and CYP3A5. 129 Hence, concomitant administration of rifampin with warfarin that is metabolized by most of these CYP450 enzymes will result in an increased risk of bleeding. Another first-line antitubercular drug isoniazid could potentially inhibit a wide range of CYP450 enzymes such as CYP1A2, CYP2A6, CYP2C19, CYP3A4, CYP2C9 and CYP2E1.130,131 Warfarin molecule exist in 2 enantiomeric forms S- and R-warfarin. The S-warfarin is mainly metabolized by CYP2C9, whereas R-warfarin is mainly metabolized by CYP1A2 and CYP3A4. 132 Isoniazid-mediated CYP450 enzyme inhibition will potentially result in elevation of the blood levels of warfarin, resulting in an increased tendency for bleeding when both the drugs are given concomitantly.
Bedaquiline is primarily metabolized in the liver by the CYP3A4 to a less active N-monodesmethyl metabolite, M2. 133 Hence, concomitant administration of CYP3A4 inhibitors with bedaquiline could result in elevated plasma concentrations of bedaquiline, increasing the risk of toxicities such as QT prolongation. 134 Ritonavir, a high-affinity type II ligand binds irreversibly to CYP3A4 and inhibits it by multiple mechanisms. These include metabolic-intermediate complex formation, competitive inhibition, irreversible type II coordination to the heme iron, heme destruction and CYP3A4-mediated ritonavir activation and subsequently form a covalent bond to the apoprotein.135,136 The mechanism of DDI between clarithromycin and various drugs that are CYP3A4 substrates such as dexamethasone, methylprednisolone, budesonide, calcifediol, ruxolitinib, losartan, warfarin, atorvastatin, lovastatin and simvastatin is attributed to mechanism-based inhibition exerted by clarithromycin. CYP3A4 metabolizes clarithromycin to form reactive nitrosoalkane via N-demethylation. This reactive nitrosoalkane generated further interacts with CYP3A4 to form an intermediate metabolite complex. This metabolic intermediate covalently bonds to the same CYP3A4 enzyme and facilitates its inactivation. Hence, a larger dose of clarithromycin can result in more potent CYP3A4 inhibition because of the larger formation of metabolite intermediates that can further inhibit CYP3A4 activity.116,137–145 Concurrent use of clarithromycin and pravastatin may increase pravastatin exposure and an increased risk of myopathy or rhabdomyolysis by clarithromycin inhibiting in a concentration-dependent manner OATP1B1- and OATP1B3-mediated uptake of pravastatin.146,147 Clarithromycin could increase the plasma exposure of dabigatran and edoxaban by P-gp inhibition and rivaroxaban by P-gp as well as CYP3A4 inhibition resulting in an increased risk of bleeding.148–150 CYP3A4 plays a significant role in the metabolism of colchicine to its inactive metabolites. Colchicine is also a substrate for P-gp. Therefore, clarithromycin, through dual inhibition of CYP3A4 and P-gp, can increase the plasma concentrations of colchicine, resulting in the toxicity of the latter drug. 151 Zinc chelates with fluoroquinolones such as levofloxacin, moxifloxacin and ofloxacin, resulting in significant reduction of their antimicrobial activity.152,153 Disruption of intestinal flora synthesizing vitamin K and inhibition of cytochrome P450 enzymes involved in the metabolism of warfarin are proposed to be the major mechanisms involved in the drug interaction between antibiotics such as fluoroquinolones, amoxicillin-clavulanate and linezolid with warfarin resulting in increased risk of bleeding.154–156 Concurrent use of amikacin and indomethacin with ibuprofen may result in increased amikacin exposure. Changes in renal blood flow is postulated to be the mechanism involved in explaining the DDI between NSAIDs and aminoglycosides. Inhibition of renal prostaglandins by NSAIDs may affect the renal blood flow and glomerular filtration rate during the development of aminoglycoside nephrotoxicity.157–160 Ascorbic acid could reduce the antibacterial actions of streptomycin and kanamycin by mechanisms that are not fully understood. 161 Since remdesivir was found to be an excellent substrate of P-gp (N.S = −2687.21) through modelling study, caution should be warranted when other P-gp inhibitors are given.
With the advent of pharmacogenomics, it becomes imperative to assess the potential impact of genetic polymorphisms of drug metabolizing enzymes and drug transporters to understand clinically critical DDIs. An interaction solely caused by drug response to CYP450 genetics is referred to as drug–gene interaction (DGI) whereas an interaction that is a cumulative effect of both a DDI and DGI is referred to as drug–drug–gene interaction (DDGI). The incorporation of gene variants of CYP2D6, CYP2C19 and CYP2C9 increased the number of potentially clinically significant interactions by ~51% in a retrospective analysis assessing drug interactions among 1143 patients. 162 Since there is a significant proportion of DDIs explained by induction and inhibition of CYP3A4 enzyme and P-gp transporter in Figure 1, it may be useful in future to identify the influence of their genetic variants to predict potentially clinically significant interactions in especially patients who are on second-line antitubercular drugs such as clarithromycin or to understand rifampin-mediated DDIs. Single nucleotide polymorphisms (SNPs) in the CYP3A4 could abolish, reduce or increase CYP3A4 enzymatic activity. The effect of SNPs in CYP3A4 on substrate binding was found to be substrate specific. 163 The ABCB1 or MDR1 gene that encodes for P-gp is highly polymorphic and the allelic variants have been reported to significantly influence the P-gp transporter activity. 164 The distribution of certain CYP3A4 and MDR1 allelic variants appears to be ethnicity dependent.163,164 In these contexts, assessing the influence of allelic variants of CYP3A4 and MDR1 on the pharmacokinetics of individual substrate as well as substrate combinations (identified to be interacting) will be required to establish the precise extent and risk of clinical consequences of DDIs in clinical scenarios in different ethnic populations.
Therefore, predicting potential DDIs related to CYP3A4, P-gp and hERG is crucial in delivering safe chemotherapeutic TB and COVID-19 care. Further computational methods such as molecular dynamic studies (MDS) and in vitro assessments would validate and refine the current research results, to scale up our findings into clinical practice. The authors will consider this in near future. The risks of DDIs identified in this study should not impede the delivery of repurposed COVID-19 drugs or antitubercular therapies in all situations. The proper precautions, monitoring and safety management could deter the adverse events arising from DDIs between these drugs in most scenarios. Summarizing these potential DDIs for clinical decision support is challenging. Further clinical data on the clinical significance of these DDIs are needed from well-designed pharmacokinetic/pharmacodynamic studies. This helps to validate the data coming from drug information databases and molecular docking studies. These data are required to rule out the possibilities of poor alert specificity and indiscriminate DDI warnings. However, the current study provides an initial thrust of alertness regarding the occurrence of possible DDIs between repurposed COVID-19 therapies and antitubercular therapies based on drug information software and molecular docking software data. This aids clinicians and medical researchers identify and predict DDIs, particularly in countries with increased incidences of TB and COVID-19 infections. DDI information generating from real-time clinical scenarios may be used to develop algorithms for managing DDIs in COVID-19 patients treated with antitubercular drugs and repurposed COVID-19 drugs. Physicians, pharmacists and other health care providers should be encouraged to report adverse drug reactions that occur while treating the co-infected patients to regional/zonal/national pharmacovigilance centres for the wide dissemination of the safety information. There is a need for a single open accessible DDIs repository that integrates the literature evidence from in silico, in vitro, in vivo and real-time clinical data and provides health care providers with more precise and reliable information for clinical decision making.
Conclusion
The data derived from DDI checker software and in silico computational methods have predicted the possibility of several potential DDIs with repurposed COVID-19 drugs. This highlights the need for a conscientious treatment plan, particularly in MDR-TB patients. The great challenge is especially when repurposed COVID-19 drugs are co-administered with second-line antitubercular drugs with the potential for QT prolongation or with antitubercular drugs that are inducers or inhibitors of CYP3A4 and P-gp. Further clinical data from well-designed, real-time pharmacokinetic and pharmacodynamic research in the TB patients co-infected with COVID-19 is required for the pragmatic picture of the DDIs reported in this work. These integrated data could then be potentially employed by clinicians, pharmacists and other healthcare providers to develop a meticulous surveillance strategy to identify and manage the DDIs. This improves the treatment outcomes, reduces healthcare expenditures and decreases related morbidities and mortalities of the affected patients.
Footnotes
Acknowledgements
Levin Thomas, Sumit Raosaheb Birangal and Rajdeep Ray are thankful to Dr TMA Pai PhD Scholarship from Manipal Academy of Higher Education, Manipal, India. All the authors are thankful to Centre for Translational Research, MAHE and Manipal Schrodinger Centre for Molecular Simulations for providing computational facility to carry out the study. Levin Thomas and Sumit Raosaheb Birangal contributed equally.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical approval and consent to participate
No ethical approval was needed as this was an integrated data analysis from drug information database and in silico computational methods.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.