
Abstract
Metastasis is a leading cause of mortality in breast cancer and is critically influenced by cell–extracellular matrix (ECM) interactions, mechanical forces, and cellular motility. In this study, we present a cell surface engineering approach using tris(2-carboxyethyl)phosphine (TCEP), a biocompatible thiol-modifying agent, to modulate the biomechanical behavior of breast cancer cells. TCEP treatment increased surface thiol availability, enhanced phosphorylation of focal adhesion kinase (FAK), and promoted the elongation of pFAK-positive focal adhesions, along with cytoskeletal remodeling and stronger cell–ECM adhesion. These molecular and structural changes corresponded with significantly reduced migration and invasion of MCF7 and MDA-MB-231 cells. Using traction force microscopy (TFM), we further observed increased intracellular tension and traction stress, providing quantitative insight into how surface modification regulates mechanotransduction. These findings highlight the potential of cell surface thiol engineering to control cancer cell adhesion and motility, providing a platform for future identification of clinically applicable redox-modulating agents.

Keywords
Introduction
Breast cancer remains one of the most prevalent malignancies worldwide, with approximately 2.3 million new cases reported in 2020, making it the most frequently diagnosed cancer globally. 1 Despite significant advances in early detection and therapeutic interventions, breast cancer continues to be the leading cause of cancer-related mortality, primarily due to metastasis. 2 Metastasis is a multistep cascade involving cancer cell detachment from the primary tumor, migration through the extracellular matrix (ECM), and invasion into distant tissues.3,4 These processes are tightly regulated by cell–ECM interactions, which are orchestrated by focal adhesion complexes, integrin-mediated signaling, and cytoskeletal remodeling. 5
To prevent metastatic progression, diverse therapeutic strategies have been explored, including the inhibition of epithelial–mesenchymal transition (EMT) and the suppression of integrin-mediated adhesion. 6 More recently, increasing attention has been directed toward the mechanical properties of the tumor microenvironment—particularly ECM stiffness—and the mechanotransduction pathways that regulate cellular responses to mechanical cues.7–9 Notably, strategies that enhance cell–ECM adhesion and modulate mechanotransduction signaling have emerged as promising approaches to limit cancer cell motility and invasiveness, even within pathologically stiff microenvironments. 10 Similarly, mechanical cues not only influence tumor progression but also play critical roles in physiological processes such as tissue regeneration, where mechanotransduction governs cellular remodeling and functional recovery. 11
The cell membrane, acting as a critical interface between the ECM and the intracellular cytoskeleton, presents a tractable target for modulating adhesion-related signaling. 12 Membrane proteins such as integrins are stabilized by disulfide bonds (S–S), which can be selectively cleaved to expose reactive thiol (–SH) groups.13–15 These disulfide bonds are essential for maintaining protein conformation and redox homeostasis, and their dysregulation is frequently implicated in cancer progression.16,17 Moreover, increased surface thiol content enhances the binding capacity of cells to functional groups like maleimide, thereby enabling a range of bioengineering applications including cell patterning, drug delivery, tissue fabrication, and biosensing.18–20
Consistent with this, several studies have reported that cancer cells exhibit elevated levels of cell surface thiol groups compared to their normal counterparts, likely due to increased reduction of disulfide bonds. This elevated thiol-disulfide exchange, partly mediated by protein disulfide isomerase (PDI), has been linked to integrin activation, endothelial adhesion, and transendothelial migration in cancer cells.21,22 These findings highlight that redox regulation at the cell–matrix interface is not only altered in cancer but also contributes to its invasive phenotypes. In glioma cells, treatment with tris(2-carboxyethyl)phosphine (TCEP) has been shown to enhance focal adhesion formation and inhibit migration and invasion by strengthening cell–matrix interactions. 23 Similarly, in human mesenchymal stem cells, TCEP-mediated surface thiol generation has been reported to activate integrin β1/focal adhesion kinase (FAK) signaling, promoting cell adhesion, spreading, and early osteogenic differentiation. 24 However, the effects of TCEP-induced thiol modification on adhesion dynamics and mechanotransduction in breast cancer cells remain largely unexplored.
In the present study, we employed TCEP as a model chemical reductant to transiently modulate the redox state of cell-surface disulfide bonds, thereby increasing cell surface thiol exposure. This strategy enabled us to systematically investigate how redox modulation affects adhesion dynamics, focal adhesion signaling, traction force generation, and migratory behavior in breast cancer cells. Given that FAK signaling and cytoskeletal tension are central to cancer cell migration, understanding how surface thiol modification affects these mechanobiological pathways is crucial for elucidating the role of redox regulation in tumor invasion.
To this end, we utilized two breast cancer cell lines, MCF7 and MDA-MB-231, which represent low and high metastatic potential, respectively. We examined the effects of TCEP-induced thiol modification on FAK activation, traction force generation, and cell motility within physiologically relevant, stiffness-tuned substrates. Importantly, this study does not aim to evaluate TCEP as a therapeutic agent, but rather as a mechanistic tool to probe how surface redox regulation modulates cell–matrix interactions and migration-associated mechanotransduction.
Materials and methods
Cell lines and modification of cell surface with TCEP
MCF7 and MDA-MB-231 human breast cancer cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF7 cells were cultured in RPMI-1640 (Cat# 10-040-CV, Corning, NY, USA), while MDA-MB-231 cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Cat#10-01-CV, Corning) supplemented with 10%(v/v) fetal bovine serum (FBS; Cat#35-015-CV, Corning) and 1% (v/v) penicillin-streptomycin solution (Cat# 15140122, Gibco, Auckland, New Zealand). Cells were incubated at 37°C in a humidified atmosphere with 5% CO₂.
For surface thiol group formation, MCF7 and MDA-MB-231 cells were detached, counted, and resuspended at a density of 1 × 106 cells/mL in sterile phosphate-buffered saline (PBS, pH 7.4) containing 2 mM TCEP (Cat# C4706, Sigma-Aldrich, St. Louis, MO, USA). Cell suspensions were incubated for 5 min at room temperature (RT) in the dark. After treatment, samples were centrifuged at 1200 × rpm for 3 min, the supernatant was removed, and cell pellets were washed once with 1 mL sterilized PBS to remove residual TCEP. Control samples were processed in parallel but incubated in PBS without TCEP. Washed cells were then re-suspended in the appropriate buffer or medium and used for thiol-labeling assays or downstream motility experiments.
Cell viability test
Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT; Cat# M6494, Invitrogen, Carlsbad, CA, USA). A total 2 × 105 cells/cm2 were seeded in 96-well plates and incubated for 24 h. Cells were then treated with TCEP for 5 min, followed by a 2 h incubation with MTT working solution prepared by a 1:10 dilution of a 12 mM stock solution in fresh growth medium. After incubation, dimethyl sulfoxide (DMSO; Cat# D8418-50ML, Sigma-Aldrich, St. Louis, MO, USA) was added to dissolve the formazan crystals, absorbance was measured at 540 nm using a microplate reader (BioTek, Winooski, VT, USA).
Additionally, cell viability was confirmed using the Live/Dead™ Viability/Cytotoxicity Kit (Invitrogen, Cat# 3L-3224, Carlsbad, CA, USA), applied to confluent cultures (5 × 10⁴ cells/cm²) according to the manufacturer’s protocol.
Quantification and visualization of thiol group on the cell surface
Cell surface thiol content was quantified using 5,5′-dithio-bis-(2-nitrobenzoic acid) (DNMT; Cat# 22582, Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Briefly, cell pellets were incubated with 10 mM DNMT in the dark for 15 min. L-cysteine (Cat# C7352, Sigma-Aldrich, St. Louis, MO, USA) was used to generate a standard curve. Absorbance was measured at 412 nm using a microplate reader (BioTek). The thiol content was normalized to cell number by dividing the optical density (OD) values by the corresponding cell count and expressed as relative fold change compared to the untreated control group.
To visualize free thiol groups on the cell surface, 3 μg/mL Alexa Fluor® 488 C5 Maleimide (Mal-488, Cat# A10254, Thermo Fisher Scientific, Waltham, MA, USA) was prepared in PBS and mixed with the TCEP-treated cell pellet. The suspended cells were then incubated in the dark for 20 min at RT. After labeling, the cells were washed with PBS, and green fluorescence was imaged using a scanning confocal microscope (LSM 710; Carl Zeiss, Oberkochen, Germany) at the Soonchunhyang Biomedical Research Core Facility of the Korea Basic Science Institute. Z-stack images were captured 50–70 slices at 30 μm intervals across the entire cell using scanning confocal microscopy. The images were visualized as 2D cross-sections and 3D reconstruction images using Image J.
Lyn-FAK FRET biosensing
The Lyn-FAK biosensor (Addgene plasmid #78299) was subcloned into the pAd/CMV/V5-DEST™ Gateway™ vector (Thermo Fisher Scientific, Cat# V49320) using the manufacturer’s protocol, generating the pAd_CMV_Lyn-FAK biosensor construct. For adenoviral production, the recombinant vector was sent to a commercial service provider (VectorBuilder, Guangzhou, China), where adenoviral packaging and amplification were carried out according to standardized protocols. The resulting adenoviral particles (1 × 10¹⁰ PFU/mL) were used to infect MDA-MB-231 or MCF7 cells at a multiplicity of infection (MOI) of 100.
Cells transduced with adenovirus encoding the Lyn-FAK FRET biosensor were detached using trypsin and incubated in suspension for 5 min in either PBS (control) or PBS containing TCEP. Following treatment, the cells were seeded onto confocal imaging dishes and allowed to attach for approximately 1 h. After this attachment period, non-adherent cells were removed by gentle washing with PBS, and adherent cells were supplied with growth medium. During live-cell imaging, cells were maintained at 37°C in a stage-top chamber equilibrated with humidified air containing 5% CO2.
Image acquisition was performed on a Leica DMi8 inverted fluorescence microscope equipped with a 40× objective lens (NA = 1.25, glycerol immersion; HC PL APO Leica, Cat# 11506422). FRET images (1100 × 1600 pixels) were acquired for ECFP (478 nm) and YPet (527 nm) channels, with a pixel size of 0.223 μm/pixel.
In vitro wound healing migration assay
In vitro wound healing assays were performed using silicone migration mold/insert (Ibidi, Cat# 80206, Gräfelfing, Germany) to generate a defined gap between two cell populations. MCF7 and MDA-MB-231 cells were seeded at a density of 5.4 × 10⁵ cells/cm² on each side of the mold in a complete growth medium containing FBS. After 18 h of incubation, upon reaching confluence, the inserts were carefully removed. Non-adherent and dead cells were removed by PBS washing, and fresh growth medium was added. Phase-contrast images of the wound area were acquired every 8 h using the EVOS™ M7000 Imaging System (Invitrogen, Carlsbad, CA, USA). The total wound area was calculated using a custom MATLAB script (R2020b, MathWorks, Natick, MA, USA), and the percentage of wound closure was determined as follows 25 :
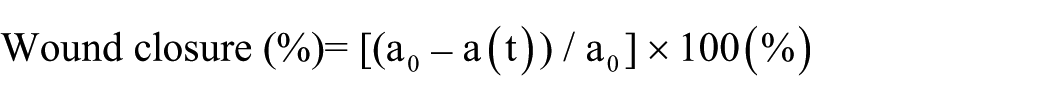
where a0 is the wound area at time zero, and a(t) is the area at a given time point.
Transwell migration and invasion assays
Transwell migration and invasion assays were performed using 8.0-μm pore size polycarbonate membrane inserts (Cat# 3422, Corning, NY, USA). For the migration assay, 3 × 10⁴ cells suspended in serum-free media and seeded into the upper chamber of each insert. The lower chamber was filled with a complete growth medium with FBS to establish a chemoattractive gradient that facilitated cell migration to the underside of the membrane. Cells were incubated at 37°C with 5% CO₂ for 24 h. After incubation, cells were fixed with 4% paraformaldehyde (Cat# CNP015-0500, Cell Nest, Jiansu, China) for 10 min and washed with PBS. Migrated cells on the underside of the membrane was stained with 0.1% crystal violet (Cat#C0775, Sigma-Aldrich) in 10% ethanol for 15 min, followed by gentle washing with deionized water. The inner surface of the upper chamber was carefully wiped with a cotton swab to remove non-migrated cells. Images of stained cells were acquired using an EVOS fluorescence microscope (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a 20× objective lens (NA = 0.45). Cell coverage on the membrane was quantified using Image J software and expressed as a percentage of the total membrane area.
For the invasion assay, 20 μL of Matrigel (Cat# 356231, Corning, NY, USA) diluted 1:8 in serum-free medium was added to the upper surface of the insert and allowed to polymerize. Cells were prepared as described for the migration assay, except they were suspended in media containing 2% FBS prior to seeding into the Matrigel-coated inserts. The assay was conducted under identical incubation conditions as the migration assay and staining and imaging were carried out using the same protocol.
Western blot analysis
For immunoblotting analysis, TCEP-treated MCF7 and MDA-MB-231 cells were seeded at a density of 5 × 104 cells/cm2 and cultured for 24 h. Cells were lysed using RIPA buffer (Cat# eba11491, Elpis-Biotech, Daejeon, South Korea) supplemented with phosphatase inhibitor (Cat# 4906837001, Sigma-Aldrich, St. Louis, MO, USA) and protease inhibitor (Cat# 5892970001, Sigma-Aldrich, St. Louis, MO, USA). Protein quantification was performed using the BCA protein assay (Cat# 23225, Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein (20 μg per lane) were separated on 8%–10% sodium dodecyl sulfate-polyacrylamide gels and transferred onto nitrocellulose membrane (Cat# 1620112, Bio-Rad, Hercules, CA, USA) using the Trans-Blot® Turbo™ Transfer System (Cat# 1704150, Bio-Rad). The membrane was blocked with 5% skim milk for 1 h at RT and then incubated with primary antibodies overnight at 4°C. This was followed by incubation with either a goat anti-mouse IgG (H+L) secondary antibody HRP or goat anti-rabbit IgG (H+L) HRP conjugate for 1 h at RT. The blots were developed using the Amersham ECL Prime Detection Reagent Kit (Cat# RPN2232, Cytiva, Marlborough, MA, USA) and visualized using a Gel Image Analyzer (Amersham Imager 600, GE Healthcare, Buckinghamshire, UK). The following antibodies were used: phospho-FAK, FAK, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The specific antibody information, including dilution factors and catalog number, is provided in Supplemental Table S1.
Immunofluorescence staining of cells and focal adhesions
TCEP-treated MCF7 and MDA-MB-231 cells were seeded on glass-bottom confocal dishes at a density of 5.3 × 103 cells/cm2. Two sets of experiments were performed to capture both early adhesion events and stabilized adhesion states. For the early adhesion kinetics study, cells were fixed and stained at 0.5, 1, and 2 h after seeding. For the long-term adhesion study, cells were incubated for 24 h at 37°C with 5% CO₂ prior to fixation and staining. After incubation, cells were washed with PBS and fixed with 4% paraformaldehyde at room temperature for 15 min, followed by three washes with PBS. For permeabilization and blocking, cells were incubated with a blocking buffer containing 3% BSA and 0.1% Triton X-100 in PBS for 1 h at RT. Subsequently, cells were incubated overnight at 4°C with primary antibody against p-FAK and F-actin. After primary antibody incubation, the cells were washed three times with PBS for 10 min per wash. After that, the cells were incubated with the appropriate fluorophore-conjugated secondary antibody for 1 h at RT. Hoechst 33342 was used for nuclear staining. Detailed antibody information, including dilution factors and catalog numbers, is provided in Supplemental Table S2.
Quantitative analysis of cell morphology, focal adhesions, and cytoskeletal organization
Quantitative image analysis of cell morphology, focal adhesion (FA) features, and F-actin fluorescence intensity was performed using custom MATLAB-coded scripts developed in-house, based on previously validated protocols.26–28 Confocal fluorescence imaging was performed using a laser scanning confocal microscope (LSM-710; Carl Zeiss, Oberkochen, Germany) equipped with a 40× oil immersion objective lens, yielding a pixel size of 0.20 µm/pixel. Focal adhesion markers (phosphorylated FAK, pFAK) and F-actin cytoskeletal structures were imaged under identical acquisition settings across all conditions.
Image analysis was conducted using MATLAB (R2020b; MathWorks, Natick, MA, USA). To minimize user bias, segmentation was automated using an Otsu histogram-based thresholding algorithm. 29 Morphological descriptors including cell area, length, and circularity were extracted from binarized cell masks. All analyses were performed in a blinded manner with uniform image processing parameters. For each condition, three biological replicates were analyzed, with 10–12 randomly selected fields per replicate and approximately 50–150 cells per replicate, yielding a total of ~120–585 cells per condition. Detailed sample sizes are provided in the respective figure legends.
Focal adhesion regions were segmented by removing background noise (lowest 0.5% pixel intensities) followed by a fixed threshold set at ~5% of the maximum intensity. Regions between 1.5–30 µm² were retained to exclude small noise artifacts and large aggregates. For each segmented FA, quantitative parameters including length, circularity, number of adhesions per cell, total intensity (sum of pixel values), and mean intensity (normalized to area, a.u./µm²) were computed. Only the normalized mean intensity was used for statistical comparisons.
F-actin fluorescence intensity was quantified using two parallel approaches: within manually traced cell boundaries and within automatically segmented cell areas from binarized images. In both cases, total F-actin intensity was normalized to the corresponding cell area (µm²) to account for differences in cell spreading. Circularity, which reflects the roundness of the object was defined by the squared ratio of perimeter of perfect circle to cell perimeter of the same area:

where: A = area and P = perimeter length.
All acquisition settings, thresholding criteria, and post-processing protocols were applied consistently across all experimental groups to ensure reproducibility and eliminate batch effects.
Quantification of FRET biosensing
Quantification of FRET was performed following a previously described method, unless otherwise noted. 30 Fluorescence images of ECFP (478 nm) and YPet (527 nm) were acquired as described in the biosensing section (Section 2.4). To reduce background noise, the lowest 1% of pixel intensities, as determined from the image intensity histogram, was removed from from both the ECFP and YPet images. FRET ratio images were then calculated as ECFP (478 nm)/YPet (527 nm) using a custom Python script. The mean FRET ratio was calculated within the cell boundaries, which were defined in the image processing section (Section 2.9). To avoid division artifacts, pixels with zero intensity in either the ECFP or YPet channel were excluded from the analysis.
Single cell migration trajectory analysis
To represent the pathological stiffness of the tumor microenvironment, polyacrylamide (PAA) gels (Young’s modulus = 19.66 kPa, AA/Bis = 8%/0.26%) were cast on the surface of glass-bottom dishes (Cat# 101350, SPL, Pocheon, Korea). The Young’s modulus of PAA was assessed by the Instron device (EZ-SX, SHIMADZU, Kyoto, Japan). In brief, the glass-bottom dish was made hydrophilic by passing the glass surface through the flame of a Bunsen burner, followed by the application of sodium hydroxide (0.1 M; Cat# 39155S0350, Junsei, Tokyo, Japan). The glass was then activated using trichloro(1H,1H,2H,2H-perfluorooctyl) silane (silane; Cat# 448931-10G, Sigma-Aldrich, St. Louis, MO, USA) and glutaraldehyde (0.5%; Cat# G5882, Sigma-Aldrich). Polymerization was initiated by adding ammonium persulfate (0.6%; Cat# AMP001, BioShop, ONT, Canada) and N,N,N′,N′-tetramethylethylenediamine (0.4%; Cat# TEM001, BioShop, ONT, Canada), and the gels were cast by sandwiching on activated glass with an inactive coverslip of 18 mm diameter (Cat# 0111580, Marienfeld, Königshofen, Germany). After 1 h of gel polymerization, the inactive coverslip was removed, and type I collagen (100 μg/mL; Cat# 354236, Corning, NY, USA) was conjugated on the substrate surface with a bi-functional cross-linker, N-sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino] hexanoate (Sulfo-SANPAH; Cat# 22589, Thermo Fisher Scientific, Waltham, MA, USA), using UV (365 nm; Blak-Ray® XX-15L, UVP, Upland, CA, USA) to ensure cell adhesion on the gel. Gels were sterilized for 10 min under UV (254 nm; Mineralight® XX-15S, UVP, Upland, CA, USA) and equilibrated with cell culture medium at 37°C with 5% CO2 for at least 10 min prior to cell seeding.
To assess single cell migration, cells were first treated with TCEP while in suspension, then seeded onto polyacrylamide (PAA) gels at a density of 3 × 10³ cells/cm² in complete growth medium containing FBS, and incubated for 2 h at 37°C in a humidified atmosphere with 5% CO₂. Before image acquisition, cells were stained with Hoechst 33342 for 10 min. Real-time phase-contrast and fluorescence images were acquired using an EVOS M700 imaging system equipped with an onstage incubator (Thermo Fisher Scientific, Waltham, MA, USA) to maintain physiological conditions, and a 10× objective lens (N.A. = 0.3), for tracking single cell migration trajectories. To track single cell trajectory, automatic tracking was performed using TrackMate in FIJI with a LoG spot detector and a Simple Lap tracker for spot tracking. MATLAB-coded programs were used to determine mean squared displacement (MSD) and mean absolute distance (MAD) following the equations published previously, as mentioned. 18 MSD is a measure of the averaged displacement of a cell travel over a specified time interval, Δt. MAD a measure of total travel distance (not displacement) is defined similarity as MSD except absolute distance is used rather than squared displacement. As the cell motion trajectory was recorded at a constant sampling time, δt, the time interval Δt is specified as a multiple of δt. Thus, when considering a cell motion trajectory having N data points (numbered from 1 to N), the number of data pairs separated by the specified time interval is N−i.
Mean squared displacement at time point i

Mean absolute distance at time point i

Where:
di,j = end-to-end distance between point i and j.
li,j = absolute distance between point i and j.
N = total number of points of migrating cell trajectory
δt = data sampling time
i = number of data points for the time step Δt (i.e. Δt = i δt)
j = number of data points for averaging along time (j = 1, 2, . . . N−i)
Diffusion coefficients D (=slope/4 of MSD), a measure of cell dispersion speed (or how far the cell moves from the original location) was calculated from the linear regression of MSD curves. Cell moving speed V (=slope of MAD), a measure of cell movement speed (or how fast the cell moves regardless of the cell location) was calculated from the linear regression of MAD curves. High R2 values close to 1.0 indicate that cell shows random-walk behavior.
Cell–ECM forces analysis using traction force microscopy (TFM)
Traction force microscopy (TFM) method was used to measure mechanical stresses between cell to ECM following previous studies, otherwise mentioned.31,32 Polyacrylamide (PAA) gels (Young’s modulus = 19.66 kPa, AA/Bis = 8%/0.26%) were synthesized on the glass-bottom dishes (Cat# 101350, SPL, Pocheon, Korea) to mimic the pathological stiffness of the tumor microenvironment. Briefly, the glass-bottom dish was made hydrophilic by passing the glass surface through the flame of a Bunsen burner, followed by the application of sodium hydroxide (0.1 M; Cat# 39155S0350, Junsei, Tokyo, Japan). The glass was then activated using trichloro(1H,1H,2H,2H-perfluorooctyl) silane (silane; Cat# 448931-10G, Sigma-Aldrich, St. Louis, MO, USA) and glutaraldehyde (0.5%; Cat# G5882, Sigma-Aldrich). As markers of substrate deformation, red fluorescent beads (0.01%, ex/em = 580/605 nm) were used in two sizes: 0.2 μm for single cell analysis (Cat# F8812, Invitrogen, Carlsbad, CA, USA) and 1.0 μm for collective cell analysis (Cat# F8821, Invitrogen, Carlsbad, CA, USA) were vortexed and added to the monomer solutions. Polymerization was initiated by adding ammonium persulfate (0.6%; Cat# AMP001, BioShop, ONT, Canada) and N,N,N′,N′-tetramethylethylenediamine (0.4%; Cat# TEM001, BioShop, ONT, Canada), and the gels were cast by sandwiching on activated glass with an inactive coverslip of 18 mm diameter (Cat# 0111580, Marienfeld, Königshofen, Germany). After 1 h of gel polymerization, the inactive coverslip was removed, and type I collagen (100 μg/mL; Cat# 354236, Corning, NY, USA) was conjugated on the substrate surface with a bi-functional cross-linker, N-sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino] hexanoate (Sulfo-SANPAH; Cat# 22589, Thermo Fisher Scientific, Waltham, MA, USA), using UV (365 nm; Blak-Ray® XX-15L, UVP, Upland, CA, USA) to ensure cell adhesion on the gel. Gels were sterilized for 10 min under UV (254 nm; Mineralight® XX-15S, UVP, Upland, CA, USA) and equilibrated with cell culture medium at 37°C with 5% CO2 for at least 10 min prior to cell seeding. MCF7 and MDA-MB-231 cells were seeded on PAA substrate at a density of 5 × 103 cells/cm2 for single-cell experiments and 5.5 × 105 cells/cm2 for collective cell experiments and incubated for 18 h. For the in vitro wound healing experiment, a silicone insert with two wells was added before the cell seeding. This insert was removed to create a wound gap of 500 μm after the cells became confluent. The cells were washed with PBS to remove unattached cells from the substrate, and the cell culture medium was added. Images of fluorescent beads, green fluorescent cell membranes, and differential interference contrast (DIC) were acquired using a laser scanning confocal microscope (LSM-710; Carl Zeiss, Oberkochen, Germany) with 40× water immersion objective lense (Plan-Apochromat, N.A. = 1.20; 6× zoom out; pixel size = 0.17 um/px) and 10× objective lense (Plan-Apochromat, N.A. = 0.45; 6× zoom out; pixel size = 0.69 um/px) for single and collective cell experiment, respectively.
Computation of substrate deformation and stresses was processed by a MATLAB-coded program and Abaqus V6.14 (Dassault Systèmes, Vélizy-Villacoublay, France). Bead displacements were calculated using the particle image velocimetry (PIV) program coded in MATLAB by comparing the images of the null force state (absence of cells) and force state (presence of cells). 32 The null-force state was determined by adding 10% sodium dodecyl sulfate (Cat# 196-08675, Wako Chemicals, Richmond, VA, USA) to remove the cells. The finite element method was used to solve partial differential equations with Abaqus. Finally, the traction stress vectors in the x and y directions were determined from the stress tensors acquired from the finite element method analysis. The traction stress magnitude (TSmag) is defined as the magnitude of TS vector:

Where: σ(x,y) = [σx, σy]: traction stress vector
Cell–cell and intracellular force analysis using intracellular force and monolayer stress microscopies
We employed intracellular force microscopy (IFM) to measure intracellular tension for single cell6,32 and monolayer stress microscopy (MSM) for collective cells unless otherwise mentioned.33,34 Both IFM and MSM used traction stress (TS) field acquired from the TFM to compute the tension and stress field.
For the single cell analysis, intracellular tension (IT) was determined by averaging the intracellular tension along the major and minor axes from the contractile moment analysis. IT along each axis was calculated by dividing the cell into two distinct domains along the intracellular section line and applying Newton’s first law to each domain. The major- and minor-axis lines were determined via eigenvalue analysis, which computed the contractile moment. 35 The magnitude of intracellular tension (ITmag) is defined as the magnitude of the IT vector as shown below.
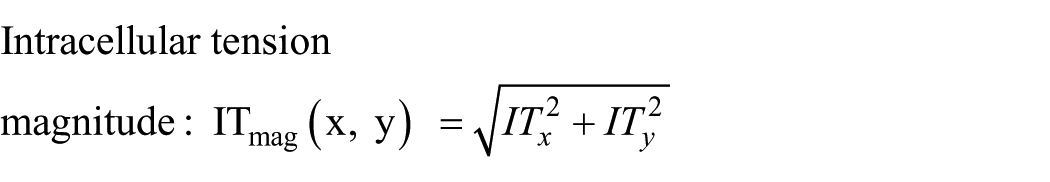
where: IT(x, y) = [ITx, ITy]: intracellular tension vector
For collective cell analysis, intracellular stresses (IS) were determined by MSM. Intracellular stress magnitude (ISmag) is defined as the magnitude of intracellular stress components as described below.

where τmax, and τmin: maximum and minimum intracellular stress components acquired from the eigenvalue analysis of stress tensor, respectively.
Statistical analysis
All results are presented as the mean ± standard error of the mean (SEM) in bar graphs and median ± quartiles in violin graphs. For two groups, statistical significance was determined using the Student’s t-test for comparisons between two groups. For multiple groups, significance was determined using the one-way analysis of variance (ANOVA), followed by Tukey’s post-hoc analysis. All experiments were independently repeated at least two biological repeats, and representative data are shown. Statistical significance is indicated by asterisks as follows: *p < 0.05, **p < 0.01, and ***p < 0.001. All analyses were conducted using the GraphPad Prism software version 10.3 (GraphPad Software, San Diego, CA, USA).
Results and discussion
TCEP induces free thiol groups on the breast cancer cell surface
To establish optimal conditions for selectively reducing disulfide bonds on the surface of breast cancer cells, we treated MCF7 and MDA-MB-231 cell lines with tris(2-carboxyethyl)phosphine (TCEP). MCF7 represents a well-differentiated, estrogen receptor-positive (ER⁺) breast cancer model, whereas MDA-MB-231 is a triple-negative subtype known for its high metastatic potential. 36 Given that TCEP cleaves disulfide bonds within and between proteins, it is critical to achieve a balance between effective thiol formation and minimal cytotoxicity, which guided the optimization of our experimental conditions. 16
A schematic representation illustrates how TCEP reduces surface-exposed disulfide bonds to generate reactive thiol groups, which were subsequently labeled with Alexa Fluor 488-conjugated maleimide for visualization (Figure 1(a)). Confocal microscopy demonstrated a concentration-dependent increase in thiol labeling in both cell types following treatment with 1–2 mM TCEP (Figure 1(b)). Quantitative analysis using Ellman’s assay confirmed these findings, revealing a dose-dependent increase in thiol levels (Figure 1(c)). These results establish TCEP as an effective agent for redox-based surface modification in both MCF7 and MDA-MB-231 cells.

Quantification and visualization of cell surface free thiol groups in breast cancer cells after TCEP treatment. (a) Schematic illustration showing that TCEP treatment reduces disulfide bonds on the cell surface, leading to the generation of free thiol groups. (i) Cells were detached, suspended at a density of 1 × 10⁶ cells/mL, treated with 2 mM TCEP for 5 min at room temperature in the dark, washed with PBS, and re-seeded onto culture plates. (ii) Free thiol groups on the cell surface were visualized using Alexa Fluor 488 C5 Maleimide labeling. (b) Confocal images of MCF7 and MDA-MB-231cells treated with 0, 1, 1.5, and 2 mM TCEP for 5 min. Free thiol groups on the cell surface were labeled with Alexa Fluro-488 C5 Maleimide (Mal-488). Upper panels show 2D-sliced views of the cell membrane, while lower panels show 3D-stacked side views (x, y, z planes). Scale bars = 10 µm. (c) Quantification of free thiol levels in MCF7 and MDA-MB-231 cells using Ellman’s assay. Data are presented as fold changes relative to untreated controls. (d) Cell viability following TCEP treatment for 5 min was assessed in both cell lines using MTT assay. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
Alexa Fluor 488–maleimide is widely used reagents for labeling thiol groups generated on the cell surface following disulfide bond reduction. 37 Consistent with this, our prior work using C2C12 myoblasts treated with 1mM TCEP for 5 min (pH 7.4) demonstrated this treatment does not impair viability, disrupt basal intracellular signaling, or induce cytotoxicity, but instead selectively enhances cell surface adhesion and promotes myogenic differentiation without evidence of intracellular thiol disruption. 15 In the present study, weak intracellular fluorescence occasionally observed, including even in untreated controls, likely reflects maleimide diffusion into the cytoplasm during fixation and permeabilization, which can transiently compromise membrane integrity. This phenomenon is well-documented in thiol-labeling protocols and is considered a technical artifact, rather than evidence of intracellular disulfide reduction by TCEP. The comparable background signal observed in control cells further supports the conclusion that TCEP-mediated reduction and thiol exposure are restricted to the cell surface. Collectively, these results indicate that TCEP treatment effectively induces surface thiol presentation that can be visualized using maleimide labeling, without disrupting intracellular redox homeostasis.
To evaluate potential cytotoxicity, MTT assays were performed at increasing TCEP concentrations (0–2 mM). No significant reduction in viability was observed in either cell line, suggesting good biocompatibility of TCEP at the tested concentrations (Figure 1(d)). This was corroborated by Live/Dead staining at 2 mM TCEP, which showed no appreciable increase in cell death At 2 mM, cell viability remained above 96%, whereas treatment with higher concentrations (3 mM or 4 mM) led to more than a 20% increase in cell death and a corresponding decrease in viability in both cell lines (Supplemental Figure S1). Based on these results, we employed 2 mM TCEP for subsequent experiments as it provided efficient thiol induction without compromising cell viability. Together, these data validate the use of TCEP for thiol induction under mild and non-toxic conditions.
The thiol-disulfide exchange reactions on the cell surface occur naturally through enzymatic activity, such as that of protein disulfide isomerase (PDI), or can be chemically induced using reducing agents such as dithiothreitol (DTT) and TCEP. 37 As a membrane-impermeable reducing agent, TCEP was applied here as a cell-surface engineering strategy to generate free extracellular thiols via redox modulation without affecting intracellular disulfide bonds.38,39 Importantly, both cell types were treated under identical conditions (5 min at pH 7.4) to ensure that the reduction was restricted to the extracellular compartment. Previous work by Kim et al. also demonstrated that TCEP-based strategies can be broadly applied to diverse cell types, including mesenchymal stem cells, Jurkat T cells, and cancer cell lines such as HeLa, for cell-surface engineering and subsequent evaluation of cell motility.18,40
While high concentrations of TCEP can disrupt essential disulfide bonds that stabilize cell membrane and intracellular protein structures—potentially resulting in cellular dysfunction or death, 41 our experimental conditions employ an optimized TCEP dose that selectively reduces surface-exposed disulfide bonds without significantly affecting intracellular architecture. This selective redox modulation enables the controlled induction of surface thiols while preserving cell viability and functionality, a critical requirement for redox-based strategies aimed at modulating cell behavior without inducing cytotoxicity. 42
TCEP-induced free thiol groups promote rapid, time-dependent enhancement of early cell adhesion
We next investigated how TCEP-mediated redox modification influences intracellular signaling related to adhesion. Previous studies have reported that thiol groups readily interact with the extracellular environment, such as the ECM or coating materials.43–45 Cha et al. demonstrated that TCEP treatment increases the formation of free thiol groups on the surface of glioma spheroids, thereby enhancing the expression and assembly of the adhesion marker vinculin. This enhancement of focal adhesion (FA) formation contributed to spheroid aggregation maintenance while suppressing cellular invasion. 23 Based on these findings, we hypothesized that TCEP-induced thiol groups could modulate FA dynamics in breast cancer cells through focal adhesion kinase (FAK), a central mediator of cell–ECM interactions and metastasis. 46
To test this hypothesis, cells were treated with 2 mM TCEP for 5 min, followed by PBS washing to induce free thiol group formation. Cells were then seeded and analyzed at 0.5, 1, and 2 h to capture the early adhesion and spreading process from the suspension state. During this period, focal adhesion formation was evaluated in both MCF7 and MDA-MB-231 cells (Figure 2(a)). As cells adhered, cell area increased, accompanied by elevated phosphorylated FAK (pFAK) and F-actin expression in TCEP-treated cells compared to controls (Figure 2(b) and (c)). Quantitative analysis revealed a time-dependent increase in cell area in both cell lines following TCEP treatment relative to controls (Figure 2(d)). Simultaneously, cytoskeletal reorganization, as indicated by F-actin expression, was enhanced during adhesion and spreading, with higher levels observed in TCEP-treated cells (Figure 2(e)). Additionally, focal adhesion area, number, and intensity were significantly increased upon TCEP treatment compared to controls (Figure 2(f) and (g)). These findings suggest that TCEP not only accelerates time-dependent FA formation but also promotes coordinated activation of pFAK alongside actin cytoskeleton remodeling, reinforcing the link between integrin signaling and FAK activation. 47 FAK clusters are known to be closely associated with actin filaments at cellular protrusions, coordinating FA formation with cytoskeletal remodeling. 48 Consistent with this model, our data indicate that TCEP-induced thiols may promote integrin clustering at ventral and peripheral regions of the basal membrane, subsequently enhancing FAK phosphorylation and F-actin polymerization. This integrin–FAK–actin axis likely underpins the observed increase in both adhesion stability and spreading capacity. 30 Building on the previously established Lyn–FAK FRET sensor developed by Wang et al., which visualizes FAK activity via Src-mediated activation in membrane microdomains,30,49,50 we specifically monitored pFAK expression as a readout of FAK activation. MCF7 and MDA-MB-231 cells were imaged for 80 min at 10-min intervals, simultaneously capturing YFP and CFP emissions. The fluorescence intensity of ECFP was normalized to YPet to generate ratiometric FRET images, which were merged with phase-contrast images to visualize the dynamic transition from suspension to matrix adhesion (Figure 3(a) and (b)). Over time, cells progressively spread, and TCEP-treated cells exhibited markedly enhanced pFAK expression. Quantitative analysis of FRET-based FAK activation (ECFP/YPet ratio) revealed a significant increase immediately after adhesion, starting from 0 min (Figure 3(d) and (e)).

TCEP treatment accelerates time-dependent cell adhesion and promotes FAK phosphorylation with cytoskeletal remodeling in breast cancer cells. (a) Schematic illustration showing that MCF7 and MDA-MB-231 cells were treated with 2 mM TCEP, washed with PBS, and seeded onto tissue culture plates (TCP). Cells were then fixed at 0.5, 1, and 2 h after seeding to assess cell–matrix adhesion dynamics during the transition from suspension to adhesion. (b, c) Immunofluorescence staining for phosphorylated FAK (pFAK, green), F-actin (red), and nuclei (Hoechst, blue) in MCF7 and MDA-MB-231 cells. Merged images show pFAK and F-actin co-localization, with enlarged insets highlighting pFAK distribution at the leading edge and associated cytoskeletal changes. Scale bars = 25 µm. (d) Morphological analysis of MCF7 and MDA-MB-231 cells. Violin plots show changes in cell area across different time points, with sample sizes as follows: Ctrl 0.5 h (n = 327), Ctrl 1 h (n = 585), Ctrl 2 h (n = 286), TCEP 0.5 h (n = 347), TCEP 1 h (n = 261), and TCEP 2 h (n = 358). (e) Quantification of F-actin fluorescence intensity in MCF7 and MDA-MB-231 cells following TCEP treatment. (f) Quantification of focal adhesion (FA) morphology, including pFAK-positive area and number of FA structures per cell. (g) Quantification of pFAK fluorescence intensity in cells. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.

TCEP treatment enhances FAK activation dynamics during early adhesion, as revealed by a FRET-based biosensor. (a) Schematic illustration showing that MCF7 and MDA-MB-231 cells were transduced with the Lyn–FAK FRET adenovirus for 24 h, treated with TCEP, and allowed to adhere for 1 h to monitor pFAK activation during the early adhesion process. Under basal conditions, cells emit yellow fluorescence at 530 nm (YFP) upon 423 nm excitation. Upon FAK activation, ATP binding induces a conformational change, leading to a shift in emission toward cyan fluorescence at 480 nm (CFP). Real-time FRET imaging of pFAK activation was performed every 10 min over an 80 min period. (b, c) Phase-contrast and corresponding ECFP/YPet FRET ratio images of Lyn–FAK biosensor-expressing MCF7 and MDA-MB-231 cells. The color bar indicates ECFP/YPet ratio values (n = 10 per group). Scale bar = 20 μm. (d, e) Time-course analysis of normalized ECFP (480 nm)/YPet (530 nm) emission ratios in MCF7 and MDA-MB-231 cells. FRET ratios were normalized to the average baseline signal prior to adhesion. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
Both immunofluorescence staining and the Lyn–FAK FRET biosensor confirmed Src-mediated FAK activation, demonstrating that TCEP-induced surface redox modulation rapidly promotes cell attachment to the matrix via FAK signaling. These findings align with the report by Zhao and Guan, which showed that FAK autophosphorylation at Tyr397 and subsequent Src recruitment facilitates early adhesion and cytoskeletal stabilization. However, they also noted that sustained or excessive FAK activity may modulate adhesion turnover and promote migration in tumor cells. 46 Taken together, our results are consistent with this context-dependent model, indicating that TCEP primarily enhances the early adhesive function of FAK during cell–matrix interactions by promoting rapid integrin–FAK–actin signaling.
TCEP-induced free thiol groups promote the focal adhesion signaling
Building on the previous observations, we next examined the effect of TCEP on long-term focal adhesion (FA) signaling and cytoskeletal organization in breast cancer cells. To this end, we assessed the level of phosphorylated FAK (pFAK), a key indicator of FA signaling activation. Treatment with 2 mM TCEP for 5 min significantly increased pFAK levels in both MCF7 and MDA-MB-231 cells without altering total FAK expression (Figure 4(a) and (b)). This increase was further corroborated by immunofluorescence staining, which revealed elevated pFAK localization at the leading edge of TCEP-treated cells (Figure 4(c)). Quantitative analysis showed that TCEP treatment significantly increased the length and number of pFAK-positive FA structures in both cell lines (Figure 4(d) and (e)), supporting the conclusion that TCEP promotes FA assembly.

TCEP treatment promotes FAK phosphorylation and induces cytoskeletal remodeling in breast cancer cells. (a) Schematic illustration showing that MCF7 and MDA-MB-231 cells were treated with TCEP for 5 min, washed with PBS, and then cultured for 24 h to evaluate pFAK expression. This experiment was designed to assess cell adhesion strength following surface reduction by TCEP compared to control cells. (b) Western blot analysis of pFAK and total FAK expression in MCF7 and MDA-MB-231 cells treated with 2 mM TCEP for 5 min. GAPDH was used as a loading control. Quantification of pFAK/FAK ratio indicates a significant increase in pFAK expression in TCEP-treated cells compared to controls. (c) Immunofluorescence staining of pFAK (green), F-actin (red), and nuclei (Hoechst, blue) in MCF7 and MDA-MB-231 cells treated with 2 mM TCEP for 5 min. Merged images show co-localization of pFAK and F-actin. Enlarged panels highlight the distribution of pFAK at the leading edge and associated structural changes. Scale bars = 25 µm. (d) Quantification of FA morphology, including pFAK-positive area, length, circularity, and number of FA structures per cell. (e) Quantification of pFAK fluorescence intensity in MCF7 and MDA-MB-231 cells. (f) Morphological analysis of MCF7 and MDA-MB-231 cells. Violin plots show changes in cell area, length, circularity, and roughness after TCEP treatment. n = 185 (MCF7 control), 200 (MCF7 + TCEP), 120 (MDA-MB-231 control), 369 (MDA-MB-231 + TCEP). (g) Quantification of F-actin fluorescence intensity in MCF7 and MDA-MB-231 cells following TCEP treatment. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
Consistent with prior studies, FA size correlates with adhesion lifetime and cellular motility.51,52 Nascent adhesions are small (<1 μm²) and highly dynamic; intermediate adhesions (~1–5 μm²) exhibit balanced turnover supporting migration, whereas large, mature, or supermature adhesions (>5 μm²) are more stable, disassemble slowly, and are typically associated with inhibited migration. In line with this paradigm, control cells predominantly exhibited adhesions in the ~1–5 μm² range, whereas TCEP-treated cells formed much larger adhesions (frequently >10 μm²) with increased number, suggesting FA hypermaturation. This adhesion hypermaturation is known to occur when several factors converge, including increased cytoskeletal tension, F-actin stabilization, and activation of FAK/ROCK signaling.53–55 Stiff ECM conditions or exogenous stabilizers have been shown to induce similar FA stabilization. 56 Such “frozen” adhesions with large size and high pFAK content restrict dynamic remodeling, impairing efficient migration. Our observations that TCEP-treated cells displayed both enlarged adhesion structures and increased F-actin polymerization strongly suggest that redox-mediated membrane changes shift the cells toward a highly adhesive, less motile phenotype.
In parallel, F-actin staining revealed an increase in fluorescence intensity following TCEP treatment, indicating increased actin polymerization and cytoskeletal remodeling (Figure 4(g)). Morphological analysis based on F-actin-outlined cell boundaries indicated that TCEP treatment significantly increased cell area and length in both MCF7 and MDA-MB-231 cells, while circularity remained largely unchanged in MCF7 cells (Figure 4(f)). These findings suggest that TCEP treatment promotes focal adhesion signaling by promoting FAK phosphorylation and inducing cytoskeletal remodeling, which synergistically support FA hypermaturation and a highly spread, adhesive morphology. Interestingly, MCF7 cells, which are typically more rounded and epithelial, showed an increase in cell area with minimal change in circularity, suggesting preserved epithelial morphology. This observation is consistent with previous reports indicating that morphological features such as cell elongation and spread area can serve as predictive markers of functional cell states across diverse cell types. 57 These cell-type-dependent differences are presented as supplementary and descriptive observations, suggesting a phenotypic shift toward reduced motility, rather than enabling direct statistical comparisons between cell lines. Thus, these findings imply that TCEP-induced redox modulation may exert cell-type-specific effects depending on baseline cellular phenotypes.
The spatial enrichment of pFAK at the leading edge aligns with its role in nascent FA formation and ECM anchoring. 58 Moreover, the elongation of pFAK-positive structures suggests FA maturation, a process often linked to stable adhesion and actin bundling. 59 Notably, previous studies have shown that strong adhesion and elevated pFAK levels can suppress the formation of invadopodia, which are actin-rich protrusions necessary for ECM degradation and invasion. These findings suggest that pFAK may act as a negative regulator of invasive structures by stabilizing focal adhesions and preventing unnecessary matrix degradation.60–62
In our study, TCEP treatment elevated both pFAK and F-actin levels, consistent with enhanced focal adhesion signaling and cytoskeletal reinforcement. Although we did not directly assess invadopodia formation, the observed increase in adhesion stability and cytoskeletal organization was accompanied by reduced cell motility. These results support the concept that TCEP-induced membrane thiol remodeling promotes a transition from dynamic, motile adhesions to stabilized, hypermature focal adhesions, ultimately suppressing migration. These findings highlight that while both ECM stiffness and TCEP-induced surface thiol modulation affect adhesion and cytoskeletal dynamics, their outcomes may differ. Whereas ECM stiffness can promote migration under certain conditions, TCEP-mediated enhancement of adhesion and cytoskeletal stability appears to suppress migration in our experiment.63,64
TCEP-induced free thiol groups reduce migration and invasion of breast cancer cells
We next evaluated whether TCEP-induced thiol modifications affect collective migration and invasion (Figure 5(a)). To assess this, we performed in vitro wound healing, transwell migration, and invasion assays. The in vitro wound healing assay assessed the collective migratory capacity of both cell lines. In the untreated control group, MDA-MB-231 cells exhibited progressive wound closure over time, indicative of strong migratory activity. In contrast, MCF7 cells, which are less metastatic, showed slower wound closure, consistent with their limited migratory potential. Notably, TCEP-treated cells displayed significantly delayed migration at 8 and 16 h (Figure 5(b) and (c)). The reduction in wound closure was more pronounced in MDA-MB-231 cells, suggesting that TCEP-induced thiol modifications effectively suppress the collective migration of highly invasive breast cancer cells. These results indicate that TCEP treatment diminishes collective migratory ability, particularly in metastatic cells, by interfering with underlying motility mechanisms. To further assess whether TCEP also inhibited single-cell migration, we performed a transwell migration assay. Compared with untreated controls, TCEP-treated MCF7 and MDA-MB-231 cells exhibited significantly reduced migration across the membrane (Figure 5(d), left panels; Figure 5(e), left graph).

TCEP treatment suppresses wound healing, migration, and invasion in breast cancer cells. (a) Schematic illustration of the in vitro wound healing assay. A defined cell-free gap was generated using an ibidi culture insert. After seeding 5.4 × 10⁵ cells/cm² cells and allowing ~18 h for adhesion, the insert mold was removed, and collective cell migration into the gap was assessed at 0, 8, and 16 h after mold removal. (b) Representative image of wound closure in MCF7 and MDA-MB-231 cells at 0, 8, and 16 h after removal of the mold. Cells were pretreated with TCEP for 5 min and seeded into ibidi culture inserts to generate a defined gap. Yellow dashed lines indicate wound edges. Scale bars = 400 µm. (c) Quantification of wound coverage (%) in MCF7 and MDA-MB-231 cells. Wound coverage was calculated as the percentage of the initial gap area occupied by migrating cells at each time point. (d) Representative images from transwell migration and invasion assays. (e) Quantification of migrated and invaded area. Scale bars = 200 µm. *p < 0.05, **p < 0.01, ***
Cancer cell invasion requires extracellular matrix (ECM) degradation and penetration—processes essential for metastatic dissemination. To evaluate the effect of TCEP-induced thiol modifications on invasive capacity, we performed a Matrigel-coated transwell invasion assay. In line with the migration data, TCEP treatment significantly inhibited the invasive ability of both MCF7 and MDA-MB-231 cells, as reflected by a reduced number of invaded cells and smaller invaded areas on the membrane (Figure 5(d), right panels; Figure 5(e), right graph). Although FAK is widely recognized for its role in promoting cell motility across various cancer models, 65 our data show that TCEP-induced FAK activation correlates with reduced migration and invasion. This seemingly paradoxical result may reflect the dual role of FAK in adhesion dynamics. Michael et al. 66 demonstrated that while FAK enhances the initial rate of adhesion strengthening through integrin activation, sustained FAK activation can inhibit migration by increasing adhesive forces and stabilizing focal adhesions over time. Therefore, the sustained FAK activation observed upon TCEP treatment may have shifted the balance toward reinforced cell–ECM adhesion, ultimately hindering the dynamic adhesion turnover necessary for mesenchymal migration.
Our observation of increased pFAK expression aligns with this model, suggesting that TCEP promotes focal adhesion maturation and stabilization. Kim and Wirtz 52 also proposed that migration speed follows a biphasic relationship with FA size, whereby motility declines once adhesions become excessively large or stable. In this context, the trend toward larger and elongated pFAK positive structures, alongside decreased migration and invasion, suggests that TCEP treated cells may exceed the optimal adhesion size required for efficient migration (Figure 6).

TCEP treatment increases mechanical force generation and altering morphology on PAA gels in breast cancer cells. (a) Schematic illustration of the experimental procedure; MCF and MDA-MB-231 cells were treated with or without TCEP and seeded on PAA gel (Stiffness = 19.66 kPa) embedded with 0.2 µm fluorescent beads for 18 h. Cells were imaged for morphology and traction force microscopy (TFM) analysis. (b–d) Representative images showing cell morphology (DIC and fluorescence) (b), traction stress distribution (c), and intracellular tension (d) for each condition. Scale bars = 30 µm. (e–g) Quantification of cell morphology on PAA gels, including area (e), length (f), and circularity (g). Quantification of cell-generated mechanical forces, including traction stress (h) and intracellular tension (i) in MCF7 and MDA-MB-231 cells. n = 108 (MCF7 control), 112 (MCF7 + TCEP), 105 (MDA-MB-231 control), 121 (MDA-MB-231 + TCEP). *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
TCEP treatment enhances mechanical traction stresses and intracellular tensions in single breast cancer cells
To further investigate the biomechanical effects of TCEP-induced thiol modifications in single breast cancer cells, we analyzed cellular morphology, intracellular stress, and traction force using traction force microscopy (TFM). Adhesion regulation is a key determinant of invasion in breast cancer cells. Breast tumors are characterized by a stiff and fibrotic microenvironment compared to normal tissue. Previous studies have shown that the mechanical properties of cancer cells within stiff environments strongly influence cell adhesion, migration, and invasion.67–69 Xu et al. 70 reported that while healthy breast tissue is very soft (approximately 0.2 kPa), malignant breast tissue is significantly stiffer (>4 kPa), and this increased stiffness promotes tumor progression, invasion, and metastasis through activation of mechanotransduction pathways such as YAP/ERK signaling and invadopodia formation. Similarly, Berger et al. 67 demonstrated that increased stiffness in 3D tumor microenvironments affects invasion: breast cancer cells encapsulated in stiff (12 kPa) hydrogels exhibited elevated EGFR and PLCγ1 signaling, leading to increased expression of the invadopodia associated protein Mena, compared to cells in soft (2 kPa) hydrogels. Based on these findings, we cultured breast cancer cells on PAA gels with a stiffness of 19.66 kPa, corresponding to the range reported for malignant breast tissue, to investigate mechanical effects at the single cell level.
The overall experimental setup is illustrated in Figure 6(a). Representative phase-contrast and fluorescent images (Figure 6(b)) reveal distinct morphological changes following TCEP treatment in both MCF7 and MDA-MB-231 cells. Specifically, TCEP-treated cells exhibited increased projected cellular area and length (Figure 6(e) and (f)), while circularity was significantly reduced (Figure 6(g)), indicating a more spread and elongated morphology. These morphological changes were accompanied by pronounced mechanical alterations. TFM-based traction stress maps and quantitative analyses showed a significant increase in cell–matrix forces (traction stress) in TCEP-treated cells compared to untreated controls (Figure 6(c) and (h)). Likewise, intracellular tension measured via monolayer stress microscopy (MSM) was elevated in both cell lines (Figure 6(d) and (i)). Notably, the increases in traction stress and intracellular tension were more pronounced in the highly metastatic MDA-MB-231 cells than in MCF7 cells, indicating a differential sensitivity to redox modulation depending on cell type.
These findings suggest that thiol modifications induced by TCEP promote cytoskeletal tension and stabilize focal adhesions, potentially via FAK activation and enhanced actomyosin contractility. Although FAK activation is typically associated with increased contractility and adhesion maturation, excessive stabilization of focal adhesions can paradoxically inhibit cell motility by reducing adhesion turnover—a key requirement for effective mesenchymal migration. 71 In this context, our results suggest that while TCEP-induced FAK activation increases intracellular tension, it does not enhance migratory capacity. Rather than facilitating dynamic movement, TCEP appears to reinforce mechanical anchorage by stabilizing the actin cytoskeleton and focal adhesions through thiol-based redox modulation. As described earlier, TCEP-treated cells displayed increased projected area and elongation with reduced circularity—morphological traits indicative of cytoskeletal reinforcement and mechanical stiffening. These features are consistent with enhanced actin organization and are commonly associated with elevated intracellular tension.
This mechanically reinforced state was further substantiated by TFM and MSM analyses, both of which revealed increased cytoskeletal tension and stronger cell–ECM coupling. Such a stiffened phenotype may limit the dynamic shape changes necessary for efficient migration. 72 Taken together, these findings indicate that thiol-based redox modifications suppress cell motility by promoting intracellular contractility while concurrently restricting the mechanical flexibility required for effective migration. This mechanistic interpretation is consistent with emerging evidence revealing the pivotal role of mechanical cues in governing cellular behavior and migratory potential.73,74 These results suggest that TCEP-induced redox modulation is associated with increased intracellular contractility and mechanical reinforcement, which may contribute to the reduced motility observed in breast cancer cells.
TCEP-induced thiol modifications suppress breast cancer cell motility
Having shown that TCEP promotes focal adhesion signaling and cytoskeletal remodeling, we next asked whether these structural changes are associated with functional changes in cell motility. Given that the tumor microenvironment is often characterized by increased matrix stiffness due to collagen accumulation and cross-linking, we utilized PAA substrates tuned to replicate the mechanical properties of breast cancer tissues. 9
Using single-cell tracking analysis, we monitored the migratory behavior of MCF7 and MDA-MB-231 cells over time. Representative time-lapse images at 0, 200, 400, and 600 min revealed that control cells exhibited active migration with long trajectories, whereas TCEP-treated cells remained more confined (Figure 7(a) and (b)). Displacement maps further highlighted this confinement of cell motility following TCEP treatment in both cell lines (Figure 7(c)). A representative video illustrating these real-time migration behaviors is provided in Supplemental Video 1. Quantitative analysis supported these qualitative observations. In MDA-MB-231 cells, TCEP treatment significantly reduced both the diffusion coefficient (D) and migration speed (V), indicating impaired directional movement, while a similar but non-significant trend was observed in MCF7 cells (Figure 7(e) and (g)). To maintain physiological relevance, all migration assays were conducted in the presence of serum, and the observation period was limited to within 24 h to minimize the effects of cell proliferation. Under these controlled conditions, TCEP-induced surface thiol modification consistently enhanced cell–ECM adhesion and promoted cytoskeletal stabilization, which corresponded with reduced motility. These findings suggest that modulation of cell surface redox status can influence migration behavior independently of proliferation or chemotactic cues, underscoring the importance of adhesion and cytoskeletal dynamics in regulating breast cancer cell motility.

TCEP treatment reduced single-cell motility in breast cancer cells. (a) Schematic illustration showing that MCF7 and MDA-MB-231 cells were treated with TCEP, incubated for 4 h to allow cell adhesion, and then imaged at 10-min intervals over a total of 10 h. (b) Time-lapse imaging of MCF6 and MDA-MB-231 cells tracked over 600 min. Cells were cultured on 19.66 kPa PAA hydrogels and treated with or without 2 mM TCEP. Scale bars = 50 µm. (c) Cell displacement maps showing individual migration trajectories of MCF7 and MDA-MB-231 cells tracked for 600 min. Trajectories are color-coded by time (blue to red), and each plot represents the pooled single-cell paths under control or TCEP-treated conditions. n = 150 (MCF7 control), 62 (MCF7 + TCEP), 63 (MDA-MB-231 control), 193 (MDA-MB-231 + TCEP). (d) MSA over time, plotted to assess overall migration behavior. (e) Quantification of diffusion coefficient (D, µm2/min) derived from MSD curves. (f) MAD from the starting point over time. (g) Quantification of cell migration speed (V, µm/min). Violin plots display median and interquartile ranges. n = 108 (MCF7 control), 112 (MCF7 + TCEP), 105 (MDA-MB-231 control), 121 (MDA-MB-231 + TCEP). *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
To further characterize the motility dynamics, we analyzed mean squared displacement (MSD) and mean absolute distance (MAD) over time. MSD plot revealed a substantial reduction in displacement following TCEP exposure in both cell lines, as evidenced by a decrease in the slope of the MSA-time curves (Figure 7(d) and (e)). Similarly, MAD values indicating cell movement speed were significantly diminished in TCEP-treated groups (Figure 7(f) and (g)), indicating an overall decrease in the ability of cells to spread and migrate.
This may reflect their greater reliance on dynamic focal adhesion turnover and cytoskeletal remodeling for cell motility. 65 These results are consistent with previous studies showing that stabilized focal adhesions can impede migration by restricting adhesion turnover, a key process in mesenchymal-like migration modes. Moreover, the increased actin polymerization observed in the earlier section may contribute to enhanced intracellular stiffness, which reduces cellular deformability during migration.75,76 Remarkably, the use of a stiffness-tuned biomimetic substrate in our study allowed for the detection of these force-dependent responses, underscoring the relevance of mechanical microenvironment control in mechanobiological investigations of cell behavior in engineered tissues. 77 Induced cell–ECM adhesion may also restrict deformation-associated processes required for effective movement, thereby further reducing motility. 78 Together, these findings indicate that TCEP modulates breast cancer cell migration by reinforcing focal adhesion and stabilizing the cytoskeleton, ultimately impairing the dynamic processes essential for cell movement.
TCEP treatment inhibits collective migration by modulating traction and intercellular stress
We next examined the effects of TCEP-induced thiol modifications on collective cell migration during in vitro wound healing using a hydrogel-based assay. Cells were seeded to form a 500-μm wound gap on a polyacrylamide (PAA) gel (19.66 kPa), and both traction stress and intracellular stress were quantified and spatially mapped. We analyzed both cell–matrix (traction force) and cell–cell mechanical stresses (intracellular tension) in migrating monolayers and compared their spatial distributions relative to the wound edge (Figure 8(a)).

The effect of TCEP treatment decreased collective cell migration by modulating traction and intracellular stress in breast cancer cells. (a) Schematic illustration of the experimental setup for measuring traction stress (TS) and intracellular stress (IS) during collective migration on PAA hydrogels. (b) Representative fluorescence images of wound closure at 0 and 4 h after wound gap formation. Scale bars = 250 µm. (c) Spatial profiles showing the distance of the leading edge from the original wound margin at 4 h. (d) Heatmaps of traction stress magnitude, showing spatial distribution of cell–substrate forces during collective migration in control and TCEP-treated conditions. (e) Intracellular stress maps derived from intracellular force microscopy (IFM), displaying stress polarity and gradients within migrating monolayers. (f) Quantification of wound coverage (%) at 4 h. (g) Quantification of average traction stress magnitude per condition. (h) Quantification of intracellular stress magnitude per monolayer. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
Representative time-lapse images and quantitative analysis revealed that TCEP treatment significantly impaired collective wound closure (Figure 8(b) and (f)). MDA-MB-231 cells exhibited the most pronounced decrease in migration speed, while MCF7 cells showed a moderate but statistically significant reduction. The migratory behavior of the collective monolayers closely mirrored the trends observed in single-cell migration described in Figure 3.
Mechanical force mapping at 4 h post-wounding revealed a broader and more elevated distribution of traction stress in TCEP-treated cells compared to untreated controls (Figure 8(d)). To further analyze these patterns, we quantified regional stress magnitudes within the leader (0–100 μm from the wound edge) and follower (100–400 μm from the edge) cell zones (Figure 8(c)). TCEP treatment significantly increased traction stress in both leader and follower regions in MCF7 and MDA-MB-231 cells (Figure 8(g)), suggesting enhanced mechanical anchorage throughout the migrating monolayer. In addition, intracellular stress mapping revealed a more intense and widespread distribution of stress magnitude in TCEP-treated cells, especially in MDA-MB-231 (Figure 8e). Quantitative analysis confirmed that intracellular stress magnitude was significantly elevated in both the leader and follower zones upon TCEP treatment (Figure 8(h)), with the highest levels observed in TCEP-treated MDA-MB-231 cells, consistent with their elevated invasive potential.
Traction forces generated at the cell–ECM interface are essential for migration, enabling cells to anchor, generate propulsion, and coordinate directional movement. 79 However, excessive traction force generation has been shown to paradoxically suppress migration. Lo et al. and others have demonstrated that optimal migration occurs at an intermediate level of adhesion and contractility, and that overly stabilized adhesions hinder the dynamic remodeling necessary for efficient motility.80–82 However, excessive traction force generation has been shown to paradoxically suppress migration. Lo et al. and others have demonstrated that optimal migration occurs at an intermediate level of adhesion and contractility, and that overly stabilized adhesions hinder the dynamic remodeling necessary for efficient motility.
Intracellular mechanical tension also plays a critical role in regulating migration. However, when excessively elevated, it can likewise impede motility. Tao et al. demonstrated via mechanical modeling that while moderate intracellular tension promotes movement, excessive tension reduces migration by limiting cellular deformability and responsiveness to directional cues—highlighting a biphasic relationship between adhesion strength and migration speed.83,84 Similarly, Sens et al. 85 reported that increased membrane tension inhibits actin-driven protrusion at the leading edge, thereby reducing migration efficiency. Although we did not directly measure membrane tension, we observed increased actin polymerization and elevated intracellular tension in TCEP-treated breast cancer cells, suggesting that thiol modification strengthens cytoskeletal tension that interferes with coordinated migration. These results are consistent with the hypothesis that excessive intracellular or membrane tension hinders the dynamic remodeling necessary for effective cell movement. Furthermore, our observations align with prior studies showing that mechanical loading regulates cellular behavior through cytoskeletal reorganization and mechanotransductive signaling. 86 Taken together, our findings demonstrate that TCEP-induced thiol modifications promote both intracellular tension and traction force generation in collectively migrating breast cancer cells. However, rather than enhancing motility, these mechanical increases were accompanied by a significant reduction in wound closure. This inverse relationship suggests that excessive mechanical stress—whether from actomyosin contractility or cell–matrix adhesion—can disrupt the force coordination required for efficient collective migration.
Conclusion
This study demonstrates that TCEP induced thiol modifications strengthen focal adhesion signaling and cytoskeletal organization, thereby suppressing breast cancer cell motility. TCEP treatment increased FAK phosphorylation, enhanced traction stress, and elevated intracellular tension in individual cells, as well as amplified intercellular tension in leader cells during collective migration. These mechanical changes contributed to stronger cell–matrix anchorage and greater mechanical stability. In parallel, TCEP treatment significantly reduced both migration and invasion. Importantly, these effects were observed in both nonmetastatic MCF7 cells and highly metastatic MDA MB 231 cells, emphasizing that TCEP was used here as a tool to investigate how modulation of cell surface redox states affects adhesion and mechanotransduction, rather than as a therapeutic agent. Taken together, our findings highlight the potential of cell surface thiol engineering as a strategy to regulate cancer cell adhesion and motility, offering a platform for future studies aimed at identifying physiologically compatible and clinically applicable agents that modulate surface redox states to influence mechanotransduction and migration.
Supplemental Material
sj-docx-1-tej-10.1177_20417314251394126 – Supplemental material for Cell surface engineering for inhibition of breast cancer cell motility through modulation of mechanotransduction and focal adhesion dynamics
Supplemental material, sj-docx-1-tej-10.1177_20417314251394126 for Cell surface engineering for inhibition of breast cancer cell motility through modulation of mechanotransduction and focal adhesion dynamics by Juyeon Kim, Gyo Jeong Gu, Sung Sik Hur, Min-Kyu Kim, Fenny Soetanto, Jiwon Son, Joo Hyun Kim, Taewan Kim, Yun Kyung Lee, Jaemoon Yang, Hyung Kwon Byeon, Jong Eun Lee, Sun Wook Han, Sung Yong Kim, Jun-Hee Lee, Ju Hun Lee, Myung Jin Ban and Yongsung Hwang in Journal of Tissue Engineering
Footnotes
Acknowledgements
The corresponding author, Prof. Yongsung Hwang, wishes to express his deepest gratitude to Mrs. Gyungsun Jo, his beloved mother, who passed away from breast cancer. Her unwavering love and kindness continue to inspire his dedication to advancing breast cancer research. This work is respectfully dedicated to her memory.
ORCID iDs
Author contributions
Conceptualization, S.S.H. and Y.H.; methodology, G.J.G., S.S.H., J.S., and Y.H.; validation, G.J.G., J.H.L., M.J.B., and Y.H.; formal analysis, G.J.G., J.K., S.S.H., F.S., and J.S.; investigation, G.J.G., J.K., S.S.H., F.S., J.S., M.K.K., J.H.K., T.K., Y.K.L., J.Y., H.K.B., J.E.L., S.W.H., S.Y.K., J.H.L., M.J.B., and Y.H.; data curation, J.K. and S.S.H.; visualization, G.J.G., S.S.H., and J.H.L.; writing—original draft preparation, G.J.G., J.K., and Y.H.; writing—review and editing, J.H.L., M.J.B., and Y.H.; supervision, J.H.L., M.J.B., and Y.H.; project administration, M.J.B. and Y.H.; funding acquisition, Y.H.; resources, Y.H. All authors have given approval to the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Soonchunhyang University Research Fund and the National Research Foundation of Korea funded by the Ministry of Science and ICT (MSIT) [grant numbers: RS-2019-NR040068, RS-2023-00284258, and RS-2024-00440151], as well as by the research fund of Hanyang University (HY-2022-2563).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data is available upon request to the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.