
Abstract
Current treatment for complex and large-scale volumetric muscle loss (VML) injuries remains a limited success and have substantial disadvantages, due to the irreversible loss of muscle mass, slow muscle regeneration, and rapid formation of non-functional fibrosis scars. These VML injuries are accompanied by denervation and the destruction of native vasculature which increases difficulties in the functional restoration of muscle. Here, reconstruction of the vascular network at the injury site was offered as a possible solution for improving the repair of muscle defects through the timely supply of nutrients and oxygen to surrounding cells. A hydrogel-based tissue construct containing various densities of the vascular network was successfully created in the subcutaneous space of mice by manipulating hydrogel properties, and then implanted into the VML injury site. One month after implantation, the mouse treated with the highly vascularized tissue had extensive muscle repair at the injury site and only spent a shorter time completing the inclined plane tests. These findings suggest that the reconstruction of the functional vascular network at the VML injury site accelerated muscle fiber repair through a timely supply of sufficient blood and avoided invasion by host fibroblasts.
Keywords
Graphical abstract

Introduction
Skeletal muscle has some capacity for regeneration following injury. However, this ability is limited in case of large volumetric muscle loss (VML; >20% of muscle by mass), which overwhelms this innate repair mechanism, leading to incomplete functional repair. Current clinical treatments of these non-regenerative defects involve using autologous muscle grafts accompanied by physical therapy. It is unable to completely restore muscle strength due to poor reconstruction and re-innervation of the injured muscle tissue.1–4 Following skeletal muscle injury, the myofibers decompose and release their intracellular contents, triggering a cascade of events leading to an inflammatory response. Subsequently, quiescent myogenic cells and non-muscle stem cells are involved in innervation, blood vessel formation, and muscle regeneration at the injury site. In severe VML injuries, there is a dense deposition of scar tissue that impairs the reconstruction of regenerating muscle, so developing an alternative treatment to reestablish the natural regenerative processes (e.g. inflammatory response, muscle progenitor cell migration, innervation, and blood vessel formation) to restore function in VML injuries is clinically necessary. The efficacy of scaffold-based regeneration critically depends on the scaffold materials and cell-material interaction. 5 Previous studies using acellular, decellularized, extracellular, matrix-based, or composite scaffolds have shown promising results, but thus far there has been limited success regarding the complete repair of VML in large animals.6–10 A promising advancement in skeletal muscle tissue engineering studies is the production of vascular networks to enable vascular perfusion in an engineered muscle construct. Pro-angiogenic cells were precultured for 3 weeks in vitro for vascular maturity and integration following implantation in an abdominal wall defect model, but these took too much time to reach limited success in the recovery of muscle strength.11–16 Efforts have been made to avoid the in vitro cultivation of pre-vascularized constructs and develop a more clinically translatable treatment for VML defects. For this purpose, microvascular fragments (MVFs) from rat epididymal fat pads, which have vascular regenerative properties, were freshly isolated immediately prior to implantation. However, perfused vessels within the defect were mainly located at the border of the construct, and the overall level of perfusion after 2 weeks was low. 17 Shandalov et al. 13 developed a host vessel infiltration technique to increase the perfusion and anastomosis of their pre-vascularized constructs using a two-step implantation protocol. The construct was cultured in vitro for 10 days and subsequently implanted around the mouse femoral arteries and veins for 1–2 weeks. Next, the construct was transferred as a pre-vascularized graft to an abdominal wall VML defect site. The study used a degradable synthetic polymer scaffold and tested differences among constructs containing mono-cultured, co-cultured, and tri-cultured cells. The results showed that implanting myoblasts into a coculture of endothelial cells and fibroblasts promoted vasculogenesis by femoral vessels. Despite differences between groups after incubation around femoral vessels, fewer differences were observed following transfer to an abdominal wall VML defect.18–21 Moreover, prior efforts to vascularize tissue grafts have mostly relied on in vitro preculture of constructs or/and in vivo incubation to form connected lumens within constructs prior to implantation, and anastomosis after implantation to form perfused blood vessels. Although the presence of these vessels improves muscle repair and restores partial functions, the formation of vascularized tissue constructs requires considerable time, and the formed networks do not provide efficient perfusion to large-scale defects. After implantation, these grafts partially integrate with host vasculature without establishing effective perfusion in a timely manner.
To combat this problem, the present study aimed to engineer large-scale pre-vascularized tissue constructs with well distributed controllable densities of perfused blood vessels. Collagen is the major structural protein in skeletal muscle, and collagen hydrogels have been confirmed as an excellent biomaterial to engineer vascularized tissues with high densities of blood vessels (~80–100/mm2). However, its capability to control vessel densities is limited, so the development of methacrylate collagen (CMA) hydrogels to create vascularized tissues with different vascular densities is necessary. First, blood-vessel-forming cells, namely human umbilical endothelial cells (HUVECs) and mesenchymal stem cells (MSCs), were encapsulated into collagen-based hydrogels with tunable crosslinking conditions. Subsequently, their viability and proliferation in vitro and their capability to form vascularized cell-laden hydrogel s at the subcutaneous site of mice were investigated. A murine VML model was established to evaluate the potential of transplantable cell-laden hydrogel constructs with high, intermediate, and low-density of vascular networks to facilitate de novo restoration of skeletal muscle function. Muscle repair and growth were systematically assessed by histological and immunostaining analyses for de novo myofiber formation, fibrosis, innervation, and anastomosis between engineered and host blood vessels. To further investigate the importance of pre-vascularized tissue on muscle repair, freshly prepared cell-laden constructs containing HUVECs and MSCs were directly implanted into VML defect sites and served as the control.
Experimental section
Preparation of acellular collagen and methacrylated collagen (CMA) hydrogels
After removing the fat layer, porcine skin tissues were cut into small pieces (1 cm × 1 cm) and further minced using a mixer grinder. To remove non-collagenous proteins, minced tissues were immersed into 0.1 M NaOH at 4°C for 3 days, rinsed with distilled water, and immersed into 10% butanol at 4°C for 24 h to remove the residual fat and debris. Collagen was extracted using 0.5 M acetic acid at 4°C for 3 days with continuous stirring. The supernatants of the extracted solutions were collected by centrifugation (7000 rpm), desalted by adding NaCl to a final concentration of 3 M, and centrifuged again at 4°C. The precipitate was collected and re-dissolved in 0.5 M acetic acid, dialyzed with 0.5 M acetic acid for 1 day and distilled water for 1 day to remove salts, and lyophilized. Freeze-dried collagen was dissolved in 0.02 M acetic acid by stirring at 4°C at the indicated concentration. 1 mL of collagen solution was mixed with 0.1 mL of cooled calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (DPBS) and 0.025 mL of (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer solution in an ice bath; the pH was adjusted to 7.4–7.6 using 1 M NaOH, and the solution was loaded into the PDMS-made mold to form disk-shaped Collagen hydrogels with 1 mm thick and incubated at 37°C for 30 min to initiate a self-assembled gelation process for the formation of a collagen hydrogel with 1 mm thick (acellular Collagen).
The CMA conjugate was synthesized by conjugating methacrylic anhydride to collagen molecules. The details were modified from the published literature.22,23 Briefly, Freeze-dried collagen was dissolved at a 0.0625% (weight/volume (w/v)) collagen solution in 0.1 M HCl aqueous solution at 4°C overnight. Morpholinoethanesulfonic acid (MES; Sigma-Aldrich, USA) was added to collagen solutions to reach a concentration of 50 mM and the pH was subsequently adjusted to 6 by adding 0.1 N NaOH solution at 4°C. Next, 0.459 g of methacrylic anhydride (Sigma-Aldrich, USA), 0.337 g of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC; Sigma-Aldrich, USA), and 0.08 g of N-hydroxysulfosuccinimide (NHS; Sigma-Aldrich, USA) were added to 100 mL of collagen solution, and the mixture was placed at 4°C for 24 h. Next, the solution was dialyzed with 0.005 M HCl for 14 days and freeze-dried to obtain the powder form of CMA conjugates. To prepare acellular CMA hydrogels, a CMA prepolymer solution was prepared by mixing the freeze-dried CMA (0.3% (w/v) final) and the photo-initiator (Irgacure 2959) (0.125% (w/v); CIBA Chemicals, USA) dissolved in PBS at 4°C. The glass spacers, 1 mm thick, were added on both sides of the glass slide to define the thickness of the hydrogel. 100 μL of CMA prepolymer solution was dropped in the middle of the glass slide and covered with polydimethylsiloxane (PDMS)-modified slides. After light exposure, a disk-shaped CMA gel was formed by removing the PDMS-modified slide.
Photocrosslinking was achieved by exposing the prepolymer to 11.42 mW/cm2 UV light (365 nm; M&R Nano Technology, Taiwan) to 15 s (acellular CMA-15) and 20 s (acellular CMA-20). The intensity of UV light was monitored using an ultraviolet (UV) intensity meter (Model 202; G&R Labs, Taiwan) at room temperature.
Characterization of hydrogels
Proton nuclear magnetic resonance (1H NMR) spectra were recorded by the NMR spectrometer (AV-400 (400 MHz); Bruker, USA) at 4°C to characterize the conjugation of the methacrylic group in the CMA conjugates (1 mg per mL in D2O). For the measurement of circular dichroism (CD) (J-815 CD; Jasco, USA), all samples were adjusted to a concentration of 0.03% (w/v). The solution was scanned at a wavelength range of 190–270 nm at 4°C. Rheological measurements of hydrogel formation were performed using a strain-controlled parallel-plate rheometer (AR-G2; TA Instruments, USA) at room temperature as previously described.24,25 Analysis of three-dimensional (3D) microstructure of the hydrogel in an aqueous environment was performed under the multi-photon confocal microscopy (TCS-SP5-X; Leica, Germany). Atto 550 NHS ester (Sigma-Aldrich, USA) was conjugated to collagen-based hydrogels, according to the instructions provided by the manufacturer, through incubation overnight at room temperature. Fluorescence-labeled hydrogels were imaged using a multi-photon confocal microscope system (TCS-SP5-X; Leica, Germany). To analyze the microstructure of freeze-dried hydrogels, the hydrogels were immersed in liquid nitrogen for 1 min and placed in a freeze-dryer for 1 day. The microstructure of the hydrogels was investigated through scanning electron microscopy (SEM) (S-4300; Hitachi, Japan). To understand its drug release profile, the preformed hydrogel was immersed in 2 mg/mL of fluorescein isothiocyanate-dextran (70 kDa) isothiocyanate-dextran (FITC-dextran; Sigma-Aldrich, USA) solution. After 24 h of absorption, the hydrogels were washed three times with PBS and soaked in PBS to measure their cumulative release profiles for FITC-dextran at 5, 10, 30, 60 min and 24 h. This was achieved by measuring the FITC intensity with a UV-visible spectrophotometer (SpectraMax Plus 384; Molecular Devices, USA) at an excitation wavelength of 495 nm.
Cell culture
HUVECs (Lonza, USA) cultured for 4–7 passages in endothelial cell growth medium (EGM-2; Lonza, USA) containing 2% (volume/volume (v/v)) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin (PS) were used. MSCs were isolated from human normal deidentified discarded white adipose tissue obtained during a clinically-indicated procedure following an Institute Review Board-approved protocol from Dr. Juan M. Melero-Martin at Harvard Medical School (Boston, MA, USA), as previous. 26 MSCs were cultured for 8–9 passages in MSC growth medium (MSCGM; Lonza, USA) containing 20% (v/v) FBS, 10 ng/mL basic fibroblast growth factor and 1% (v/v) PS and subsequently used. NIH-3T3 fibroblasts were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS and 1% (v/v) PS.
Two-dimensional (2D) cell viability assays
Cells (2000 cells per well) were cultured in 96-well plates in regular culture medium. After 1 day of culture, photo-initiator at various concentrations (0.0625-1%) was added to the culture medium, and the wells were exposed to UV light for 0–60 s. Furthermore, cells were cultured in Collagen or CMA hydrogels in regular medium. After an additional 2 days, cell viability was calculated as the number of live cells and expressed as a percentage of the control using a live/dead viability kit (Invitrogen, USA) according to the instructions provided by the manufacturer.
3D cell encapsulation
Prepolymer Collagen and CMA solutions were gently mixed with MSCs alone (1 × 106 cells/mL) or a mixture of MSCs and HUVECs (1:1 ratio; 1 × 106 cells/mL). The 100 μL of cell-laden Collagen hydrogels were formed at 37°C for 30 min into the PDMS-made mold to form disk-shaped cell-laden Collagen hydrogels with 1 mm thick. The cell-laden CMA gel formation occurred under UV irradiation for 15 (CMA-15) or 20 (CMA-20) seconds. In this process, a Petri dish (Corning Inc., USA) was used as the substrate. The glass spacers, 1 mm thick, were added on both sides of the glass slide to define the thickness of the hydrogel. 100 μL of cell-laden prepolymer solution was dropped in the middle of the glass slide and covered with polydimethylsiloxane (PDMS)-modified slides. After light exposure, a disk-shaped cell-laden CMA gel was formed by removing the PDMS-modified slide.
The cell-laden gels were cultured in 24-well dishes for 2 days. After staining with a live/dead viability kit, the spreading area of live cells (green-labeled cells) was imaged using a multiphoton confocal microscope system (TCS-SP5-X; Leica, Germany). Serial optical sections were captured in the central depth plane with respect to the surface of the hydrogel structures, and the resultant z-stacks were merged into an image using ImageJ (National Institutes of Health (NIH), USA) to analyze the cell spreading area inside the hydrogel. Cell spreading was quantified by measuring the area of cells from z-stacks confocal images (n = 3 to 5 for each group).
In vivo vasculogenic assay
All animal experiments were approved by the institutional animal care and use committee of National Tsing-Hua University, Hsinchu, Taiwan (Protocol number: 108030). Male BALB/cAnN.Cg-Foxnlnu/CrlNarl mice (aged 5–7 weeks) were purchased from the National Laboratory Animal Center, Taiwan. The formation of vascular networks in vivo was evaluated using a xenograft transplantation model in immunodeficient mice.24,27 HUVECs alone or MSCs alone cannot support vessel formation in collagen hydrogels (Figure S1). Therefore, MSCs and HUVECs were directly co-cultured at a 1:1 cell ratio in the cell density of 1 × 107 cells/mL, a suitable combination to generate vascular networks.26,28–30 For the cell-laden Collagen group, the collagen prepolymer solutions (100 μL) were mixed with MSCs and HUVECs (1:1 ratio; 1 × 107 cells/mL) before gelation. The cell-laden CMA mixture (100 μL) was pipetted onto a polydimethylsiloxane-coated glass slide, with aliquots separated using a 1 mm spacer following 15 and 20 s of UV light exposure to form the cell-laden CMA-15 and CMA-20 groups, respectively. Two hydrogels were surgically implanted into the subcutaneous sites of a mouse. Each experimental condition was performed in 5–7 replicates. On day 7 after implantation, the mice were sacrificed and the hydrogel constructs were removed from the mice, and a 3D laser scanning confocal microscope (VK-X1000; Keyence, Taiwan) was used to scan and determine the 3D surface and topography of tissue constructs. This allowed the measurement of the thickness, horizontal area, and volume of the structures prior to tissue processing for histological analysis. The microvessels were quantified by evaluating five randomly selected fields under 100× magnification of H&E-stained horizontal sections taken from the middle part of the implants. The microvessels were identified as luminal structures containing red blood cells. The values reported for each experimental condition are the mean values ± the standard deviation, obtained from five to seven individual hydrogel for each group.
VML injury model and implantation of cell-laden hydrogels pre-formed at the subcutaneous space for 7 days or freshly prepared cell-laden hydrogels
BALB/cAnN.Cg-Foxnlnu/CrlNarl mice (aged 7–8 weeks) were anesthetized through inhalation of 1% isoflurane. Hair was removed from the incision site on the hindlimb, and the skin was disinfected using ethanol and iodine solution. An incision was performed to expose the quadriceps muscle on the left foot of mice. Using a biopsy punch tool (diameter: 4 mm) (VWR, USA) to make consistent VML full-thickness defects on the rectus femoris and vastus intermedius of the quadricep 31 2 mm above the knee (Figure S6(b)–(c)). The muscle (4 mm in diameter and 3 mm thick) was removed from the bone to have the VML defect (Figure S6(c)–(d)). Cell-laden tissue constructs pre-formed at the subcutaneous site of mice for 7 days, or freshly prepared cell-laden hydrogels were implanted into the defect sites. Each mouse received one implant for cell-laden or acellular hydrogel groups, and the VML was left untreated in the non-treated empty group. After the injury, the skin was closed using sutures.
Histological and immunofluorescence analyses
Tissue constructs were fixed in 4% paraformaldehyde overnight. Staining of tissue sections using hematoxylin and eosin (H&E) and Masson’s trichrome was performed to investigate the formation of the vascular network and repair of muscle tissue. For immunofluorescence staining, tissue constructs were permeabilized with 0.5% Triton X-100 in DPBS for 25 min. The hydrogels were subsequently blocked with 5% bovine serum albumin (Sigma-Aldrich, USA) in DPBS for 2 h and incubated overnight with rabbit anti-alpha-smooth muscle actin (anti-α-SMA; 1:100 dilution; Clone 1A4; Sigma-Aldrich, USA), mouse anti-human-CD31 (1:100 dilution; Clone JC70A; Dako Cytomation, USA), rabbit anti-laminin (1:200 dilution; Sigma-Aldrich, USA), mouse anti-myosin heavy chain (anti-MHC; 1:100 dilution; DSHB, USA), mouse anti-human nuclear (1:100 dilution; Clone 235-1, Abcam, USA), anti-mouse Ly6 G antibodies (1:50 dilution; Clone1A8, eBioscience, USA), anti-mouse F4/80 antibodies (1:50 dilution; Clone CI A3-1, BD Pharmingen, USA), anti-mouse CD45 antibodies (1:50 dilution; Clone 30-F11, BD Pharmingen, USA), and mouse anti-β-Tubulin III (1:100 dilution; Abcam, USA) antibodies at 4°C. After washing three times with DPBS, the cell-laden hydrogels were incubated with Alexa Fluor 488-conjugated or Fluor 647-conjugated secondary antibodies (1:200 dilution; Invitrogen, USA) at room temperature for 2 h. Finally, sections were stained with Hoechst solution (Invitrogen, USA) and examined using a confocal microscope (TCS-SP5-X; Leica, Germany). For human CD31 immunohistochemical analysis, sections (thickness: 7 mm) were deparaffinized; this was followed by antigen retrieval, and the sections were blocked for 30 min at room temperature. After incubation with mouse anti-human CD31 antibodies (1:100 dilution; Clone JC70A; Dako Cytomation, USA) for 1 h, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies (1:100 dilution in blocking buffer; Vector Laboratories, USA) for 1 h at room temperature. The bound antibody was detected using an ImmPACT DAB substrate kit (Vector Laboratories, USA). Finally, the sample was counterstained with hematoxylin and mounted. After 1- and 2-month, repaired tissue constructs were gently excised from the site of the muscle defect for assessment of muscle revascularization and repair. To investigate the types of tissues grown in tissue constructs, parafilm tissue sections (thickness: 5 μm) in front- and side-view were prepared through H&E and Masson’s trichrome staining. To quantify myofiber regeneration within the constructs, tissue sections were immune-fluorescently double-stained with anti-laminin (1:200 dilution; Sigma-Aldrich, USA) and anti-MHC (1:100 dilution; DSHB, USA) antibodies, stained with Hoechst solution, and mounted with mounting media (Thermo Fisher Scientific, USA); z-stacked fluorescent images were analyzed through confocal microscopy (TCS-SP5-X; Leica, Germany). The total number, thickness, and area of each muscle bundle that dually expressed MHC-positive and laminin-positive myofibers in the tissue constructs were counted using the multipoint tool in ImageJ. The uninjured group at the same age of mice were used as positive control and set as 100% of the tissue area. To quantify staining within the defect regions, the defect region was identified using DAPI staining to manually demarcate an area within the muscle section. A percentage of positive tubulin III in the sections was calculated by the numbers of positive β-tubulin III cells divided by the total numbers of cells into the repaired muscle region.
Statistical analysis
Three independent experiments were performed for each experimental group, with 3–5 replicates per experiment. The results were analyzed using Origin Pro 8.5 (OriginLab, Northampton, MA, USA) and presented as the mean ± standard deviation. Statistical analysis was performed using one-way variance (ANOVA) followed by Tukey’s honest significant difference test for equal variance and Tamhane T2 test for groups with unequal variance. Statistical significance was assumed at p-values < 0.05, 0.01, and 0.001 (indicated as *, **, and ***, respectively).
Results
Synthesis and characterization of acellular collagen and CMA hydrogels
In this study, high yield of acid-soluble collagen was obtained from porcine skin with high purity and bioactivity of type I collagen molecules according to our optimal conditions (Figure S2). To attain a photocurable collagen hydrogel, CMA conjugates were synthesized via direct reaction between the free lysin amino groups in collagen molecules (Collagen) and methacrylic anhydride through nucleophilic substitution using low-concentration (0.0625%) collagen aqueous solutions at pH 6 and 4°C. The characterization of CMA using 1 H-NMR spectroscopy (Figure 1(a)) displayed the presence of a peak at δ = 1.4, 1.9, and 2.8 ppm characteristic of the methyl group of methacrylates,23,31,32 implying the successful synthesis of CMA conjugates. The structural integrity of CMA was verified using CD analysis. The CD spectra of unmodified collagen (Collagen) (Figure 1(b)) showed negative absorption in the region of 190–200 nm. We prepared Collagen hydrogels that are physically crosslinked via hydrogen bonds between native collagen molecules with concentrations of 0.3% and 0.6%, and CMA hydrogels that are chemically crosslinked via covalent bonds between modified MA groups through free-radical polymerization reaction with exposure to light for various periods. These preparations were conducted to understand the tunability of the properties of these collagen-based hydrogels (i.e. acellular Collagen and CMA hydrogels). The mechanical properties of hydrogels were evaluated through rheological measurements. With increasing concentration of collagen molecules from 0.3% to 0.6%, the gelation time of the Collagen hydrogel was markedly decreased from 710 s to 180 s, whereas its storage modulus (G’) (Figure 1(c)) was increased by 50% from 48 Pa to 72 Pa. Furthermore, the prepolymer of Collagen with a concentration >0.6% was excessively gelling, thereby impairing the preparation of uniform cell-laden gels. Under 0.3% collagen concentration, the G’ of CMA hydrogels (Figure 1(d)) increased by approximately four-fold (from 47 to 187 Pa) through an increase in the time of exposure to light from 10 s (CMA-10) to 20 s (CMA-20). Collectively, the adjustability of the gelation time and storage modulus of Collagen hydrogels was limited and highly dependent on the concentration of collagen molecules. In contrast, CMA hydrogels were more tunable in terms of stiffness, which is independent of collagen concentration compared to the collagen hydrogels. We examined the effects of hydrogels on nutrient transport within the hydrogel by monitoring the diffusion rate of 70 kDa FITC-dextran in an aqueous solution (Figure 1(e)). The results showed that the cumulative release rate of 50% of the dextran from the 0.3% Collagen hydrogel was approximately 2.5-fold faster than that noted from the 0.6% Collagen and CMA-15 hydrogel, and four-fold faster than that of CMA-20 hydrogel. Interestingly, the permeability of the hydrogel was highly dependent on the stiffness of the hydrogel instead of the crosslinking methods in these soft gels (G’ <200 Pa) (Pearson coefficients of correlation, r = 0.792) The microstructure of collagen-based hydrogels was investigated by confocal microscopy (Figure 1(f)) and scanning electron microscopy (Figure 1(g)). The results showed numerous low-density and interconnected fibrils in Collagen, and a densely packed honeycomb-like structure in CMA hydrogels, which possessed distinct microstructures. An increase in collagen concentration significantly increased collagen fibrils’ density in Collagen hydrogels. The CMA-10 is too soft to encapsulate cells to form a cell-laden CMA-10 hydrogel, so was not used for the following studies. In CMA-15 and CMA-20 hydrogels, the pore size and density decreased with increasing time of exposure to light. In CMA-20 hydrogels, pores were barely visible. In this study, we successfully synthesized Collagen and photocurable CMA hydrogels precisely without substantially altering the native secondary structure and bioactivity of collagen. Both resultant hydrogels exhibited tunable physio-biochemical properties over the mechanical properties, microstructure, and permeability. To successfully create vascularized tissues with tunable vessel density in vivo, we selected the 0.3% Collagen, CMA-15 (exposure to UV: 15 s), and CMA-20 (exposure to UV: 20 s) hydrogels with distinctly different levels of stiffness and permeability for the subsequent experiments.

(a) 1H NMR spectra of Collagen and CMA macromers. Successful conjugation of MA groups was labeled with blue arrows. (b) CD spectra of Collagen and CMA prepolymer solutions at 4°C. (c and d) The mechanical strength of (c) acellular Collagen and (d) acellular CMA hydrogels can be tailored by altering the protein concentrations and the time of exposure to UV, respectively. (n = 3) (e) Cumulative release profiles of 70 kDa FITC-dextran from acellular Collagen and CMA hydrogels with various crosslinked conditions (n = 3–4). The microstructures of acellular hydrogels were studied using (f) laser confocal microscopy and (g) scanning electron microscopy (SEM).
Hydrogel-mediated encapsulated cell behavior in vitro
HUVECs and MSCs commonly used in engineering three-dimensional (3D) vascularized constructs were used as model cells to investigate cellular responses inside collagen-based hydrogel scaffolds. Crosslinkers were always considered harmful to cells, so the conditions used for the photo-crosslinking process (0.125% (w/v) photoinitiator (PI) and 0–60 s of UV irradiation) were first confirmed nontoxic for both HUVECs and MSCs 29 (viability >90%; Figure S3(a)–(d)). To evaluate the biocompatibility to of our collagen-based hydrogels to cells, NIH3T3, HUVECs and MSCs cells were seeded on the top of preformed Collagen and CMA-15 gels and cultured in a two-dimensional environment. After 2 days, there is no difference in cell proliferation rates between the Collagen and CMA-15 groups among three kinds of cells (proliferation rate >90%; Figure S3(e)) compared to that on the standard tissue culture plates. To explore the 3D cell behavior and capability of vasculogenesis in different collagen-based hydrogels, HUVECs, and MSCs were comixed with Collagen and CMA prepolymer solutions to form Collagen, CMA-15, and CMA-20 cell-laden hydrogels. After 2 days of culture (Figure 2(a) and (b)), HUVECs (hCD31 + (green) and/or α-SMA+ (red), yellow and transparent yellow arrows in Figure 2(b)) significantly elongated and well-spread along MSCs (α-SMA+ (red), white arrows in Figure 2(b)) into Collagen and CMA-15 hydrogels, while both cells in CMA-20 hydrogels were kept round in both bright field and fluorescence-stained images. To verify the cell behavior, we acquired the fluorescent images by a confocal microscope to quantify the area for HUVEC and MSCs using ImageJ software. There is no significant difference in the spreading area of the embedded HUVEC cells (Figure 2(c)) between the Collagen and CMA-15 hydrogels, but it significantly decreased in the CMA-20 gel exhibiting only about 60% of the cell spreading area observed in the Collagen gel. The same trend was also observed on embedded MSCs (Figure 2(d)) in hydrogels, probably due to the stiffer, lower permeability in CMA-20 hydrogel. No lumen structures are observed in any cell-laden hydrogels after 2 days of co-culture. The soft hydrogels no matter the crosslinked methods, that is, Collagen and CMA-15, with better oxygen permeability can support cell spread in vitro. Here, collagen-based hydrogels provided a permissible environment to support cell viability and spreading in 2D and 3D culture systems.
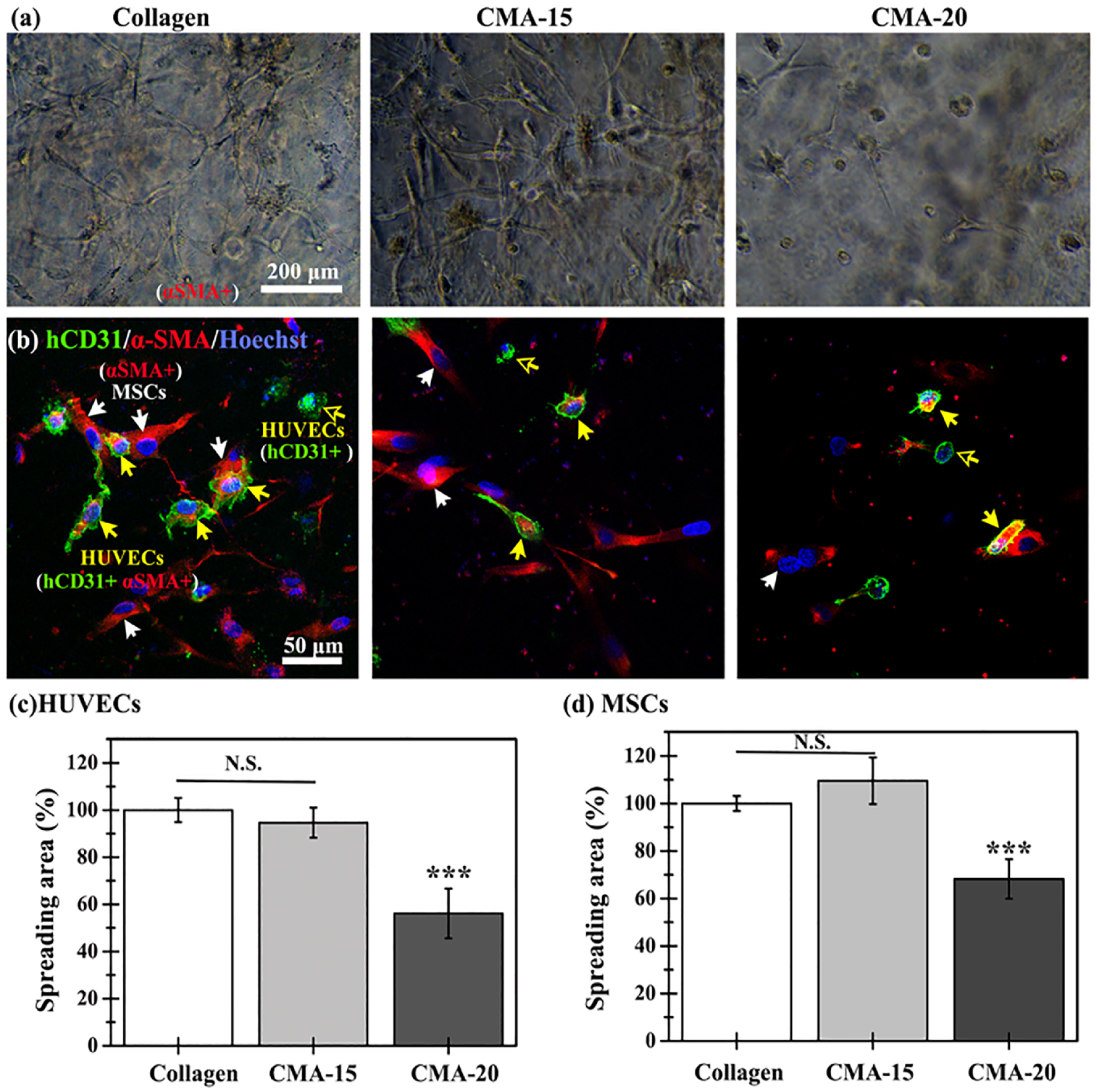
Representative (a) optical and (b) confocal fluorescence images of co-encapsulated HUVECs and MSCs in the Collagen, CMA-15, and CMA-20 hydrogels. The cells were immunostained with hCD31 (green) and α-SMA (red), and counterstained with Hoechst for nuclei (blue). The labeling of HUVECs is as follows: HUVECs exhibiting both hCD31+ (green) and α-SMA+ (red) staining are denoted by yellow arrows, while HUVECs solely displaying hCD31+ (green) staining are indicated by transparent yellow arrows. MSCs, on the other hand, are identified by α-SMA+ (red) staining alone, and these cells are pointed out by white arrows. Cell spreading area of the co-culture of (c) HUVECs and (d) MSCs inside the indicated hydrogel group. Data are presented as the mean ± SD. *** p < 0.001 indicates significant differences versus the Collagen group at the same time point (n = 3–5).
Control of perfused vascular formation in the cell-laden hydrogels at a subcutaneous site through tailoring of the physical properties of hydrogels
The timely and adequate supply of blood is critical to tissue repair and regeneration. Therefore, the formation of functional blood vessels in the large-sized construct within a week is crucial for increasing the survival rate of transplanted tissues or regeneration rate at the site of injury. To examine the effect of acellular hydrogel on the formation of a vascular network, we implanted acellular Collagen, CMA-15, and CMA-20 hydrogels (diameter: 10 mm; thickness: 2 mm) into a subcutaneous site in mice. After 7 days, no vessel ingrowth or formation inside the implanted gels was observed in any of the groups (Figure S4).
Simultaneous improvement of vasculogenesis and angiogenesis is the strategy for shortening the necessary time for the formation of functional vascular networks inside constructs. Thus, HUVECs and MSCs were co-encapsulated into Collagen, CMA-15, and CMA-20 hydrogels, and implanted cell-laden hydrogels subcutaneously into mice to engineer tissue-engineered functional blood vessels. After 7 days, cell-laden hydrogels were removed to analyze their volume and formation of stable vascular networks. In Figure 3(a), the thicknesses of the cell-laden Collagen and CMA-20 hydrogel were similar (Collagen: 1.55 mm; CMA-20: 1.61 mm), and only ~55 % of that observed in CMA-15 structures (2.86 mm). The horizontal-sectional area of the cell-laden Collagen hydrogels was the smallest (~57 mm2), while those of the other hydrogels were similar (~81 mm2). The volumes of explants were: cell-laden Collagen (~40 mm3), CMA-20 (~68 mm3), and CMA-15 (~103 mm3) (Figure 3(e)). The chemical-crosslinked covalent bonds inside CMA hydrogels allowed 1.5-2.5-fold better retention of the thickness of hydrogels by compared with the physical-crosslinked Collagen hydrogels. Moreover, the spacer allowed two-fold better retention of the thickness of the Collagen hydrogels versus that of others produced without using the spacer previously reported in the literature.24,33–35
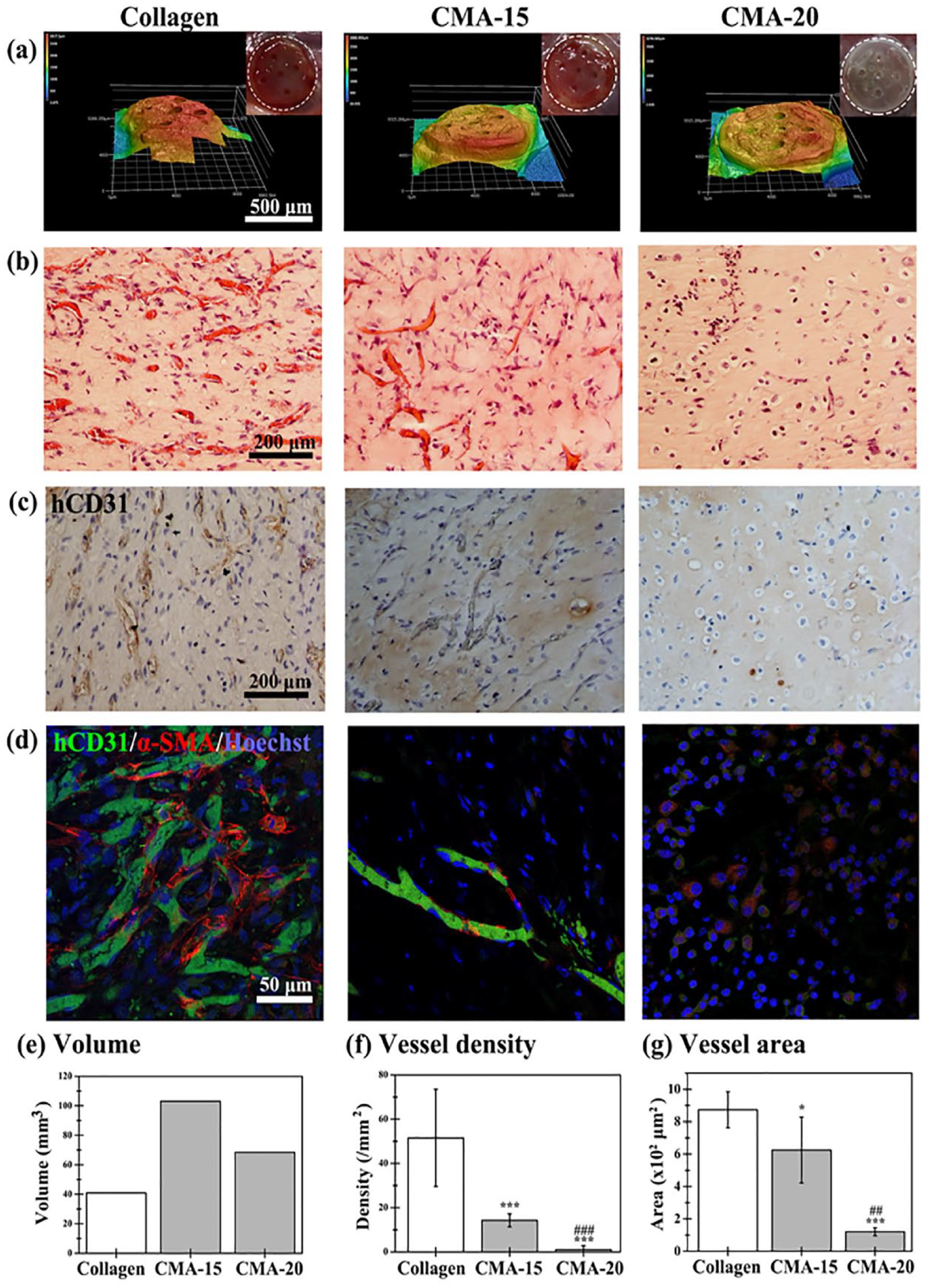
Cell-laden hydrogels formed in the subcutaneous space of mice for 7 days. (a) Macroscopic images (inset) and surface topography scanned using laser scanning confocal microscopy of the cell-laden hydrogels. (b) Representative horizontal-sectional H&E images of the cell-laden hydrogel revealing distribution of perfused blood vessels (blue arrows) in the structures. (c) Representative images of sections stained with human CD31+HUVECs identified by immunohistochemistry. (d) Mature perfused human vessels were lined exclusively with hCD31-expressing HUVECs (green) and surrounded by αSMA-single positive expressing MSCs (red). Nuclei (blue) were labeled with Hoechst. The extent of vascular network formation was quantified by measuring the entire explant volume, and counting the numbers and areas of erythrocyte-filled lumens. (e) Explant volume, (f) vessel density, and (g) vessel area in the engineered cell-laden tissue constructs removed 7 days after implantation. *p < 0.1 and *** p < 0.001 indicate significant differences versus the Collagen group. Hash symbols (## p < 0.01 and ### p < 0.001) indicate significant differences versus the CMA-15 group (n = 5–7).
Histological examination via H&E staining of both horizontal (Figures 3(b) and S5) and cross sections (Figure S6) revealed numerous mature and functional blood vessels containing erythrocytes in Collagen and CMA-15 explants, which were evenly distributed throughout the whole explants (Figures 3(b)–(d) and S5). Fewer perfused blood vessels were observed in the CMA-20 hydrogels; these vessels were negative for human CD31 and mainly distributed on the periphery of the constructs, showing the ingrowth of vasculature in mice through angiogenesis. In Collagen and CMA-15 explants, most micro-vessels were stained positive for human CD31 (Figure 3(c) and (d)), confirming that the vessels were lined with implanted HUVECs. A close examination of the human micro-vessels revealed the presence of murine erythrocytes (Figure 3(c)), indicating that functional anastomoses with the existing murine vasculature had occurred. These results demonstrated that the formation of microvascular vessels within the hydrogels was induced by the implanted cells, and that this formation was not merely due to invasion by host blood vessels. Furthermore, most human micro-vessels were completely covered by α-SMA-single positive perivascular cells (Figure 3(d)), indicating vessel maturation. Quantitative evaluation revealed that the density of perfused blood vessels was: Collagen hydrogel (51.5 ± 21.1 vessels/mm2), CMA-15 hydrogel (14.2 ± 2.0 vessels/mm2), and CMA-20 hydrogel (1 ± 1.8 vessels/mm2) (Figure 3(b) and (f)). Importantly, the area covered by the perfused blood vessels was larger in the Collagen hydrogel, followed by CMA-15 and CMA-20 hydrogels (Figure 3(b) and (g)). In addition, the density of the perfused blood vessels in the horizontal sections was 2-threefold larger than that noted in the cross sections. This indicated that the growth of the engineered vascular network within the constructs occurred parallel to the skin (Figures 3(f) and S6). The present results showed that pre-vascularized constructs with three degrees of functional vascular networks were successfully created into collagen-based hydrogels within 1 week through precise control of the stiffness and permeability of hydrogels, corresponding to low (CMA-20), intermediate (CMA-15), and high (Collagen) density of vascular networks in vascularized soft tissues. These constructs can be used to investigate the effects of vascularized tissue graft on repair of VML defects.
Acellular hydrogels possess limited capability for repair of VML in muscles
To investigate the muscle repair and regeneration on the critical defect size, we established a VML model at the left rectus femoris in mice through the creation of a non-recovery muscle defect (Figure S7). Acellular CMA-20, CMA-15, and Collagen grafts with no microvessels pre-formed at the subcutaneous site for 7 days (Figure S4) were implanted into the muscle’s injury site. The average area of tissues in the uninjured group was set as 100%. After 1 month, the void created by surgery at the defect site remained visible from gross appearance in all groups (left panel in Figure 4(a)–(c)). There are no remaining hydrogels at the injury site in all groups. In all acellular groups, there was <60% of whole reconstructed tissues and <40% muscle tissues compared with those observed in the uninjured group. Furthermore, there was no significant difference in the area of repaired connective and muscle tissues between the groups, as shown by collagen staining with Masson’s trichrome stain (middle and right panels in Figure 4(d)–(f)). These results demonstrated that acellular Collagen, CMA-15, and CMA-20 hydrogels possess the limited capability to repair VML injured muscle.

Volumetric skeletal muscle tissue reconstruction by implanting the acellular CMA-20, CMA-15, and Collagen hydrogels in the injury VML model, at 4 weeks after implantation. (a-c) Macro-images with different perspectives at the defect site after repair are presented in the left column (yellow-dashed line). Representative Masson’s trichrome staining images of the repaired construct in the front-view and side-view sections are shown in the middle and right columns. The repaired muscle (M) and connective tissue (C) were labeled. Quantification of (d) whole reconstructed area, (e) muscle, and (f) whole tissue area in the reconstruction at the defect site (the uninjured group was set as 100%). *p < 0.1 indicate significant differences versus the acellular Collagen group. (n = 4–5).
Muscle construction at the critical defect in VML injured muscles by various degrees of vascularized cell-laden hydrogel
Next, to investigate the effects of various degrees of pre-vascularized tissue constructs on restoring VML, pre-formed perfused vascularized soft tissue grafts of various degrees (i.e. low, intermediate, and high) were implanted into the muscle defect site of mice. The non-treated and uninjured groups served as the negative and positive control, respectively; the uninjured group was set as 100% for calculating the repair of tissues (Figure S8). After 1 month, in the non-treated group, the void at the defect area created by surgery remained visible from gross appearance (Figure 5(a)). And, there are no remaining hydrogels at the injury site in all cell-laden hydrogel groups. In the low (Figure 5(b)) and intermediate groups (Figure 5(c)), the atrophic appearance of the muscle partially attenuated compared with that observed in the non-treated group. Surprisingly, in the high pre-vascularization group, it was found that the created void was filled, and the surface gloss of the muscle was also restored (Figure 5(d)). To investigate the histological changes at the defect site 1-month post-injury, the front- and side-view sections of the repaired tissue were stained with Masson’s trichrome staining. Consistent with the observation of gross appearance, in the non-treated empty and low pre-vascularization group (right panel in Figure 5(b)), VML defect could still be seen and there was little tissue repair (right panel in Figure 5(a)). In the intermediate pre-vascularization group (right panel in Figure 5(c)), the repair diameters with more regenerated muscle and less dense connective tissues were larger by 1.5 times than that in the non-treated and low pre-vascularization groups. A significant increase in the volume of reconstructed tissue was observed in the high pre-vascularization group (reconstruction tissue and muscle ~ 90%). This treatment exerted a greater effect on muscle regeneration and reduction of the adipose and fibrosis connective tissues. (right panel in Figure 5(d)–(g)).

Volumetric skeletal muscle tissue reconstruction by implanting the vascularized cell-laden hydrogels pre-formed at the subcutaneous space for 7 days in the injury VML model at 4 weeks after implantation. (a) The non-treated empty group served as the negative control. According to the density of the engineered functional vascular networks inside, the pre-vascularized cell-laden hydrogels were classified as (b) CMA-20 (Low), (c) CMA-15 (Intermediate), and (d) Collagen (High) groups. Macro-images with different perspectives at the defect site after repair are presented (yellow-dashed line in the left column). Representative Masson’s trichrome staining images of the repaired construct in the front-view and side-view sections are shown in the middle and right columns. The inset was the enlarged region of connective tissue in the cell-laden hydrogel. The repaired muscle (M) and connective tissue (C) were labeled. Quantification of (e) whole reconstructed area, (f) muscle tissues area, and (g) distribution of connective and muscle tissues in the whole reconstructed area compared with those determined in the uninjured group at the defect site (the tissue areas in the uninjured group were set as 100%). (n = 4–5) *p <0.05, ** p < 0.01, and *** p < 0.001 indicate significant differences versus the CMA-20 (Low) and non-treated group. Hash symbols (#p < 0.05) indicate significant differences versus the CMA-15 (Intermediate) group. $p < 0.05 and $$p < 0.001 indicate significant differences versus their respective acellular groups.
Preformed perfused and well-connected vascular networks supported the formation of mature muscle fibers
Histological analyses were performed by double-staining, namely laminin (red) and MHC (green), to quantitatively calculate the distribution and average size of repaired muscle fiber bundles after 1 month (Figure 6(a)). According to the front-view section of the repaired muscle region in construct (Figure 6(b)), the non-treated group presented significantly higher numbers (1100 #/mm2) of myofibers, but with reduced size (<500 μm2 each), while the non-injury group showed fewer numbers of the bundle (~530 #/mm2) with the largest bundle area (>1500 μm2 each). The low and high pre-vascularization groups showed similar numbers of myofiber bundles (~900 #/mm2), while only ~600 #/mm2 myofiber bundles were noted in the intermediate group. The intermediate and high pre-vascularization groups demonstrated similar size distribution of myofiber bundles, which was larger than that recorded in the low pre-vascularization group. Based on the side-view section of the repaired construct (Figure 6(c)), there was no significant difference in the number and size of myofiber bundles between the groups; notably, the number in the low pre-vascularization group was slightly lower than in the other groups. Taken together with the results on repair muscle size (Figures 5 and 6), these results indicate improvement of muscle repair in the high pre-vascularization group and impairment in muscle regeneration in the non-treated and low pre-vascularization group.

Muscle regeneration at the repaired muscle region in VML defects at 4 weeks after implantation. (a) Representative laminin (red)/MHC (green)/nuclei (blue) staining images of the repaired muscle in VML defect in the front view (top row) and side view (second row) implanted by the non-treated, cell-laden CMA-20(Low), cell-laden CMA-15 (Intermediate), cell-laden Collagen (High), and non-injury groups. The non-injury group served as the positive control. Scale bar: 50 μm. (b and c) Quantification of muscle fiber bundle areas and numbers in the (b) front-view and (c) side-view sections of the repaired tissue construct. ***p < 0.1 indicate significant differences versus the non-treated group (n = 4–5).
Freshly prepared cell-laden hydrogels impeded muscle repair at the VML injury site
We sought to determine whether the improvement in regenerative capacity achieved using the high-degree perfused vascularized cell-laden hydrogel s for the treatment of muscle defects was due to a connection between the preformed well-connected vascular network and host vasculature. For this purpose, instead of subcutaneously implanting pre-vascularized tissue grafts, freshly prepared cell-laden Collagen (Fresh cell-laden Collagen) and CMA-15 (Fresh cell-laden CMA-15) hydrogels were directly implanted into the injury site after the creation of VML (serving as non-pre-vascularized groups). After 1 month, the size and area of the repaired connective and muscle tissues formed at the injury site and myofibers were analyzed (Figure 7). And, there are no remaining hydrogels at the injury site in both fresh prepared cell-laden hydrogel groups. Direct implantation of freshly prepared cell-laden Collagen hydrogels (Fresh cell-laden Collagen group) significantly impeded the repair in connective and muscle tissues compared with the cell-laden Collagen group. Moreover, the same effect was observed in the Fresh cell-laden CMA-15 groups, although it did not reach statistical significance (Figure 7(c) and (d)). This may be attributed to insufficient muscle repair in the cell-laden CMA-15 group. Furthermore, no significant difference was found in the area and number of repaired myofibers between the groups (Figure 7(e) and (f)).
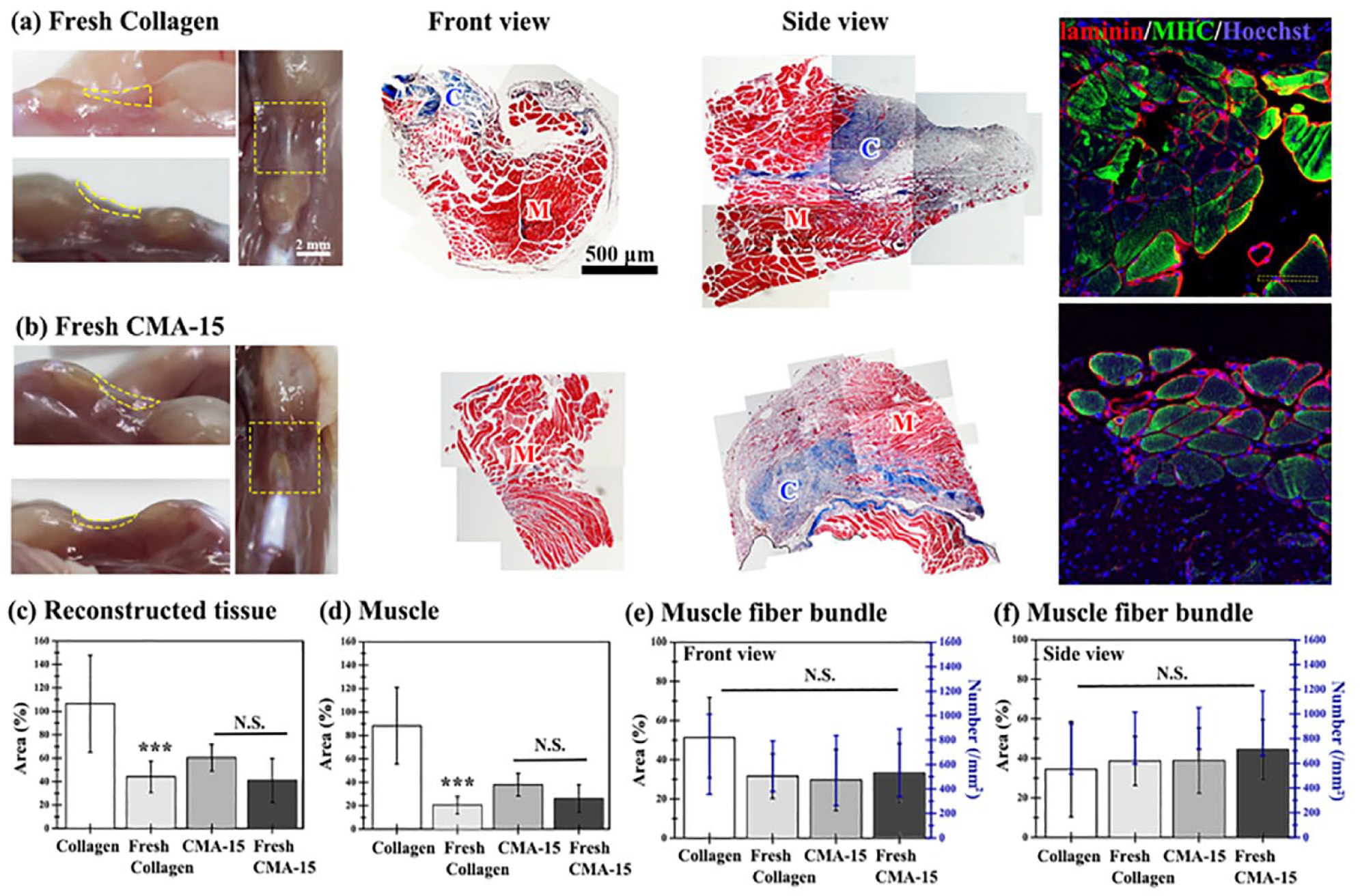
Volumetric skeletal muscle tissue reconstruction by directly implanting the freshly prepared cell-laden Collagen (Fresh Collagen) and CMA-15 (Fresh CMA-15) hydrogel in the injury defect site at 1 month after implantation. (a and b) Macro-images with different perspectives at the defect site after repair are presented (yellow-dashed line in the left column). Representative Masson’s trichrome staining images of the repaired construct in the front-view and side-view sections are shown. The repaired muscle (M) and connective tissue (C) were labeled. Representative laminin (red)/MHC (green)/nuclei (blue) staining images of implants in the front view (in the right column). (Scale bar: 50 μm) Quantification of (c) whole reconstructed area, (d) muscle, and muscle fiber bundles areas and numbers in the (e) front-view and (f) side-view sections of the repaired tissue construct (the uninjured group was set as 100%). *** p < 0.001 indicate significant differences versus the Collagen group (n = 3–4).
Fast anastomosis of vascularized cell-laden hydrogel with host micro-vessels at the site of injury to improve muscle repair
To further investigate the promotive effect of vascularized cell-laden hydrogel on muscle repair, the pre-vascularized cell-laden collagen hydrogels and the non-pre-vascularized freshly prepared cell-laden collagen groups were analyzed at different time points immediately after surgery. The findings revealed the progression of vasculogenesis, angiogenesis, and anastomosis at the injury site during the repair process (Figure 8).

Pre-vascularized cell-laden hydrogel construct formed at the subcutaneous site supported volumetric skeletal muscle tissue reconstruction through the successful connection between engineered vascular networks and host vasculature. (a) The representative H&E and hCD31+ images of the cell-laden Collagen group (Collagen) pre-formed at the subcutaneous site for 7 days before (pre-D7) and after 3 days of implantation in the muscle defect site. (b) The representative H&E and hCD31+ images of the freshly prepared cell-laden Collagen group (Fresh Collagen) at different time points after direct implantation into the muscle defect site (n = 3). Representative immunofluorescence staining of (c) Ly6G+(green) CD45+(red) neutrophils and (d) F4/80+ (green) CD45+(red) macrophages. Nuclei are stained with DAPI. (e) Quantification of neutrophils and macrophages in the Collagen group and the Fresh Collagen on day 3 (D3) after implantation.
In the pre-vascularized cell-laden collagen hydrogels group (Collagen group; Figures 8(a) and S9(b)), on day 3 after implantation (D3), all lumen structures were perfused and uniformly distributed in the constructs (~45 vessels/mm2). This indicates that the lumen structures were unchanged on day 3 compared to before implantation (51 vessels/mm2 on Day 0). This implied that angiogenic sprouting from the host vasculature and anastomosis with the engineered perfused lumens had occurred within 3 days after implantation. In the freshly prepared cell-laden Collagen group (Fresh cell-laden Collagen group; Figure 8(b)), on day 3 (D3), there were some perfused blood vessels (~20 lumens/mm2) at the peripheral site and some non-perfused lumen structures in the central region of the constructs. By day 5 (D5), the lumen density was at a similar level; however, a larger number of perfused vessels was observed in the central region. These findings demonstrate that the processes of vasculogenesis and angiogenesis were initiated together, whereas anastomosis from the edges of the constructs began later. By day 7 (D7), almost all the lumen structures located in the central regions had been perfused, thus increasing the density of perfused vessels (~53 vessels/mm2); these mature vessels were stabilized by day 10 after implantation (55 vessels/mm2; Figure 8(b) and S9(a)). To understand whether host myeloid cells, neutrophils, and macrophages, participate in the revascularization of vascularized cell-laden hydrogel here, immunofluorescent staining was performed at the injury site on day 3 after implantation (D3). More Ly6G+ CD45+ neutrophils were observed into the Collagen group compared to that in the Fresh Collagen group, while the densities of F4/80+ CD45+ macrophages were similar in both groups (Figure 8(c)–(e)). And, the area of perfused micro-vessels in the prevascularizd cell-laden collagen hydrogels (cell-laden Collagen PreD7+D3, pre-formed in the subcutaneous space of mice for 7 days, and exercised and implanted into the other mice for 3 days, Figure S9(b)) was three-fold larger than that observed in the non-prevascularized group (Fresh cell-laden Collagen D10, Figure S9(a)) for 10 days in animals. Moreover, the total density of perfused lumens in the freshly prepared cell-laden collagen group after 1-week (Fresh cell-laden Collagen D7: ~53 vessels/mm2) was similar to that recorded for the vascularized group at the subcutaneous site (Collagen PreD7: ~45 vessels/mm2); nevertheless, the average area of micro-vessels remained significantly smaller. In the freshly prepared cell-laden group, there was a time delay in forming blood vessels and the perfusion process. To clarify whether this time delay significantly affected muscle repair, we extended the observation time to 2 months after surgery (Figure S10). Results showed in the prevascularization group (cell-laden Collagen group; Figure S11(a)–(c)), the total area of reconstruction tissue remained constant, but the muscle area increased by 1.5 times from 2 to 3 mm2 after 2 months. In the non-prevascularization group (Fresh cell-laden Collagen group; Figure S11(a)–(c), the total area of reconstruction tissue significantly increased from 0.5 to ~ 3 mm2, and the area of muscle significantly increased to 3 mm2, which is similar to that in the prevascularization cell-laden Collagen group.
Neuromuscular regeneration after critical VML
To characterize the innervation of motor neurons and repaired myofibers at the injury site, we performed immunofluorescent staining sections for β-Tubulin III on the repaired tissue construct to visualize the presence of neurons (Figure 9(a) and (b)). The healthy uninjured group of the same age of mice was used as the positive control. At 1 month after implantation, similar percentages of β-Tubulin III-positive neurons surrounded by repaired myofibers were observed in the uninjured group and pre-vascularized cell-laden Collagen group, which are more than that in the freshly prepared cell-laden Collagen group (Figure 9(b)). Besides, the total repaired muscle tissue in the pre-vascularized cell-laden Collagen group is 4 times larger than that in the Fresh cell-laden Collagen group (Figure 7(d)). To confirm these differences in muscle repair, the walking behaviors of mice treated with the uninjured healthy, non-treated group, cell-laden Collagen group and Fresh Collagen groups were recorded after 1 month of treatment (Video S1). In the Fresh cell-laden Collagen group, mice displayed abnormalities in exploratory behavior, such as insufficient strength in the back-left leg. The results were in line with the quantification data on motor neuron innervation and the amount of repaired muscle volume observed at the injury site 1-month post-surgery. These experiments suggested that the cell-laden Collagen group pre-formed at the subcutaneous site for 7 days compared to the freshly prepared cell-laden Collagen group, promoted functional recovery resulting from accelerated tissue regeneration.
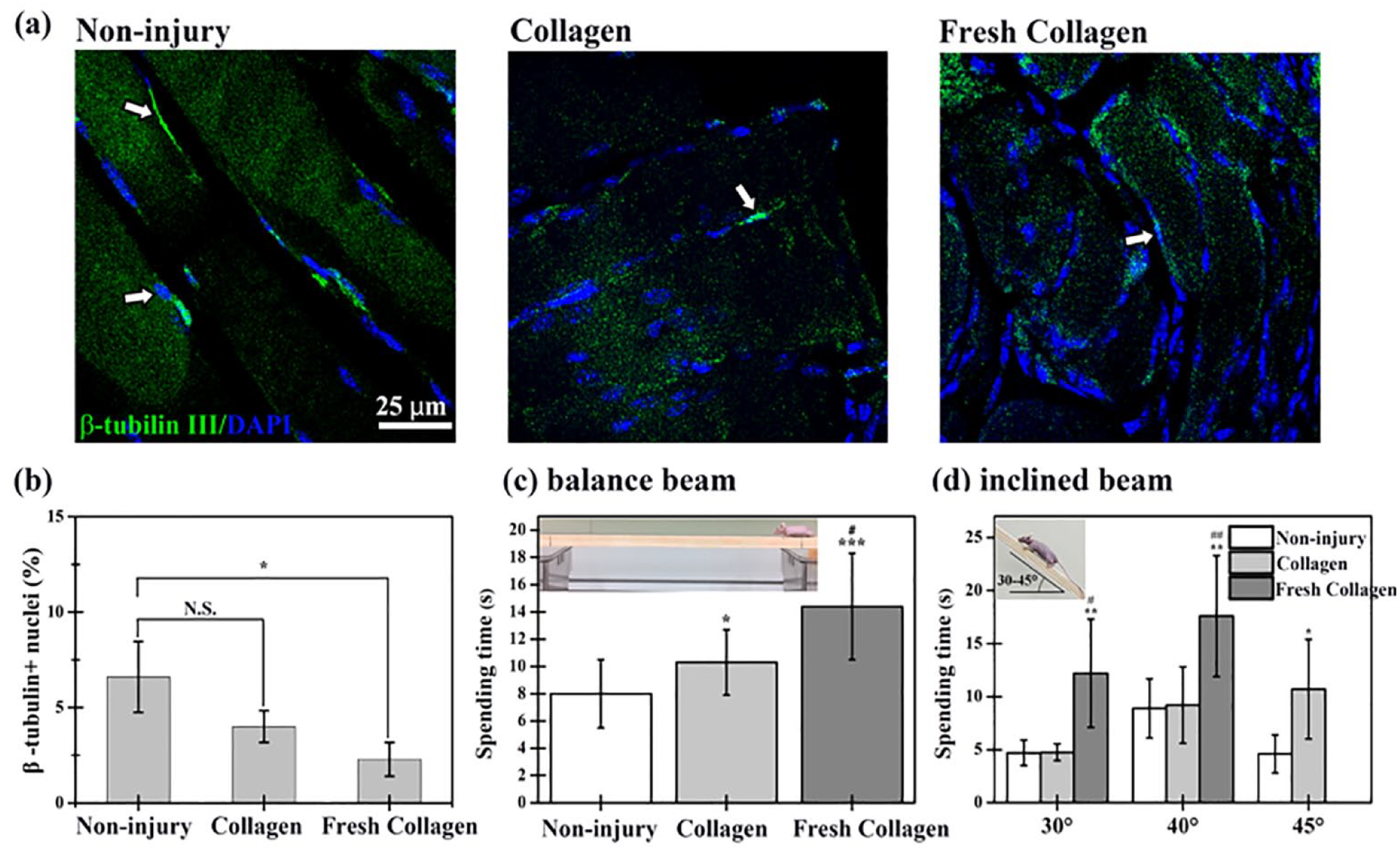
Pre-vascularized cell-laden hydrogel promoted the repair of damaged skeletal muscle and the recovery of motor function in mice. (a) Representative immunofluorescent staining for innervation (β-Tubulin III, green) in the front-view sections of the non-injury group, the pre-vascularized cell-laden Collagen group (Collagen), and the freshly prepared cell-laden Collagen group (Fresh Collagen) at 1-month after implantation. (b) The quantitation analysis of the β-Tubulin III-positive neurons in repaired muscle regions for 1-month post-surgery. White arrows indicate β-Tubulin III-positive neurons. (c) Motion functional deficits were assessed by the balance beam and inclined plane test (n = 6). (*p < 0.05, **p < 0.01, and ***p < 0.001 as compared to the non-injury group. #p < 0.05 and ##p < 0.01 as compared to the Collagen group. N.S. means no significant difference).
These results demonstrated that the time delay of muscle repair was observed in the freshly cell-laden group compared to that in the vascularized tissue graft for muscle repair. Moreover, uninjured muscle tissues at the quadriceps muscle on the right foot of injured mice were also investigated to understand the injured effects on the other uninjured foot (Figure S11(d)–(e)). Results showed that the muscle area on the right uninjured quadriceps significantly increased with time after injury (Figure S10(e)), implying that the VML injury caused extra weight loading on the other uninjured foot to increase its strength during recovery, like rehabilitative therapies. The use of well-connected perfused vascular networks flap at the defect site immediately after injury plays an important role in the following muscle remodeling and repair process. These findings demonstrate that our pre-engineered human vascular networks connected and revascularized with host vasculature within 3 days after implantation and further supported muscle tissue repair and regeneration. In summary, these results show that the prevascularized cell-laden hydrogels provided a permissive environment for the formation of blood vessels, which further supported muscle repair at the damage site.
Discussion
Acellular scaffolds and decellularized extracellular matrix-based scaffolds are more clinically translatable with a lower regulatory burden. However, their use in the treatment of VML defects showed significant deposition of fibrotic tissue post-transplantation, resulting in limited regeneration and incompletely restore of muscle strength.36,37 Lack of a timely and sufficient supply of blood after implantation of engineered tissues leads to graft failure with tissue necrosis; hence, pre-vascularization of large-sized and thick implanted tissue is expected to enhance its long-term survival.38–41 Functional skeletal muscle regeneration also greatly depends on the ability of scaffolds to rapidly initiate revascularization through the induction of vasculogenesis, angiogenesis, and anastomosis with host vasculature to meet the high metabolic demands of muscle. To address vascularization, pro-angiogenic cells alone or together with stem cells were seeded on or encapsulated into scaffolds. These scaffolds were subsequently directly implanted into the injury site to induce vascularization at the VML defect site.10,42,43 These findings implied that genetically engineered cell-laden scaffolds enhanced host neovascularization and myogenesis compared with the non-secreting control. However, vascularization required considerable time and the perfused vessels were mainly located at the border region of scaffolds, with only a few or no vessels present in the central region. Moreover, the preculture of constructs in vitro and/or incubation in vivo prior to implantation typically requires >1–2 weeks, prolonging muscle defect recovery. The contribution of these pretreatments to muscle repair may not be required, so there is a strong need to develop large-sized vascular tissue constructs, which can be formed quickly to improve the repair of muscle tissue in VML defects. Studies cultured capillary-like tubular structures in vitro to examine the distribution of cells and vascular networks to the biomimetic organ or tissue-like structures. The results demonstrated that these lumen structures were perfused by host vasculatures after implantation.16,44 Furthermore, the transplanted stem cells remained present and even integrated with host tissues after 2–3 months. However, this observation raised some concerns regarding the risk of the transformation of stem cells into cancer cells. Thus, vascularized grafts should improve tissue repair by providing a temporary network to supply sufficient nutrients and oxygen to surrounding progenitors or stem cells for repair, which will eventually be replaced by host blood vessels within a short period to reduce the risk of carcinogenesis.
Here, we propose an alternative approach to improve the oxygen supply and diffusion in thick tissues by engineering large-scale vascular networks in vivo instead of in vitro. The subcutaneous space is an attractive alternative site for engineering large-scale vascular networks due to the minimally invasive approach for implantation, transplantation, graft monitoring, and retrieval. 45 However, the subcutaneous tissue is not extensively vascularized, and cell encapsulation devices deployed in this tissue often fail to achieve the level of vascularization necessary to maintain the function of thick grafts.46,47 Several studies24,27,48 demonstrated that manipulation of the physical properties of hydrogels (i.e. collagen- or gelatin methacrylate-based hydrogels) could regulate the extent of cell-mediated vascular network formation in vivo at a subcutaneous site. The non-inflammatory host neutrophils at the implantation site play an important role in improving the engraftment of bioengineered tissues. 27 Most studies conducted thus far focused on the creation of engineered vascularized tissue at the subcutaneous site (i.e. not at the injury site), while few studies further investigated the functions of these perfused vascularized tissue constructs in muscle repair. Shandalov et al. 13 prefabricated grafts by in vitro culture and implanted them around the veins in vivo as a pre-vascularized graft to an abdominal wall VML defect site. Nevertheless, they observed limited improvement in the abdominal wall VML defect, which may be due to the preculture of explants for 10 days in vitro prior to implantation. Recently, our group 24 showed that these engineered vascular networks could support the in situ formation of vascularized adipose tissue at the subcutaneous site in the absence of exogenous cytokines using a covalently crosslinked cell-laden hydrogel after 1 month by manipulating the polymerization properties of a hydrogel.
Organic solvents, high pH values, and temperatures during the modification process easily denature collagen molecules. Therefore, the chemical reaction in water with neutral pH values is required to maintain physiological stability and preserve the native structure of proteins. The CD spectrum of CMA was similar to that of native collagen, indicating the retention of the triple helical assembly after the modification of collagen’s lysins. Moreover, none of the samples exhibited a positive peak in the region around 222 nm—the typical collagen fingerprint in the α helix conformation. This finding is consistent with other studies on isolating collagen from various sources and through different extraction methods.49–51 The modification of collagen using methacrylate functional groups has been previously reported in the literature.23,31,32,52 However, the low modification rate achieved, the alkaline environment, and the use of organic solvent in the modification process resulted in excessive use of crosslinkers, unreactive residues of reactants/solvents, and changes in the conformation of collagen molecules. These effects limited the use of this method in cell-mediated blood vessel formation for vascular tissue engineering. So far, the potential of photocrosslinked methacrylated collagen as a functional scaffold to produce controllable vascular networks has not been investigated.
Indeed, it’s intriguing to note that a subgroup of HUVECs indeed displayed varying degrees of α-SMA expression, as now clarified by the staining results (Figure 2(b)). Notably, the yellow arrows pinpoint the presence of cells that are both hCD31+ (green) and α-SMA+ (red) among HUVECs. In contrast, the transparent yellow arrows highlight HUVECs that exclusively display hCD31+ (green) staining. These observations validate the existence of different levels of α-SMA expression in HUVECs, which concurs with the findings of the published article. 53
In the present study, we successfully engineered large vascularized tissues with high, intermediate, and low densities of blood vessels inside the cell-laden collagen, CMA-15, and CMA-20 hydrogel groups, respectively. Following the transplantation, the effects of these perfused vascularized tissues as transferable vascular tissue construct were evaluated in the VML defect in mice. Using acellular CMA-20, CMA-15, and Collagen grafts without microvessels (Figure S4) as implants resulted in limited muscle repair, accounting for less than 60% of the entire reconstructed tissues and 10-40% of the repaired muscle tissues when compared with the uninjured group (Figure 4). When engineering various densities of perfused vessels in CMA-20, CMA-10, and Collagen hydrogels (Figure 3), the results indicated that the CMA-20 hydrogel with (CMA-20 (low)) or without (acellular CMA-20) vessels only supported 20% ± 10% of muscle repair (Figures 4(a), (e), 5(b) and (e)). In contrast, there was a significant increase of twofold to 40% ± 8% and 85% ± 35% in the area of repaired muscle in the CMA-15 (Intermediate) and Collagen (High) groups, respectively, compared to their respective acellular hydrogel groups (Figures 4(b), (c), 5(c) and (d)). These results demonstrated that incorporating perfused vessels improved muscle repair in collagen-based hydrogels, regardless of their crosslinking degrees and methods. Applying freshly prepared cell-laden collagen hydrogel directly at the injury site hindered muscle repair compared to the Collagen group with preformed vessels (Collagen (High) group). This was likely due to a time delay in forming blood vessels and initiating the perfusion process (Figure 8). Furthermore, both the CMA-15 (Intermediate) and the freshly prepared CMA-15 hydrogel groups showed comparable muscle repair levels of 20-40% (as shown in Figure 7). This similarity could be attributed to the low vessel density in the CMA-15 groups (14.2 ± 2.0 vessels/mm²). Indeed, these results suggest that a high vessel density (Collagen (High) group: 51.5 ± 21.1/mm2) and t the capability to anastomose with the host vasculature at the injury site rapidly play crucial roles in achieving muscle repair up to 85 ± 35% (Figure 5(c) and (d)) and facilitating the recovery of motor function (Figure 9).
Previously, our group have shown that host neutrophil engagement improves the engraftment of implants containing unassembled vascular cells. For the implants containing assembled human lumen structure cultured in vitro for 7 days fail to engraft owing to their inability to engage non-inflammatory host neutrophils upon implantation into mice. 27 Here, in the perfused vascularized cell-laden hydrogel group, well-preformed vascular networks caused more non-inflammatory neutrophils to rapidly integrate with the host vasculatures within 3 days compared with the freshly prepared cell-laden hydrogel (Figure 9(c)). Moreover, 1 month after implantation, the transplanted human cells in the Collagen (High) group were only 2.6% ± 0.7% existed (positive for human nuclei, Figure S12) at the site of VML injury, implying that the vascularized cell-laden hydrogel served as a temporary graft with vascular networks at the defect site and accelerated the migration of surrounding progenitor or satellite cells for repair. We demonstrated the capability and function of the vascularized cell-laden hydrogel construct in the repair of VML defects and showed that its use as a temporary vascular network could avoid the need for the transfer of large volumes of tissue from another site and eliminate postoperative injury of the donor site.
Conclusions
Vascularized tissues with high, intermediate, and low densities of blood vessels inside the cell-laden collagen-based hydrogel were successfully engineered through tailoring hydrogel properties. Following the transplantation into VML injury site, the effective muscle repair is largely determined by the degree of vascularization to enhance host neovascularization, and time delay of integration is observed in the freshly cell-laden group. The results suggest that using highly pre-vascularized cell-laden Collagen group for constructing muscle defects immediately after injury will be more beneficial than the implantation of freshly prepared cell-laden constructs (Fresh Collagen) in terms of neovascularization, which is crucial for successful engraftment. Vascularized cell-laden hydrogel constructs became more vascularized by host blood vessels through more rapid and effective integration within the host tissue than the freshly prepared cell-laden constructs. Additionally, the formation of vascularized constructs at a subcutaneous site provides transfer and scale-up capabilities as a free tissue to reconstruct defects in other muscle areas of larger animals and humans.
Supplemental Material
sj-docx-1-tej-10.1177_20417314231201231 – Supplemental material for Reconstructing vascular networks promotes the repair of skeletal muscle following volumetric muscle loss by pre-vascularized tissue constructs
Supplemental material, sj-docx-1-tej-10.1177_20417314231201231 for Reconstructing vascular networks promotes the repair of skeletal muscle following volumetric muscle loss by pre-vascularized tissue constructs by Chih-Long Chen, Shih-Yen Wei, Wei-Lin Chen, Ting-Lun Hsu and Ying-Chieh Chen in Journal of Tissue Engineering
Footnotes
Acknowledgements
The authors acknowledge the core facility of the Multiphoton and Confocal Microscope System at the College of Biological Science and Technology, National Yang Ming Chiao Tung University, Hsinchu, Taiwan for cell imaging. We would like to thank Uni-edit (![]() ) for editing and proofreading this manuscript.
) for editing and proofreading this manuscript.
Author contributions
Wei-Lin Chen and Ting-Lun Hsu contributed to designing and characterizing materials, in vitro cell culture work, establishing the VML animal model, and data analysis. Shih-Yen Wei performed histological staining and animal surgery on the formation of vascularized cell-laden hydrogel at the subcutaneous site of mice. Ying-Chieh Chen and Chih-Long Chen conducted research, interpreted results, and wrote the manuscript. All authors discussed the results and provided suggestions for the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 109-2628-E-007-001-MY3 and MOST 111-2221-E-007-017-MY3) and the Shin Kong Wu Ho-Su Memorial Hospital of Taiwan grant (2022SKHADR024).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.