
Abstract
Introduction
Drug-induced liver injury (DILI) is a major safety risk for new drugs that are submitted for regulatory approval. This safety concern has been associated with withdrawal or restrictions on use of approved drugs [FDA, 1999a].
A common approach in evaluating a new drug belonging to the same class as a known hepatotoxin is to compare their preapproval safety data. Whether such comparisons improve predictions of hepatic safety and whether such data should be used in applying class warning have not been systemically investigated.
This study assessed drugs within therapeutic classes that included at least one drug either withdrawn from the market because of liver toxicity or with a regulatory warning of liver toxicity. Drugs subject to regulatory action were matched within class with drugs not subject to regulatory action to create two groups. A within-class comparison was undertaken of drugs with and without regulatory action for potential DILI.
Methods
The published literature was systematically searched. The drug-approval packages were obtained from the websites of major regulatory agencies for drugs belonging to therapeutic classes that include at least one drug either withdrawn from the market because of liver toxicity or with a regulatory warning of liver toxicity, and at least one drug free from such regulatory action. The drugs were categorized into two groups: with action (i.e. drugs with regulatory action) or without action.
For the selected drugs, both nonclinical and clinical data were systemically reviewed and abstracted from the published literature and regulatory reports, using liver toxicity as the primary search term. The data included the rates of more than threefold, fivefold, eightfold, 10-fold, or 30-fold elevation of alanine aminotransferase (ALT), the rate of combined elevation of both ALT and bilirubin, the rate of withdrawal from clinical trials due to liver toxicity, the number of patients that developed liver failure in the preapproval clinical trials, the chemical structures and metabolic pathways of the drugs, and the significant findings in nonclinical studies. The data were compared to see if there were markers that explain the differences in the severity of hepatotoxicity.
For proportional data, relative risks (RRs) were calculated, defined as the ratio of the percentage of patients treated with the drug of interest who had ALT elevation divided by the percentage of patients treated with placebo or a comparator drug who had ALT elevation, with the uncertainty expressed using 95% confidence intervals (CIs). To compare the risks of drugs with regulatory action and those without action, the ratio of relative risks (RRR) was calculated to indirectly compare the rate against a control or placebo drug, as described by Bucher and colleagues [Bucher et al. 1997]. If any of the rates equaled 0, risk differences (RDs) and relative risk differences (RRDs) rather than RRs and RRRs were calculated. Data analyses were performed with SAS 9.2 (SAS Institute, Cary, NC, USA).
If a drug had an excessive amount of missing data (e.g. because it had been marketed for a long time and the original clinical trial data were not available in the public sector, the drug was never marketed because of severe hepatotoxicity noted in early drug development), it was excluded from the analysis.
Results
A total of 17 drugs belonging to six classes were identified for this study, including thiazolidinediones for diabetes, cyclooxygenase-2 (COX-2) inhibitors for arthritis, fluoroquinolones for infectious diseases, catechol-O-methyltransferase (COMT) inhibitors for Parkinson’s disease, leukotriene receptor inhibitors for asthma, and endothelin receptor antagonists for pulmonary hypertension (Table 1). Of 17 drugs, eight had been either withdrawn from the market due to liver toxicity or had been associated with liver toxicity. Other hepatotoxins were also identified, such as ibufenac, bromfenac, and benoxaprofen, but these drugs were excluded from the analysis because of lack of data.
Drug selection for the study.

A dash indicates none in the designated category.
COMT, catechol-O-methyltransferase; COX-2, cyclooxygenase-2; NSAID, nonsteroidal anti-inflammatory drug; TZD, thiazolidinedione.
The safety data collected for each of the six classes of drugs are summarized in Tables 2–7. Table 2 presents the safety data for thiazolidinediones [Tolman, 2003; Kostrubsky et al. 2000; FDA, 1999b, 1999c, 1999d, 1999e]. Of the thiazolidinediones, only troglitazone, with a γ- tocopherol side chain, is metabolized to a hepatotoxic quinone. Significant increases in ALT were noted in dogs treated with high doses of pioglitazone and rosiglitazone. In preapproval clinical trials, the rate of elevation of ALT of more than threefold was 1.9% in patients treated with troglitazone versus 0.6% in patients treated with placebo (RR 3.03; 95% CI 0.95–9.68). The rates were 0.33% (RR 0.825) and 0.25% (RR 1.40; 95% CI 0.18–10.79) in patients treated with pioglitazone and rosiglitazone, respectively. The RRRs were 0.27 and 0.46 (95% CI 0.04–4.85) for pioglitazone versus troglitazone and rosiglitazone versus troglitazone, respectively. Although not statistically significant, higher rates of more than fivefold, eightfold, and 10-fold elevations of ALT were also noted in patients treated with troglitazone. In addition, 0.2% of patients treated with troglitazone developed an elevation of ALT of more than 30-fold, which was not reported in any patient taking either pioglitazone or rosiglitazone. Two patients treated with troglitazone developed a combined elevation of ALT and bilirubin, whereas no patient treated with pioglitazone and rosiglitazone developed this clinical abnormality. The rate of withdrawal due to liver toxicity was 2.4% in patients treated with troglitazone, compared with only 0.3% and 0.2% of patients treated with pioglitazone and rosiglitazone, respectively.
Summary of safety data for thiazolidinediones.
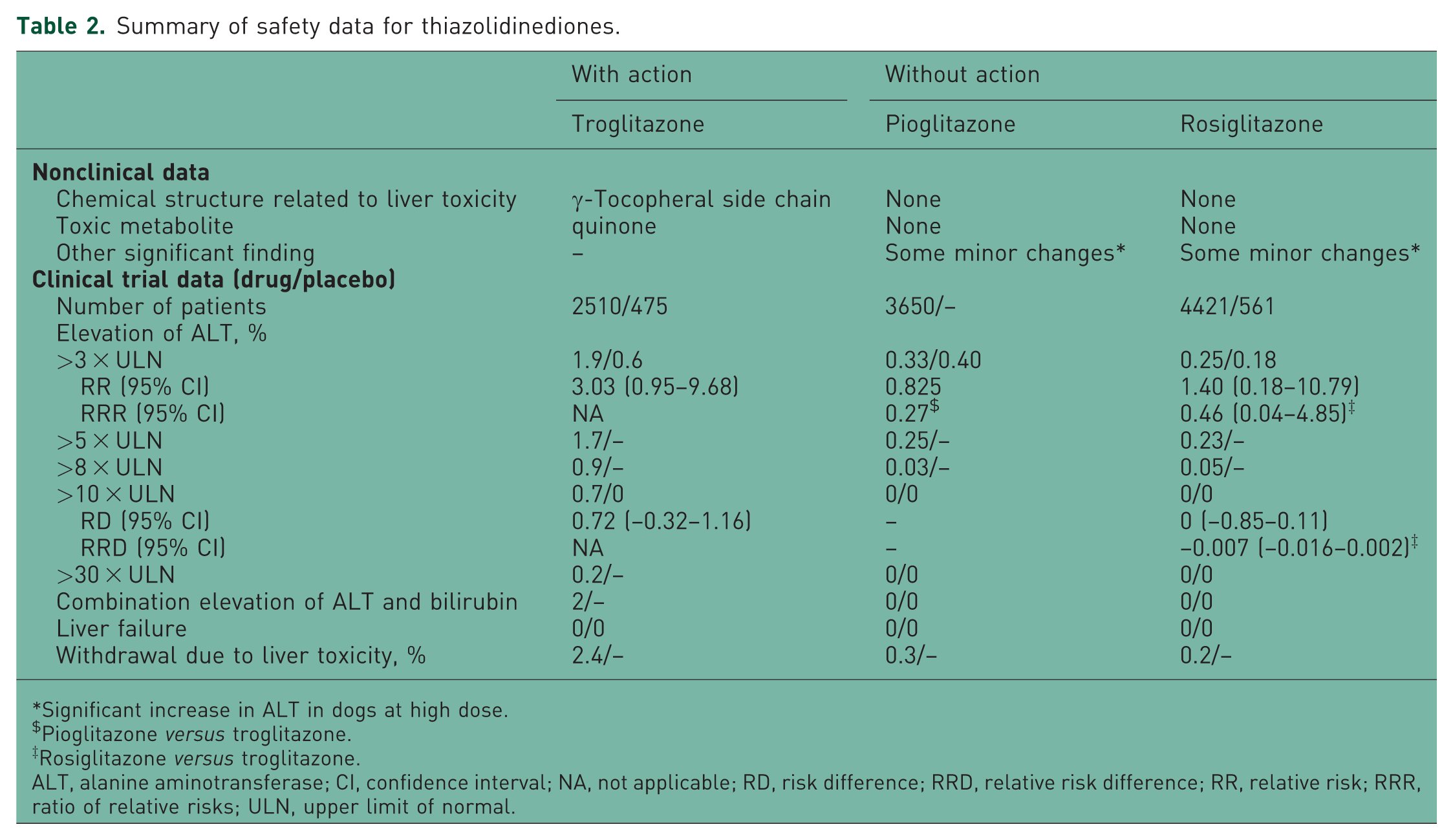
Significant increase in ALT in dogs at high dose.
Pioglitazone versus troglitazone.
Rosiglitazone versus troglitazone.
ALT, alanine aminotransferase; CI, confidence interval; NA, not applicable; RD, risk difference; RRD, relative risk difference; RR, relative risk; RRR, ratio of relative risks; ULN, upper limit of normal.
Summary of safety data for cyclooxygenase-2 inhibitors.
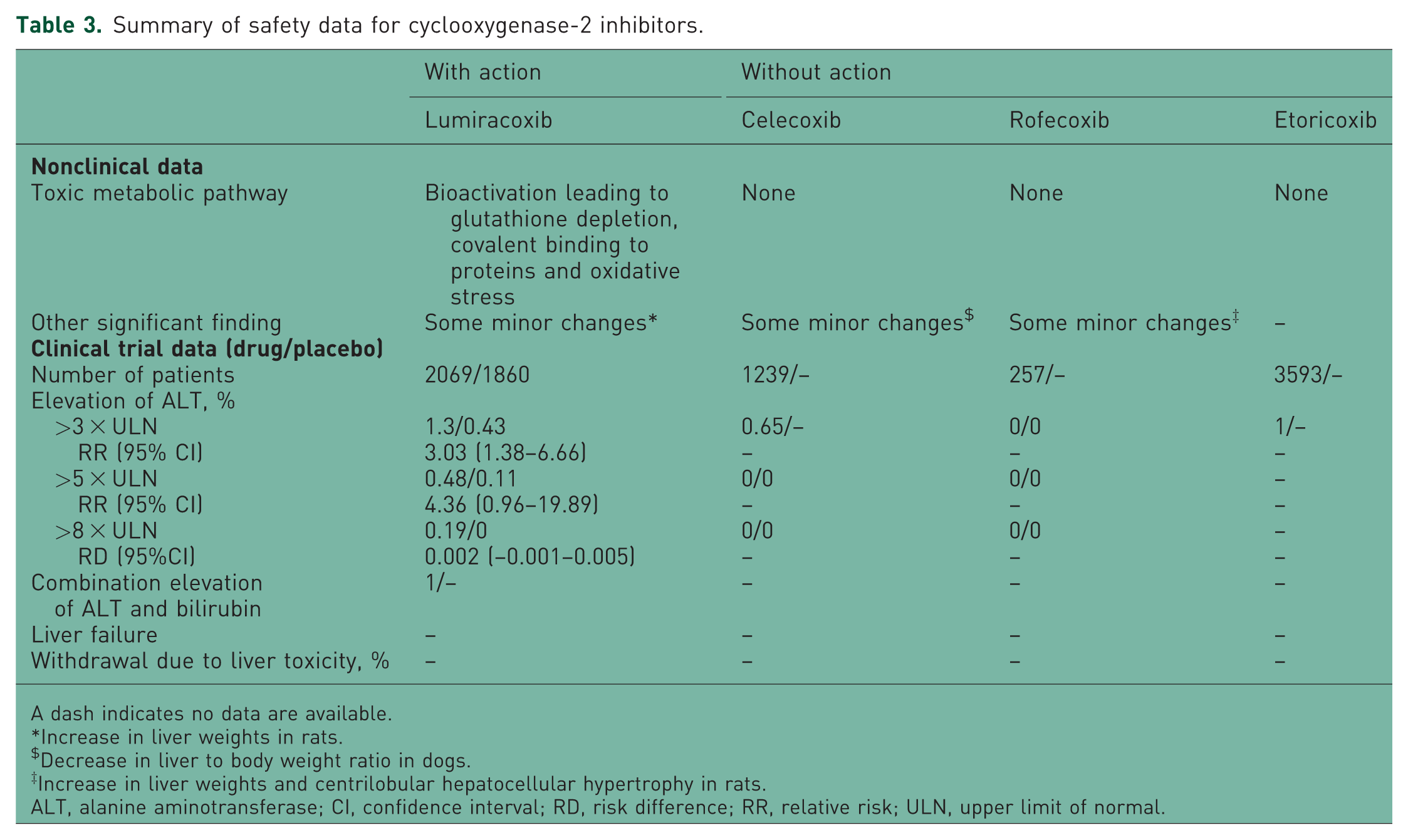
A dash indicates no data are available.
Increase in liver weights in rats.
Decrease in liver to body weight ratio in dogs.
Increase in liver weights and centrilobular hepatocellular hypertrophy in rats.
ALT, alanine aminotransferase; CI, confidence interval; RD, risk difference; RR, relative risk; ULN, upper limit of normal.
Summary of safety data for fluoroquinolones.
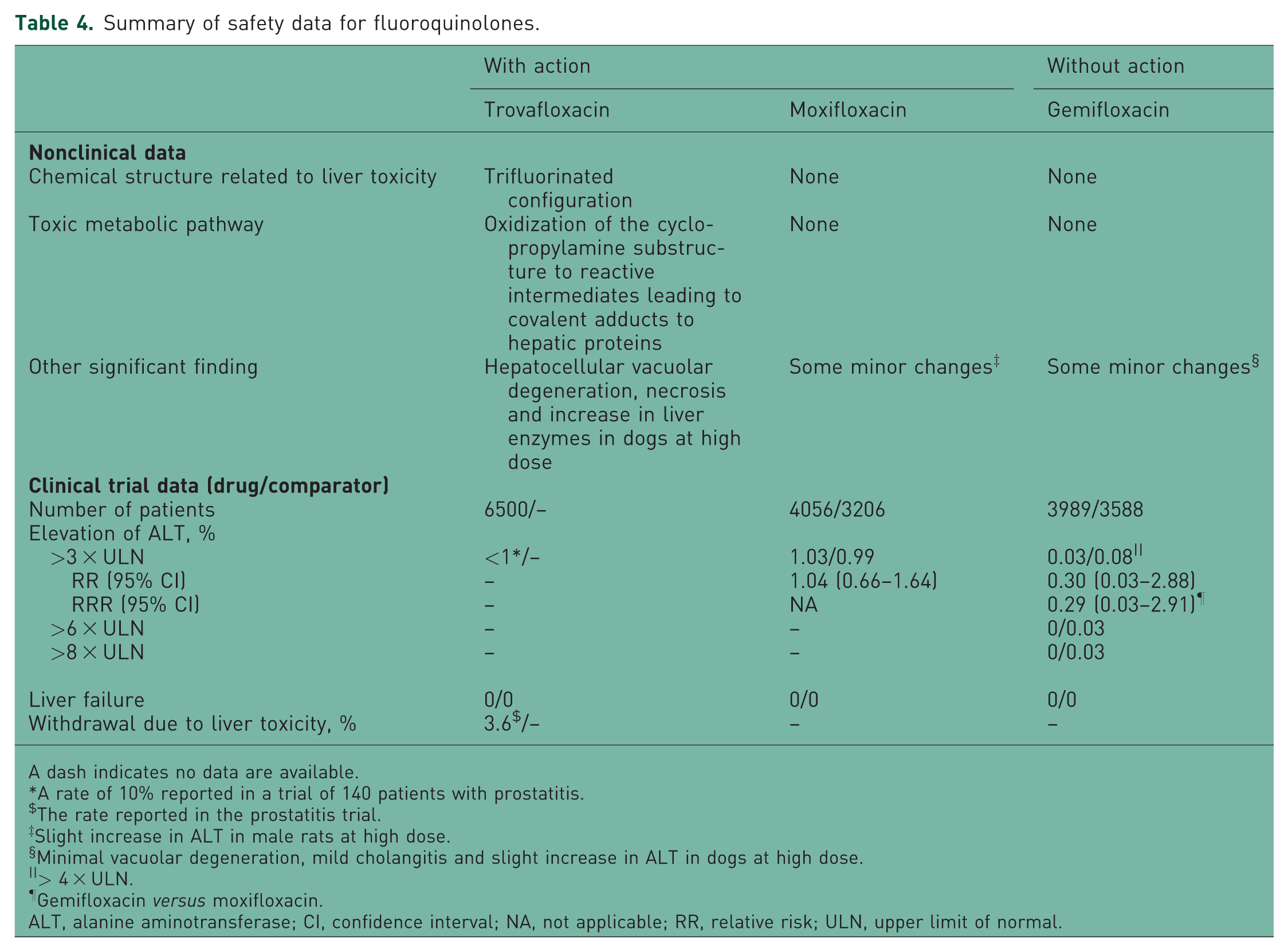
A dash indicates no data are available.
A rate of 10% reported in a trial of 140 patients with prostatitis.
The rate reported in the prostatitis trial.
Slight increase in ALT in male rats at high dose.
Minimal vacuolar degeneration, mild cholangitis and slight increase in ALT in dogs at high dose.
> 4 × ULN.
Gemifloxacin versus moxifloxacin.
ALT, alanine aminotransferase; CI, confidence interval; NA, not applicable; RR, relative risk; ULN, upper limit of normal.
Summary of safety data for catechol-O-methyltransferase inhibitors.

A dash indicates no data are available.
Slight increase in ALT in rats.
Entacapone versus tolcapone.
ALT, alanine aminotransferase; CI, confidence interval; NA, not applicable; RD, risk difference; RRD, relative risk difference; ULN, upper limit of normal.
Summary of safety data for leukotriene receptor inhibitors.

A dash indicates no data are available.
Increase in ALT in monkeys at high dose.
Montelukast versus zafirlukast.
ALT, alanine aminotansferase; CI, confidence interval; NA, not applicable; RR, relative risk; RRR, ratio of relative risks; ULN, upper limit of normal.
Summary of safety data for endothelin receptor antagonists.

A dash indicates no data are available.
Increase in ALT in rats at high dose.
Ambrisentan versus bosentan.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CI, confidence interval; RR, relative risk; NA, not applicable; RRR, ratio of relative risks; ULN, upper limit of normal.
The safety data for COX-2 inhibitors are summarized in Table 3 [Medicines and Healthcare products Regulatory Agency; Bessone 2010; FDA, 1999f, 1998a]. Among them, only lumiacoxib had a toxic bioactivation pathway, which led to glutathione depletion, covalent binding to proteins, and therefore, oxidative stress. In nonclinical studies, minor changes in liver weight and centrilobular hypertrophy were noted in some study animals treated with lumiracoxib, celecoxib, and rofecoxib. In clinical trials, the rate of elevation of ALT of more than threefold in patients treated with lumiracoxib was 1.3% (RR 3.03; 95% CI 1.38–6.66), but it was only 0.65% and 1% in patients treated with celecoxib and etoricoxib, respectively. A total of 0.19% patients treated with lumiracoxib had an eightfold elevation of ALT. In contrast, no patients treated with either celecoxib or rofecoxib had such an elevation. Only one patient in the lumiracoxib group developed a combined elevation of ALT and bilirubin. A similar change was not observed in any patients treated with the other three drugs.
The safety data for fluoroquinolones are summarized in Table 4 [Sun et al. 2008; FDA, 2003a, 2003b, 1999g, 1998b, 1997; O’Donnell and Gelone, 2000; Owens and Ambrose, 2000; Lewis, 2000; Domagala, 1994]. Among them, trovafloxacin has a chemical structure, a trifluorinated configuration, which could account for its toxicity, and contains a cyclopropylamine moiety. This moiety can be oxidized to reactive intermediates that could form covalent adducts to hepatic proteins, resulting in damage to liver tissue. In nonclinical studies, hepatocellular vacuolar degeneration and necrosis were noted in rats treated with trovafloxacin. Only some minor changes in ALT and vacuolar degeneration were noted with moxifloxacin and gemifloxacin. In clinical trials, the rates of elevation of ALT of more than threefold were less than 1% and less than 1.03% (RR 1.04; 95% CI 0.66–1.64) in patients treated with trovafloxacin and moxifloxacin, respectively. In comparison, the rate was 0.03% (RR 0.30; 95% CI 0.03–2.88) in the gemifloxacin group. In a separate study of trovafloxacin, a clinical trial for prostatitis, 14 of 140 patients (10%) developed elevation of ALT of more than threefold after approximately 3–4 weeks of therapy. In that trial, 3.6% of patients discontinued treatment due to increased liver enzymes.
The safety data for COMT inhibitors are summarized in Table 5 [Brooks, 2004; Smith et al. 2003; Tasmar, 2000; Assal et al. 1998; FDA, 1998c, 1998d, 1998e, 1998f, 1996; Nissinen, et al. 1997]. In contrast to entacapone, tolcapone undergoes methylation and oxidization to form amine and acetylamine metabolites that are further metabolized to reactive intermediates that form covalent adducts to hepatic proteins. As a result, damage to liver tissue occurs. In nonclinical studies, a slight increase in ALT was noted with both tolcapone and entacapone. In clinical trials, the rate of elevation of ALT of more than threefold in patients treated with tolcapone was 3.1% (RD 0.03; 95% CI 0.02–0.05). In contrast, only 0.5% (RD –0.018; 95% CI –0.022–0.014) of patients taking entacapone experienced such an elevation in ALT. The RRD was –0.05 (95% CI –0.07 to –0.02) for entacapone versus tolcapone. A total of 1.7% (RD 0.02; 95% CI 0.01–0.03) of patients receiving tolcapone discontinued treatment due to liver toxicity. No patients receiving entacapone discontinued treatment during the trial.
Leukotriene receptor antagonist safety data are presented in Table 6 [Kassahun et al. 2005; Wooltorton, 2004; FDA, 1999h, 1998g]. Zafirlukast is metabolized to a toxic electrophilic alpha, beta-unsaturated iminium intermediate. In nonclinical studies, minor changes in ALT were noted after montelukast administration. In clinical trials, the rate of elevation of ALT of more than threefold was 1.2% (RR 2.22; 95% CI 1.38–3.57) in patients treated with zafirlukast and 0.5 % (RR 1.51; 95% CI 0.47–4.81) in patients treated with montelukast. The RRR was 0.68 (95% CI 0.19–2.38) for montelukast versus zafirlukast. The rates of an elevation of ALT of more than fivefold were 0.46% (RR 2.46; 95% CI 1.11–5.45) and 0.2% (RR 2.42; 95% CI 0.27–21.6), with a RRR of 0.98 (95% CI 0.10–10.13).
Endothelin receptor antagonist safety data are summarized in Table 7 [Letaris product label, 2008; Leslie et al. 2007; Tracleer, 2007; FDA, 2007, 1998h; Barst et al. 2002; Barst et al. 2006; European Medicines Agency-a, b]. Among them, bosentan and sitaxentan significantly inhibit bile salt export pump and organic anion transport, respectively, which are considered the causes for the observed severe toxicity [Hartman et al. 2010; McRae et al. 2006]. In nonclinical studies, bosentan was associated with an increase in ALT and hepatocyte degeneration in rats. Centrilobular hypertrophy (and occasional necrosis), an increase in prothrombin time, and coagulopathy were observed following the administration of sitaxentan. Only minor changes in ALT were noted with ambrisentan. The rates of elevation of ALT of more than threefold were 11% (RR 5.11; 95% CI 2.25–11.61) and 7% (RR 1.4) in patients treated with bosentan and sitaxentan, respectively. The same rate was only 0.8% (RR 0.36; 95% CI 0.08–1.61) for patients treated with ambrisentan. The RRR was 0.07 (95% CI 0.01–0.39) for ambrisentan versus bosentan. In addition, two patients treated with bosentan and seven patients treated with sitaxentan developed a combined elevation of ALT and bilirubin, but only one patient treated with ambrisentan had an elevation of both ALT and bilirubin.
Variables of interest were assessed to determine if it was possible to distinguish between drugs with regulatory action and those without regulatory action. If the variable demonstrated a statistically significant difference between the two groups (i.e. the upper limit of the CI of RRR was <1 or RRD was <0) or the variable was statistically different for the hepatotoxic drug compared with the placebo (i.e. the lower limit of the CI of RR was >1 or RD was >0), and no difference between the nonhepatotoxic drug and the placebo was noted (i.e. the lower limit of the CI of RR was <1 or RD was <0), the variable was considered ‘able’ to make a distinction. Otherwise, the variable in question was considered ‘unable’ to make a distinction. As summarized in Table 8, the rate of elevation of ALT of more than threefold could distinguish between drugs with and without regulatory action for four of eight drugs. The rate of withdrawal due to liver toxicity and the number of patients developing a combined elevation of both ALT and bilirubin could also distinguish drugs with and without regulatory action for three drugs. The other variables could not be used to make a distinction or had insufficient data to draw any conclusions.
Variables assessed to determine if it was possible to distinguish between drugs with regulatory action and those without regulatory action.
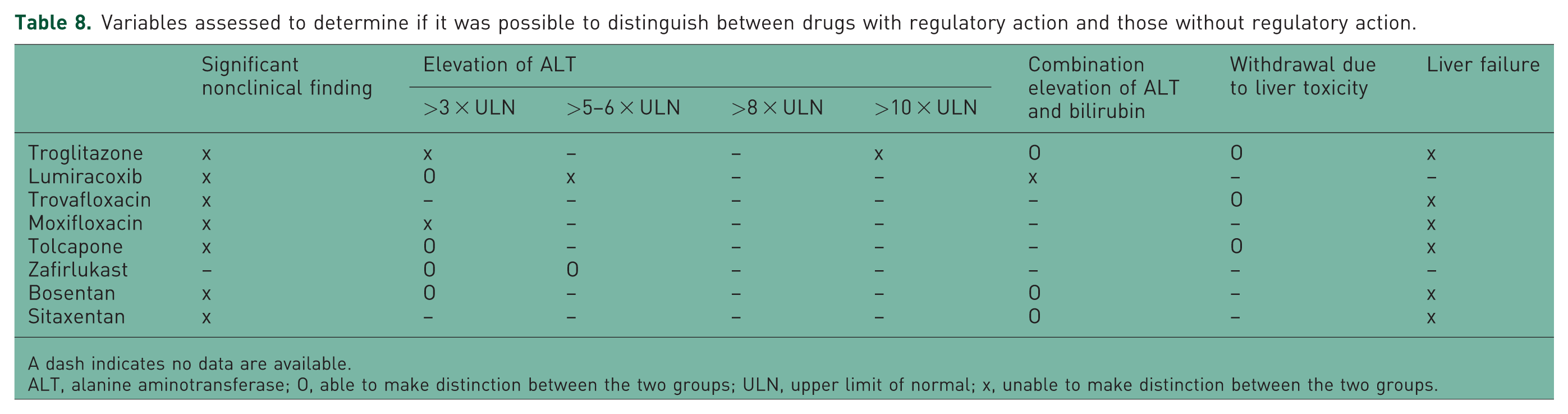
A dash indicates no data are available.
ALT, alanine aminotransferase; O, able to make distinction between the two groups; ULN, upper limit of normal; x, unable to make distinction between the two groups.
Discussion
In this study, the authors undertook a within-class comparison of preapproval data for drugs with and without regulatory action for hepatotoxicity to examine whether postapproval hepatic safety could be better predicted. McBurney previously compared drug pairs with similar chemical structures and therapeutic mechanisms to produce biomarker sets that predict the liability of a compound for generating liver toxicity [McBurney, 2010]. In this study, the authors selected a number of variables for comparison that are widely used to evaluate hepatic safety (e.g. rates of elevation of ALT, numbers of patients developing a combined elevation of ALT and bilirubin, numbers of patients with liver failure noted in clinical trials). Of note, the number of patients developing combined elevations of ALT and bilirubin is highly predictive of severe hepatotoxicity and is used by regulatory agencies to identify drugs likely to cause severe liver injury [Andrade et al. 2005; Björsson and Olsson, 2005; FDA, 1999a]. To minimize bias arising from comparisons of pooled data from multiple studies, a statistical method that indirectly compared the data was employed to preserve the randomization of the originally assigned patient groups.
As noted in the results, the rates of elevation of ALT were different for drugs with regulatory action compared with those without action. For example, the rates of elevation of ALT of more than threefold for all of the drugs without regulatory action were not different from the rates of either the placebo or comparator drugs (i.e. the CIs of the RR included 1 or the RD included 0). Four of the eight drugs with regulatory action (lumiracoxib, tolcapone, zafirlukast, and bosentan) had significantly higher rates of elevation of ALT of more than threefold than the placebo (i.e. the lower limit of the CI of RR was >1 or RD was >0). In two drug classes, the COMT inhibitors and the endothelin receptor antagonists, the risks of entacapone and ambrisentan were significantly lower than for tolcapone and bosentan, respectively (i.e. the upper limit of the CI of RRR was <1 or RRD was <0). This finding indicates that the rate of elevation of ALT of more than threefold can be used to predict postapproval hepatic safety. The available data for fivefold, eightfold, 10-fold, and 30-fold elevations of ALT were limited, although the rates among drugs with regulatory action appeared to be higher than those without regulatory action; however, the differences were not significant.
The rates for combined elevation of ALT and bilirubin were different for drugs with regulatory action compared with those without. Of the nine drugs without regulatory action, only one patient treated with ambrisentan developed a combined increase in ALT and bilirubin. Of the eight drugs with regulatory action, four of these drugs had a combined increase in ALT and bilirubin. These drugs were troglitazone (two patients), lumiracoxib (one patient), bosentan (two patients) and sitaxentan (seven patients). This variable clearly differentiates hepatotoxic drugs from those that are not hepatotoxic, and supports the rule described by the US Food and Drug Adminstration that ‘Finding one case in the clinical trial database is worrisome. Finding two is considered highly predictive that the drug has the potential to cause severe DILI’ [FDA, 1999a p. 5].
Based on the available nonclinical data, the authors were unable to identify any factors that could help distinguish drugs with regulatory action from those without regulatory action. Even in the two drug classes that had some differences in the nonclinical findings (i.e. the class of fluoroquinolones in which hepatocellular vacuolar degeneration, necrosis, and an increase in liver enzymes were noted in dogs treated with troglitazone, but only slight increases in ALT, minimal vacuolar degeneration, and mild cholangitis were noted with moxifloxacin and gemifloxacin; and the class of endothelin receptor antagonists in which centrilobular hypertrophy, occasional necrosis, an increase in prothrombin time, and coagulopathy were noted in dogs treated with sitaxentan, but only mild increases in ALT were noted with ambrisentan) the significance of those findings was obviously not emphasized based on the reviews that recommended approval of these drugs. In addition to the conclusion that there is still room for improvement in the interpretation and use of nonclinical data toward a better correlation with clinical data, this finding reflects the fact that preapproval prediction of postapproval safety is a difficult job and retrospective analysis is much easier.
The study results also demonstrate that preapproval data would have correctly identified the lack of hepatotoxicity for nine drugs without regulatory action. They present fairly compelling evidence that preapproval data facilitates decision making about hepatotoxicity without resorting to class warning. Based on the study findings, it appears that the legitimacy of applying class warnings was refuted in all six classes of drugs included in this study. Therefore, preapproval class warnings may not be justified, and new additions to a drug class should not be labeled as potentially hepatotoxic at the time of approval simply because one or two previously approved drugs in the same class have been classified as hepatotoxic.
This study suggests that preapproval safety data of new drugs belonging to a drug class with an already recognized hepatotoxin can be used by systematically reviewing the nonclinical and clinical data, including chemical structure, metabolic pathways and metabolites, positive findings in animal studies, and other factors such as genetics (if available) potentially related to toxicity presentation [Shah, 1999; Daly et al. 2009]. The clinical data can then be compared quantitatively (e.g. by comparing the rate of ALT elevation and other frequently used parameters and further comparing the RR between the new drug and the hepatotoxin). Finally, all of this information can be evaluated as a whole to judge whether there is any difference in hepatotoxic potential and whether there is any need to apply class warnings.
Like all studies, this study has a number of limitations. The first is that only six classes of drugs were included in the study and the number of drugs in each class was very small. Several other drugs were excluded from the study because the available data in the public sector were inadequate for analysis. These limitations could limit the general application of the study findings. Second, clinical trials are limited in their ability to predict safety because they are highly efficacy oriented. The study populations are highly selected populations that could differ from patient populations that would be included in uncontrolled, widespread clinical use. The sample sizes in these trials are often limited, and patients potentially at risk may drop out early for other reasons [Shah, 1999]. Third, this study does not evaluate the effect of observing severe hepatotoxicity in preapproval clinical trials because it is likely that such drugs would not be developed any further, would not reach regulatory submission, and would not be used clinically. Under such circumstances, the only possible outcome of a preapproval versus postapproval comparison for hepatotoxicity is that an alleged nonhepatoxic drug turns out to be hepatotoxic in the postapproval period. Fourth, this study did not evaluate other markers of hepatotoxicity such as genetic factors (e.g. the strong association between flucloxacillin-induced liver injury and the class I allele B*5701), chirality, and duration of drug exposure [Shah, 1999; Daly et al. 2009]. The differences in the severity of toxicity could therefore potentially arise from differences in factors other than those examined in this study.
Conclusions
Preapproval safety data may help predict postapproval hepatic safety and can be used to assess the legitimacy of applying class warnings.
Footnotes
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
We declare that we have no conflict of interest.