
Abstract
Over the last decade, the number of clinical pharmacogenetic tests has steadily increased as understanding of the role of genes in drug response has grown. However, uptake of these tests has been slow, due in large part to the lack of robust evidence demonstrating clinical utility. We review the evidence behind four pharmacogenetic tests and discuss the barriers and facilitators to uptake: (1) warfarin (drug safety and efficacy); (2) clopidogrel (drug efficacy); (3) codeine (drug safety and efficacy); and (4) abacavir (drug safety). Future efforts should be directed toward addressing these issues and considering additional approaches to generating evidence basis to support clinical use of pharmacogenetic tests.
Introduction
It has been estimated that more than 770,000 people are injured or die each year in hospitals from adverse drug events (ADEs), costing millions of dollars in healthcare costs each year [Classen et al. 1997; Lazarou et al. 1998]. The field of genomic medicine presents one potential solution to reduce health care costs associated with ADEs and poor response to pharmacotherapy. Specifically, the field of pharmacogenetics involves using a patient’s genetic makeup in combination with other clinical information to create a personalized medication regimen with greater efficacy and safety for the individual patient. Many medications currently prescribed have pharmacogenetic data to support appropriate dosing or selection. In addition, pharmacogenetic analyses are routinely performed during drug development [Liou et al. 2012].
Although it has long been understood that genes play a role in drug response [Scott, 2011], the explosion of new discoveries from genome-wide association studies (GWASs) and large population-based cohort studies has driven pharmacogenetic testing to the forefront of the personalized medicine movement. Advances in genomic technologies, enabling more accurate, faster, and cheaper tools for data generation and clinical testing, largely account for the rapid pace of discovery and development. In fact, the substantial drop in cost from these new technologies has created somewhat of a dilemma in that it may be cheaper to perform a genome-wide analysis instead of a single gene test, generating much more information than is needed.
Despite the rapid pace of discovery and test development, the routine use of pharmacogenetic testing is stymied by the lack of data demonstrating clinical utility, or evidence that use of the test will improve health outcomes for a given patient [Lesko et al. 2010; SACGT, 2000]. While randomized controlled trials (RCTs) remain the gold standard for clinical evidence, very few have been performed in pharmacogenetics. More than 2000 papers on pharmacogenetics have been published annually over the last several years [Scott, 2011], demonstrating a huge growth in discovery. In comparison, a search of the PubMed database reveals that 212 RCTs involving pharmacogenetics have been published over the past 10 years (Figure 1). However, this number may be misleading, as it is likely that only a minority of publications are RCTs with a primary pharmacogenetic endpoint and others are likely RCTs with secondary pharmacogenetic endpoints or exploratory studies. A search of the US-based clinical trials database (ClinicalTrials.gov) for ‘pharmacogenetic OR pharmacogenomic’ returned 312 studies, 147 of which were open (defined as recruiting, not yet recruiting, or available for expanded access; as of 18 September 2012). It is highly unlikely that many RCTs will be conducted and decisions to use pharmacogenetic testing will need to be based on other types of evidence [Frueh, 2009]. Of the 147 studies found in our search of ClinicalTrials.gov, only 30 were RCTs. Several reasons may account for the relatively small number of clinical trials in pharmacogenetics: lack of funding, lack of interest in conducting clinical trials (particularly for approved drugs), failure to validate initial discoveries, and small patient populations. Indeed, current use of pharmacogenetic testing is based in large part on observational and retrospective studies.
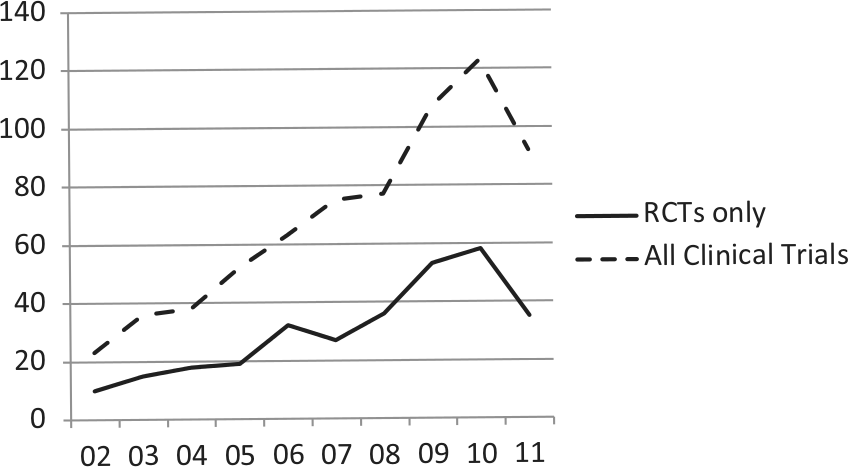
PubMed search of clinical trials for pharmacogenetic testing compared with clinical trials of drugs for heart disease and cardiovascular disease between 2002 and 2011. Search parameters: randomized controlled trial (RCTs) or clinical trials + RCTs, pharmacogenetics, between 1 January 2002 to 31 December 2011. Search conducted on 16 April 2012.
Beginning in 2003, the US Food and Drug Administration (FDA) has approved revisions to package inserts for several drugs to include information about pharmacogenetics [FDA, 2012c; Frueh et al. 2008]. Although testing is not recommended in the revised package inserts, the importance of genetic variation to risk of adverse drug reactions (ADRs) or outcome is significant enough to warrant prominent inclusion in the boxed warnings for some medications. In 2003, an estimated 1.5% of the top 200 medications included information in the package insert concerning pharmacogenomics [Zineh et al. 2006]. Repeating their analysis with the most current listing of top 200 medications sold in the US [Bartholow, 2011], we estimate that 11% of medications now include pharmacogenetic information. This 10-fold increase reflects the rapid accumulation of data about the role of genetic polymorphisms on drug safety and response.
In this review, we describe the current state of evidence of clinical utility of pharmacogenetic testing in four medications (see Table 1 for a summary). Given the many stakeholders in healthcare including the provider, patient, institution, and third-party payer, the determination of clinical utility and factors considered can vary substantially. Therefore, we also discuss other factors such as availability of clinical guidelines that may impact overall use of the test. The selected examples highlight the variability of available evidence and other factors that may impact clinical uptake in various clinical settings.
Summary of pharmacogenetic tests.
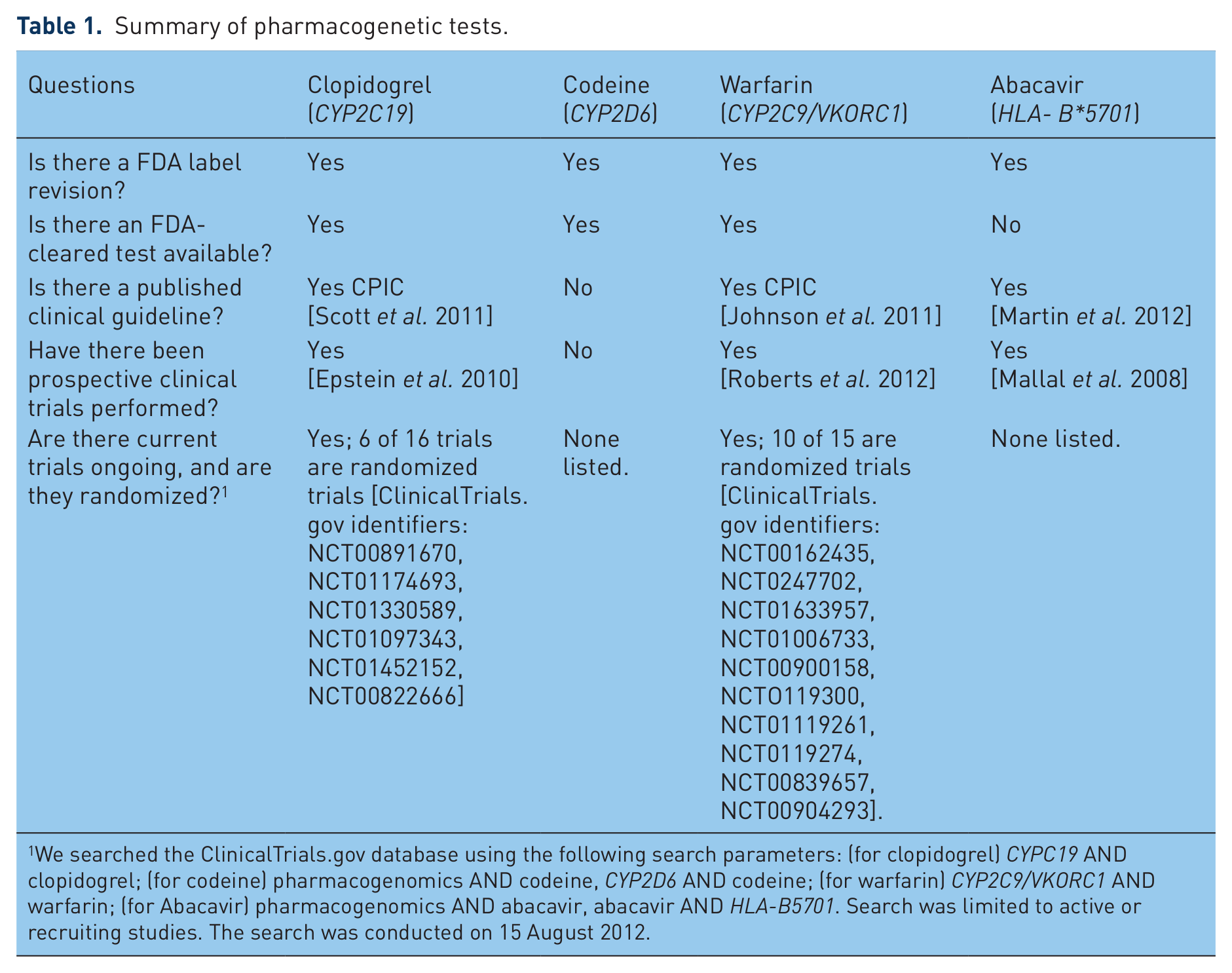
We searched the ClinicalTrials.gov database using the following search parameters: (for clopidogrel) CYPC19 AND clopidogrel; (for codeine) pharmacogenomics AND codeine, CYP2D6 AND codeine; (for warfarin) CYP2C9/VKORC1 AND warfarin; (for Abacavir) pharmacogenomics AND abacavir, abacavir AND HLA-B5701. Search was limited to active or recruiting studies. The search was conducted on 15 August 2012.
Warfarin
State of evidence
Warfarin is one of the most commonly prescribed medications worldwide, used for many indications including prophylaxis and treatment of thromboembolic disorders, atrial fibrillation, or cardiac valve replacement, and systemic embolism after myocardial infarction (MI). Approved in the US in 1954, the high efficacy of warfarin is challenged by the high risk of ADRs due to its narrow therapeutic window, requiring careful monitoring and strict compliance. The international normalized ratio (INR; ratio of a patient’s prothrombin time to a normal sample) is the standard test to assess therapeutic range. New patients on warfarin require frequent monitoring of INR levels until the optimal level is attained and monthly thereafter [Guyatt et al. 2012; Johnson, 2012]. It has been reported that approximately 60% of anticoagulation clinics are able to reach desired INR levels for patients, while the remaining clinics continually need to monitor their patients until they achieve the optimal INR goals [Marin-Leblanc et al. 2012].
Treatment variability of warfarin is attributed to a combination of factors including age, race, weight, diet (specifically intake of vitamin-K-rich foods) [Rasmussen et al. 2012], alcohol consumption, and numerous drug interactions [Johnson, 2012]. Achieving the desired INR range can take several months, raising the risk of ADRs. In 2000 and 2004, polymorphisms in two genes were reported to be associated with risk of ADRs and drug response, cytochrome P450 2C9 (CYP2C9) [Taube et al. 2000] and vitamin K epoxide reductase complex subunit 1 (VKORC1), respectively [Aithal et al. 1999; Bodin et al. 2005; Li et al. 2004; Rieder et al. 2005; Rost et al. 2004; Steward et al. 1997]. CYP2C9 is involved in the metabolism of warfarin; VKORC1 is the molecular target of the drug.
In 2009, an international collaboration published a landmark paper defining appropriate warfarin doses based on a validated dosing algorithm of clinical biomarkers and VKORC1/CYP2C9 genotypes [Klein et al. 2009]. In 2010, Epstein and colleagues conducted a prospective study of the utility of warfarin and demonstrated a reduction in hospitalizations when starting doses were informed by patient genotypes for VKORC1/CYP2C9 [Epstein et al. 2010]. Several large studies are underway (Coumagen II, Clarification of Optimal Anticoagulation Through Genetics [COAG] [French et al. 2010] and Genetics Informatics Trial of Warfarin Therapy [GIFT] trial [Do et al. 2011]) to assess the benefits of pharmacogenetic testing prior to warfarin dosing [Carlquist and Anderson, 2011; ClinicalTrials.gov identifier: NCT00927862]. A handful of groups have demonstrated the utility of genotype-guided warfarin initiation therapy [Anderson et al. 2012; Caraco et al. 2008; Gong et al. 2011].
Revisions to package insert
In 2005, an FDA advisory committee reviewed the published literature on the association of genetic variants of VKORC1 and CYP2C9 and outcomes to warfarin treatment (Figure 2) [FDA, 2005]. In 2007, the package insert was revised to include information about the impact the impact of the genetic variants in CYP2C9/VKORC1 on treatment outcomes [FDA, 2007a]. In 2010, the FDA approved additional revisions to the package insert to include a table of warfarin starting doses based on patient genotypes of VKORC1/CYP2C9, but did not recommend testing prior to treatment [FDA, 2010a]. The package insert revisions in 2007 were primarily based on retrospective studies [Higashi et al. 2002; Lindh et al. 2005; Taube et al. 2000], while the second round of revisions in 2010 were informed by data from both retrospective and prospective study outcomes [Anderson et al. 2007; Caraco et al. 2008; Jneid et al. 2012; Limdi et al. 2008; Wadelius et al. 2009; Wen et al. 2008].
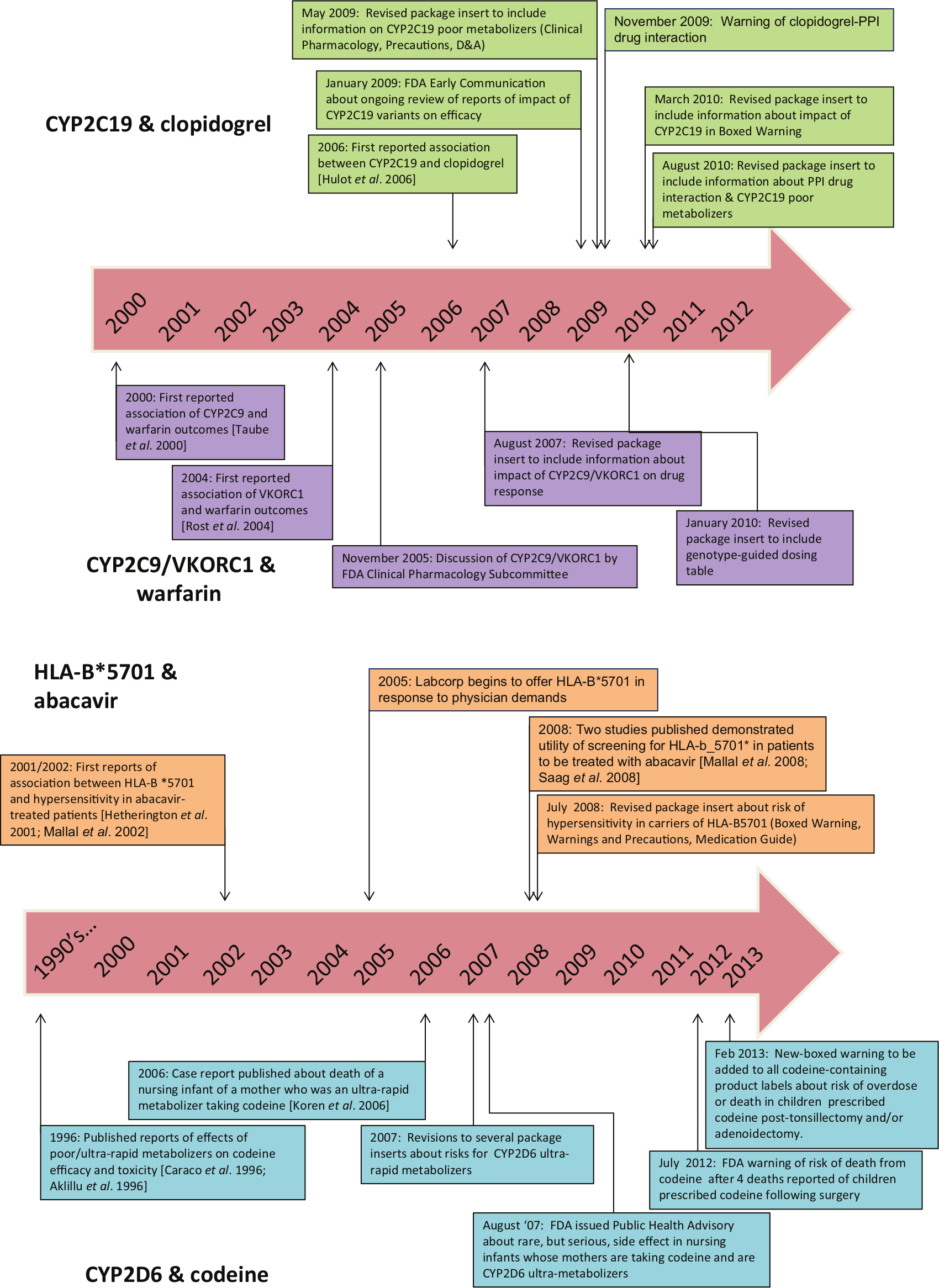
Timeline of major developments of pharmacogenetic testing for the drugs clopidogrel, warfarin (top), abacavir and codeine (bottom).
Clopidogrel
State of evidence
Like warfarin, clopidogrel is a commonly prescribed drug, with sales estimated to exceed US$6 billion in 2010 in the US [Alazraki, 2011]. Approved in 1997, clopidogrel is another cardiovascular medication intended to reduce the rate of atherothrombotic events in patients with recent MI or stroke or established peripheral arterial disease, unstable angina (UA) or non-ST-segment elevation (NSTEMI) managed medically or with percutaneous coronary intervention (PCI; with or without stent) or coronary artery bypass graft (CABG), and ST-segment elevation MI (STEMI) managed medically.
Clopidogrel is a prodrug that requires biotransformation to an active metabolite. Catalyzed by the enzyme CYP2C19, the active metabolite irreversibly blocks the P2Y12 component of adenosine diphosphate receptors on the platelet surface, which prevents activation of the glycoproteinIIb/IIIa receptor complex, thereby reducing platelet aggregation [Mega et al. 2009; Shuldiner et al. 2009]. In 2006, Hulot and colleagues reported that carriers of the reported loss-of-function allele CYP2C19*2 were significantly more likely to experience reduced platelet responsiveness to clopidogrel [Hulot et al. 2006]. Similarly, Simon and colleagues reported that patients found to be poor CYP2C19 metabolizers were almost four times more likely to experience a subtherapeutic antiplatelet response when treated with clopidogrel, resulting in a higher risk for cardiovascular adverse events [Simon et al. 2009]. Most poor metabolizer phenotypes are due to the CYP2C19*2 allele, where a splicing variant in exon 5 results in a truncated protein. Approximately 1–7% of Caucasians and African-Americans, and 13–23% of Asians are CYP2C19 poor metabolizers [Cavallari et al. 2011; Desta et al. 2002].
Several clinical trials have been initiated to assess the utility of testing to determine the proper dose of clopidogrel for CYP2C19-compromised patients [Simon et al. 2009]. However, the published findings are conflicting, suggesting other factors may be involved in predicting efficacy and adverse clinical outcomes. The ELEVATE-TIMI56 study demonstrated that CYP2C19*2 heterozygotes treated with threefold higher doses showed significantly reduced platelet reactivity, comparable with that seen in noncarriers on standard maintenance doses [Mega et al. 2011]. In addition, nonresponse in CYP2C19*2 heterozygotes was significantly reduced from 52% to 10% with the higher doses. CYP2C19*2 homozygotes did not respond to higher doses. Similarly, the Accelerated Platelet Inhibition by a Double Dose of Clopidogrel According to Gene Polymorphism (AACEL-DOUBLE) trial reported that the use of high-maintenance dose clopidogrel in high-risk patients with PCI and carriers of CYP2C19 polymorphisms showed higher platelet measures and increased risk of high post-treatment platelet reactivity than noncarriers [Jeong et al. 2010]. In contrast, Cuisset and colleagues reported that patients with CYP2C19*2 given an increased dose of 150 mg once daily did not show significant benefit and alternative medication was suggested [Cuisset et al. 2011]. Several recent meta-analyses have reported conflicting conclusions as well regarding the impact of CYP2C19 genotype on drug efficacy and/or cardiovascular events [Bauer et al. 2011; Holmes et al. 2011; Mega et al. 2010].
Revisions to the package insert
In 2009, the FDA announced that they were conducting a safety review of reports about the impact of CYP2C19 variants on efficacy and potential drug–drug interactions [FDA, 2009]. In 2010, the package insert for clopidogrel was revised to include information about the effect of CYP2C19 genetic variants on ADRs and response. Specifically, a boxed warning was added about the reduced effectiveness of clopidogrel in patients who are CYP2C19 poor metabolizers, and to inform healthcare professionals about the availability of genetic tests for CYP2C19 [Ellis et al. 2009; FDA, 2010c].
In addition, the insert was revised a second time in 2010 to include a warning about the concomitant use of proton pump inhibitors (PPIs) and clopidogrel and increased cardiovascular risk, since the majority of PPIs are metabolized by CYP2C19 [FDA, 2010b]. However, the evidence of an association between cardiovascular risk and PPIs in patients treated with clopidogrel is conflicting. Early reports suggested that concomitant use of PPIs reduced clopidogrel’s efficacy [Gilard et al. 2006, 2008], while other reports suggested no such effect [Bhatt et al. 2010; Charlot et al. 2010; O’Donoghue et al. 2009; Ray et al. 2010; Siller-Matula et al. 2009; Small et al. 2008]. Some argue that the increased cardiovascular risk linked to PPIs may be attributed to the lack of data on baseline characteristics such as smoking status, lipid levels, and body mass index [Charlot et al. 2010] or the effect is specific to select PPIs [Juurlink et al. 2009].
Codeine
State of evidence
Codeine is a commonly used drug to treat mild to moderate pain. Like clopidogrel, codeine is a prodrug; the majority of codeine (50–70%) is converted to the active metabolite codeine-6-glucuronide by the enzyme UGT2B7and a smaller proportion (0–15%) is converted to morphine by the enzyme CYP2D6 [Thorn et al. 2009]. CYP2D6 is a highly polymorphic gene, resulting in a range of phenotypes [Bernard et al. 2006; Bradford, 2002; Cascorbi, 2003]. In 1996, two reports were published about the effects of poor and ultra-rapid metabolizers on codeine efficacy and toxicity [Aklillu et al. 1996; Caraco et al. 1996]. CYP2D6 ultra-rapid metabolism is generally due to duplication of the slightly reduced activity (
Two vulnerable populations in which codeine is highly prescribed are postpartum women and children. It is estimated that 90–99% of women take some type of pain medication in the first week postpartum [Anderson, 1991; Hale, 2004; Ito and Lee, 2003] and as many as three to four drugs during breast-feeding [Stultz et al. 2007]. Oral analgesics have been reported to be the second most commonly used drug after vitamins during the postpartum period [Schirm et al. 2004]. In 2006, a case report was published of the death of an infant caused by exposure to high levels of morphine; the mother was determined to be a CYP2D6 ultra-rapid metabolizer [Koren et al. 2006]. Another paper reported that two mothers of infants with severe toxicity were also CYP2D6 ultra-rapid metabolizers [Madadi et al. 2009]. During lactation, use of higher doses of codeine in a CYP2D6 ultra-rapid metabolizer mother would likely result in the rapid transfer of drug metabolites to the milk and infant exposure. The slow metabolism and elimination by the infant during the newborn period can compound the toxic effect [Bouwmeester et al. 2003, 2004].
More recently, reports have been published of children under the age of 5 years experiencing a life-threatening event or death following use of codeine post-surgery. Specifically, four children have died from codeine toxicity; three of which were tested to be CYP2D6 ultra-rapid metabolizers and the fourth was an extensive metabolizer and all exhibited toxicity that led to these adverse events [Ciszkowski et al. 2009; Kelly et al. 2012].
Revisions to the package insert
In 2007, package inserts for approved drugs with codeine as a major component were revised to include information about ultra-rapid metabolizers of CYP2D6 in the package insert [FDA, 2007c]. Newly approved versions of codeine drugs, such as codeine sulfate, also included information about ultra-rapid metabolizers of CYP2D6 [FDA, 2012a]. In addition, two statements have been released regarding codeine use and the impact of CYP2D6 variants in children. In August 2007, the FDA recommended that physicians prescribe the lowest possible doses of codeine to nursing mothers [FDA, 2007b]. In August 2012, the FDA issued a statement warning of the danger of codeine use in children post-surgery [FDA, 2012b]. It is recommended that the ‘lowest effective dose be used for the shortest period of time’ or use of other analgesics for children undergoing tonsillectomy and/or adenoidectomy for obstructive sleep apnea syndrome. In 2013, the FDA announced it was revising all codeine-containing product labels to include a new boxed warning about the risk of codeine in pain management in children post-tonsillectomy and/or adenoidectomy. The FDA is recommending use of alternative pain relievers for pediatric patients [FDA, 2013]. Of the four drugs described here, codeine is the only one for which no evidence from prospective studies exist.
Abacavir
State of evidence
Approved in 1998, abacavir is a potent nucleoside reverse transcriptase inhibitor used to reduce viral load in HIV patients. It is indicated as a first-line therapy in combination with other HIV antivirals including lamivudine and/or zidovudine. Approximately 5–8% of patients will develop hypersensitivity, usually during the first 6 weeks after initiation of therapy [Hetherington et al. 2001]. Patients may experience a range of symptoms including fever, rash, gastrointestinal tract symptoms (abdominal pain, diarrhea, nausea, vomiting), respiratory symptoms (pharyngitis, dyspnea, cough), and potentially life-threatening hypotension. Careful monitoring was required during the initial phase of therapy as no clinical tests were available to identify patients at risk for developing hypersensitivity.
In 2002, Mallal and colleagues reported that three alleles involved in major histocompatibility complex-I antigen presentation were associated with abacavir-induced hypersensitivity [Mallal et al. 2002]. The presence of HLA-B*5701 can lead to restricted CD8+ cytotoxic T-cell activation which results in the secretion of the inflammatory mediators of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gammas [Chessman et al. 2008]. This cascade of inflammatory mediators leads to a delayed type of hypersensitivity reaction [Chessman et al. 2008; Mallal et al. 2002].
The clinical utility of pharmacogenetic screening for HLA- B*5701 was demonstrated in two major studies. The PREDICT-1 study was a large randomized double-blind trial that demonstrated a significantly reduced risk of hypersensitivity in the prescreened arm [Mallal et al. 2008]. The incidence of clinically diagnosed hypersensitivity reaction and immunologically confirmed hypersensitivity reaction was reduced by 56% and 100%, respectively, through HLA-B*5701 screening compared with no screening [Mallal et al. 2008]. The SHAPE study was a retrospective, case-control study, which also demonstrated the sensitivity of the HLA-B*-5701 marker in Black and White populations [Saag et al. 2008].
Revisions to the package insert
In July 2008, based on the outcomes of the PREDICT-1 study [Mallal et al. 2008] and the SHAPE study [Saag et al. 2008], the FDA announced that the package insert for abacavir would be revised to include information about the increased risk of hypersensitivity in patients who are carriers of the HLA-B*5701 allele [FDA, 2008a]. Specifically, it was recommended that HLA-B*5701 testing be performed prior to initiation of abacavir [FDA, 2008b].
Clinical facilitators and barriers: gradual movement toward clinical integration
Reports suggest that the use of pharmacogenetic testing is gradually increasing [Fargher et al. 2007; Faruki et al. 2007; Faruki and Lai-Goldman, 2010; Higgs et al. 2010; Hoop et al. 2010]. In particular, the use of HLA-B*5701 substantially increased following announcement of evidence of clinical utility [Lai, 2008]. However, as with any new clinical application, particularly during this challenging economy, the utility of pharmacogenetic tests has been debated considerably. As demonstrated with the four examples, the number and type of studies conducted vary substantially between the drugs. Warfarin appears to be the most well-studied of the four drugs. As the utility of pharmacogenetic testing for warfarin continues to be debated, larger studies are ongoing. The package insert for warfarin contains specific genotype-based dosing recommendations not included or required for the other drugs. Thus, RCTs are likely to be conducted for drugs for which consensus has not been reached about use of testing, particularly for very common drugs. In contrast, these expensive and lengthy studies are unlikely to be performed for drugs that have more robust data (e.g. abacavir), their side effects more severe (e.g. codeine), or comparable treatment alternatives are available (e.g. codeine or clopidogrel).
Despite the increasing scientific understanding of pharmacogenetics, anticipated benefit and patient interest, which may all drive eventual clinical uptake, two major hurdles for physicians and insurers are the absence of robust data and education/knowledge of pharmacogenetics [Lesko and Johnson, 2012; Schnoll and Shields, 2011]. In addition, other issues pose practical challenges to routine clinical use of pharmacogenetic testing such as test turnaround time (resulting in potential delay of treatment), lack of reimbursement, and lack of clinical guidelines [Lunshof and Gurwitz, 2012; Mrazek and Lerman, 2011; Schnoll and Shields, 2011; Shah, 2004; van Schie et al. 2011]. We discuss in further detail here these and other issues critical to the widespread use of pharmacogenetic testing.
Physicians may choose to avoid the use of drugs that warrant pharmacogenetic testing for various reasons. If alternative drugs are available, such as prasugrel and ticagrelor instead of clopidogrel, which have no known evidence of pharmacogenetics impacting therapy, physicians may simply switch their prescribing practices. However, since clopidogrel is the only generic medication currently available, many physicians may be compelled to prescribe it due to robust data and cost savings. In the case of warfarin, physicians are likely to be very comfortable using the time-tested INR monitoring to achieve the optimal warfarin dose compared with pharmacogenetic testing, despite the expense and inconvenience to patients due to frequent testing during the initial treatment period. Although the data have been conflicting, a recent meta-analysis reported that self-testing reduces the incidence of thromboembolic events, but not hemorrhagic events or death [Heneghan et al. 2012]. For codeine, due to the complex metabolism of the drug and availability of alternative pain medications, it is uncertain whether testing will be routinely used. Without testing, failed attempts at pain management may necessitate treatment with non-CYP2D6 analgesics [Brousseau et al. 2007].
An alternative approach to ordering pharmacogenetic testing per drug at the point of care is the use of preemptive testing, perhaps as part of an annual exam in young adults or even children that require multiple treatments. As a result of the increasing number of drugs with pharmacogenetic data, the preemptive use of testing could significantly optimize drug outcomes [Schildcrout et al. 2012]. Regardless of when ordered (at time of treatment or prior), due to the continuing decline of costs of genomic testing technologies, a broad-based pharmacogenetic screen may yield the greatest cost savings.
One of the primary drivers of test use is insurance coverage. A recent review by one of the authors [Hresko and Haga, 2012] of coverage determinations of top US insurers shows that coverage is generally low, although some insurers are covering pharmacogenetic testing for warfarin and clopidogrel. Large pharmacy benefit managers in the US (Caremark and Medco) offer testing for patients prescribed warfarin [Caremark, 2010; Medco, 2012], further evidence of the perceived importance of testing [SACGHS, 2006].
For the four tests described here, cost-effectiveness studies have not been overly convincing regarding their routine use of some pharmacogenetic tests with the exception of abacavir [Eckman et al. 2009; Hughes et al. 2004; Meckley et al. 2010; Patrick et al. 2009; Perlis et al. 2009; Rosove and Grody, 2009; Wolf et al. 2010]. One important part of cost-effectiveness analysis is the prevalence of the phenotypes with increased risk of poor or no response or adverse effects [Flowers and Veenstra, 2004]. As noted, the prevalence of phenotypes for a given allele can substantially vary between populations. As a result, the risks posed by testing will be weighed against the odds that a given patient is likely to carry an allele associated with poor response or increased risk of adverse events. As most of the alleles follow Hardy–Weinberg distributions, the proportion of patients with intermediate phenotypes will be far more prevalent than the extreme phenotypes (ultra-rapid or poor). However, the evidence basis for patients with the extreme phenotypes is greater, resulting in uncertainty for the larger proportion of patients with the intermediate phenotype.
Several professional medical organizations, federal committees, and experts have released position statements, technology assessments, or guidelines for and against the use of pharmacogenetic testing [De Leon et al. 2006a; Relling and Klein, 2011; Scott, 2011; Zineh et al. 2011]. Other groups including the Clinical Pharmacogenetics Implementation Consortium (CPIC; http://www.pharmgkb.org/page/cpic) and the Dutch Pharmacogenomics Working Group (see http://www.pharmgkb.org/page/dpwg) have developed several documents describing how pharmacogenetic test results should be used [Relling and Klein, 2011]. Of the four tests discussed, CYP2C9/VKORC1 appears to be the most controversial amongst medical organizations [CTAF, 2008; Flockhart et al. 2008; Holbrook et al. 2012]. In general, the statements concluded that while the evidence for analytical and clinical validity of these tests has been met, there is insufficient evidence at this time to recommend for or against routine CYP2C9 and VKORC1 testing in warfarin-naive patients [CMS, 2009; Hirsh et al. 2008]. For clopidogrel, one recent guideline recognized the potential of reduced efficacy of patients with CYP2C19 variants, but concluded that use of testing is uncertain [Jneid et al. 2012]. In contrast, due to its delayed response of the adverse event and strong evidence of clinical validity and utility, there is professional consensus to test all abacavir-naive patients prior to initiation [Aberg et al. 2009; Martin et al. 2012; PAGAA, 2012].
In the early days of pharmacogenetics, there was some lag between the time that package inserts were revised to include pharmacogenetic information following publication of evidence on clinical validity and utility (Figure 2). However, the FDA has responded much more quickly in disseminating information about the impact of genetic polymorphisms on drug safety and response through announcements and revisions to package inserts, likely due to increased familiarity with the science and availability of expert staff to review the data. Of the four examples, warfarin was the first to undergo review and have its package insert revised, 8 years after the initial publication of the association between CYP2C9 and risk of over-anticoagulation. Even though the most studies have been published on the pharmacogenetics of warfarin, the benefits of the test remain highly debatable. In contrast, for clopidogrel, the package insert was changed 4 years after the initial report of an association between CYP2C19 and drug response. For abacavir, the revised package insert happened even more rapidly, the same year the two major studies of clinical utility were published. Likewise, the warnings on codeine were issued relatively quickly following the reported deaths and the in the absence of any clinical trial data.
Other potential factors that may impact physician uptake are availability of FDA-approved tests and test turnaround time. Clinicians may be reluctant to use nonapproved tests (although most genetic tests in the US are laboratory-developed tests and do not currently require FDA approval). The first pharmacogenetic test, the Amplichip CYP450, was approved by the FDA in 2004, testing for 30 common mutations in CYP2D6 and CYP2C19 [de Leon et al. 2006b]. With the increasing number of companion diagnostic tests (developed and approved concurrently with the drug), however, we anticipate that as many tests will be developed in this manner going forward as those developed following drug approval. For the tests described here, many of the major national laboratories in the US (e.g. Labcorp, Quest, ARUP, and Mayo) offer testing. The turnaround time of testing is another important consideration depending on the urgency of treatment and decisions regarding dosing and initiation of therapy must be balanced with risk of adverse response in the absence of testing.
Conclusion
The incorporation of genetic information obtained from pharmacogenetic testing holds substantial promise to improve therapeutic decision making through improved efficacy and reduced adverse events [Liou et al. 2012]. As discussed here, many of the factors essential to the uptake of testing are gradually being addressed, but a major obstacle remains the absence of data demonstrating clinical utility. In addition to ongoing randomized clinical trials to address this data gap, other modes of assessment will need to be employed to more rapidly determine whether testing is useful, particularly for commonly used drugs. Although the routine use of these tests will likely remain slow for some time, this may be beneficial to allow health professionals and patients alike to increase their knowledge of testing and become more comfortable. The use of preemptive testing may address some of the practical barriers in the integration of pharmacogenetic information to avoid delays in treatment. In addition, innovative delivery models may need to be developed to insure the safe and appropriate use of pharmacogenetic testing.
Footnotes
Acknowledgements
We thank Ms. Rachel Mills for her assistance in preparing the manuscript.
Funding
This work was supported by the US National Institutes of Health (grant number5R01-GM081416-05).
Conflict of interest statement
The authors declare no conflict of interest in preparing this article.