
Abstract
Due to a relative paucity of studies on human lymphatic assembly in vitro and subsequent in vivo transplantation, capillary formation and survival of primary human lymphatic (hLEC) and blood endothelial cells (hBEC) ± primary human vascular smooth muscle cells (hvSMC) were evaluated and compared in vitro and in vivo. hLEC ± hvSMC or hBEC ± hvSMC were seeded in a 3D porous scaffold in vitro, and capillary percent vascular volume (PVV) and vascular density (VD)/mm2 assessed. Scaffolds were also transplanted into a sub-cutaneous rat wound with morphology/morphometry assessment. Initially hBEC formed a larger vessel network in vitro than hLEC, with interconnected capillaries evident at 2 days. Interconnected lymphatic capillaries were slower (3 days) to assemble. hLEC capillaries demonstrated a significant overall increase in PVV (p = 0.0083) and VD (p = 0.0039) in vitro when co-cultured with hvSMC. A similar increase did not occur for hBEC + hvSMC in vitro, but hBEC + hvSMC in vivo significantly increased PVV (p = 0.0035) and VD (p = 0.0087). Morphology/morphometry established that hLEC vessels maintained distinct cell markers, and demonstrated significantly increased individual vessel and network size, and longer survival than hBEC capillaries in vivo, and established inosculation with rat lymphatics, with evidence of lymphatic function. The porous polyurethane scaffold provided advantages to capillary network formation due to its large (300–600 μm diameter) interconnected pores, and sufficient stability to ensure successful surgical transplantation in vivo. Given their successful survival and function in vivo within the porous scaffold, in vitro assembled hLEC networks using this method are potentially applicable to clinical scenarios requiring replacement of dysfunctional or absent lymphatic networks.
Keywords
Introduction
Human blood endothelial cells from various sources (including primary microvascular endothelial cells, HUVECs and human induced pluripotent stem cell-derived endothelial cells (hiPSC ECs)) have been used successfully1–4 to assemble capillary networks in vitro, with and without parenchymal cells, and are generally seeded into scaffolds or matrices (called a construct). In vitro formation of blood capillaries is termed pre-vascularization. These constructs can be transplanted in vivo and the construct’s capillary-like structures have been shown to functionally join (inosculate) with host vessels providing a blood flow in the construct within 2–3 days. 5 This is a more efficient approach to providing an in vivo blood supply throughout the construct than if vascularization relies entirely on capillary ingrowth from surrounding host issues, which is very slow and generally results in partial construct necrosis through delayed vascularization. The pre-vascularization approach is used in neo-organoid tissue engineering models to enhance vascularization and maturation of the neo-organ.6,7 However lymphatic endothelial cell assembly in vitro into lymphatic capillaries and their fate in vivo has been studied rarely.8,9 The laboratory formation of lymphatic capillary networks has a number of purposes including for drug studies and in lymphatic disease modelling. It also has potential in regenerative medicine approaches to replace deficient or damaged lymphatics.10,11
The lymphatic vascular network is an essential second vascular system responsible for homeostatic fluid drainage from the extracellular space back to the blood vascular system, immune cell surveillance, immune cell movement throughout the body and lipid absorption. 12 Lymphatic vessels are present in most mammalian tissues and organs, with some exceptions including the eye lens, cornea, cartilage, bone marrow, central nervous system12,13 and skin epidermis. Compared to the blood vascular system, the lymphatic system has a lower fluid flow, lower pressure and is less coagulable because it generally conveys very few platelets and red blood cells. 13 Lymphangiogenesis occurs during foetal development via budding from cardinal vein endothelium, and formation of a branching lymphatic network that is separate from the arterial and venous blood vessel system 13 although independent lymphatic vessel formation from lymphangioblasts in the mesenchyme has also been proposed. 13 Lymphangiogenesis also occurs in a number of pathophysiological conditions in adults including in wound healing, and tissue inflammation. Lymphangiogenesis is controlled by a number of growth factor pathways – the most commonly recognized being VEGF-C activating VEGFR-3 to promote proliferation, migration and survival of LECs. 14 However, other growth factors including FGF-2 and FGFR can also promote LEC proliferation and migration. 14 The Angiopoietin 2/Tie2 pathway is an important signalling pathway in lymphatic differentiation and maturation, but not lymphatic sprouting 10 and PDGFR and PDGF-BB also have roles in LEC migration and lymphatic vessel formation. 14
The lymphatic system comprises specialized blind ending capillaries (termed initial lymphatics, terminal lymphatics or lymphatic capillaries) 12 which drain into larger pre-collectors and then collecting lymphatics that take the interstitial fluid and immune cells back to lymph nodes and the central venous system. The lymphatic capillaries and the collecting ducts are lined internally by lymphatic endothelium which share some cell markers with blood vascular endothelium (CD31, vWF, VE-cadherin). 15 LECs also express cell markers not expressed by blood microvascular endothelial cells including PROX1, LYVE-1, podoplanin and VEGFR3.14–16 Structurally the lymphatic capillary differs from the blood capillary in that they are wide blind ending sacs and have a discontinuous basal lamina, and overlapping endothelial cells.13,17
Approaches for lymphatic network regeneration have focused on models of skin wound healing or more commonly lymphoedema (chronic swelling of the limbs because of interstitial fluid accumulation due to lymphatic capillary loss or disfunction). Apart from surgical techniques such as lymphatic-venous anastomoses to restore lymphatic function in cases of damaged, non-functioning lymphatic networks in lymphoedema, 18 a variety of mostly experimental techniques have been investigated to stimulate lymphangiogenic growth in vivo, including injection of lymphangiogenic growth factors such as VEGF C, PDGF, FGF, Angiopoietin, 14 or growth factor encoded gene delivery.19–21
Specific scaffolds such as collagen gels in a mouse tail wound promoted lymphangiogenesis, 22 and a nanofibrillar collagen material (Biobridge) – positioned in a region of lymphatic obstruction in the pig increased lymphatic collector ducts over 3 months. 23 Scaffolds incorporating fibrin-binding VEGF-C have (exclusively) promoted lymphangiogenesis in wound models. 24 Engineering of collecting ducts has also been attempted, with decellularized human arteries seeded with adipose derived lymphatic-like cells used as replacements for collecting lymphatic vessels. 25
Direct injection of lymphatic cells of various types including human induced pluripotent stem cell-derived LECs in wounds, 26 and lymphoedema, 27 and primary human LECs in rat tail lymphoedema in nude rats 28 have had positive effects on lymphatic tissue growth.
In vitro tissue engineering studies have produced three-dimensional interconnected lymphatic networks in a variety of support materials.8,9,29–32 Two studies have included blood and lymphatic vessels in a human skin equivalent assembled in vitro.8,32 Marino et al. 8 took the skin equivalent into an in vivo model. Landau et al. 9 assembled hLEC with fibroblasts or dental pulp stem cells and blood endothelial cells in collagen sheets or PLLA/PLGA scaffolds and investigated the effects of cyclic stretch on lymphatic growth, and also transplanted the constructs into the abdominal wall where they integrated well with anastomosis to host lymphatics. Tissue engineering studies especially those where constructs are assembled in vitro could potentially accelerate the functional recovery of diseased or damaged lymphatic vessels in vivo. 14
This study has focused on in vitro assembly of human LECs into lymphatic capillaries and the efficiency in which these lymphatics can be transplanted and survive in vivo. Human LEC (hLEC) ± human vascular smooth muscle cells (hvSMC) in human fibrin were seeded within a porous polyurethane scaffold and lymphatic capillaries assembled. These three-dimensional (3-D) lymphatic networks were compared to human blood microvascular endothelial networks (hBEC) ± hvSMC assembled under identical conditions. The study compared various microscopic observations of 3-D assembly, and morphometric parameters of hLEC and hBEC in 3-D culture, their vascular markers in vitro and in vivo, and their survival and vessel morphometric parameters, and linkage to host vessels when implanted in vivo in sub-cutaneous wounds. The study has established that human primary lymphatic endothelial cells can readily assemble into lymphatic vascular networks in vitro, with potential applications in regenerative medicine as these engineered lymphatics demonstrated good survival in vivo.
Materials and methods
Materials
Primary human dermal microvascular endothelial cells were obtained from Lonza (HMVEC-dBlAd; Basel, Switzerland) and Cascade (HMVECad; Portland, USA) respectively, and were both cultured in Endothelial Basal Media-2 supplemented with EGM2-MV Bulletkit (Lonza). Human dermal lymphatic endothelial cells (hDLEC) and human cardiac microvascular endothelial cells (HCMEC) were from PromoCell (Heidelberg, Germany) and were cultured in Endothelial Basal Media-2 supplemented with EGM2-MV Bulletkit (Lonza). hDLEC and HCMEC were used as positive controls to determine the Lonza primary cells (HMVEC-dBlAd) were blood microvascular endothelial cells (subsequently called hBEC) and the Cascade Biologics cells (HMVECad) were lymphatic microvascular endothelial cells (subsequently called hLEC). Primary human coronary artery smooth muscle cells (hvSMC) and media (SMC Growth Medium 2) were from PromoCell (Heidelberg, Germany). All cell types were used up to passage 6.
Sterile, circular NovoSorb™ polyurethane porous scaffolds of 6 mm diameter, and 1.3–1.6 mm thickness with interconnected pores of 300–600 μm diameter were manufactured by PolyNovo Biomaterials Ltd. (Port Melbourne, Australia). Fibrinogen and thrombin from human plasma were from Sigma-Aldrich (St Louis, USA).
Animal experiments were approved by St Vincent’s Hospital, Melbourne Animal Ethics Committee (Protocols 32/13, 20/17 and 25/20). Twenty-five healthy male nude (CBH-rnu/Arc) rats were purchased from Animal Resources Centre, Perth, Western Australia. Rats underwent the first operative procedure between 8 and 12 weeks of age. Rats were housed individually in Tecniplast (Tecniplast Australia Pty Ltd, Lane Cove, New South Wales) ventilated isolator cages in a PC2 certified room in the Experimental and Medical Research Unit at St Vincent’s Hospital, Melbourne. Rats were kept in a 12 h light/12 h dark cycle, at a temperature of 19°C–21°C, and humidity of 35%–45%. Temperature and humidity were recorded daily. Rats were fed Barastoc rat and mouse cubes (irradiated) (Ridley AgriProducts Pty Ltd., Melbourne, Australia) and water ad libitum. No wild animals were used in the study and no field collected samples were obtained. All animal procedures were conducted in accordance with National Health and Medical Research Council of Australia guidelines.
Methods
Immunocytochemistry of plated primary human endothelial cells
Endothelial cells were detached with TrypLE (Life Technologies, Carlsbad, USA) and 5000–6000 cells replated into 8-well Millicell EZ slides (Merck, Darmstadt, Germany). Forty-eight hours later, cells were fixed with 4% paraformaldehyde (PFA) for 30 min, (blocked with (10% (v/v) goat serum, 30 min) and overlaid with mouse anti-human CD31 antibody for 60 min (clone JC70A, 1:100, Dako, Santa Clara, USA), or mouse anti-podoplanin (clone D2-40, 1:100, Dako). After washing with PBS-T (PBS + 0.05% (v/v) Tween20), secondary anti-mouse IgG-AlexaFluor488 conjugate (1:200, Life Technologies, CA, USA) was applied for 60 min followed by DAPI (10 ng/mL, Sigma-Aldrich) for 10 min before being washed and coverslips mounted with Fluorescent mounting media (Dako).
Seeding of NovoSorb scaffolds with primary ECs and hvSMCs and in vitro culture
Scaffolds were seeded using methods previously described. 4 Briefly, 6 mm diameter sterile circular scaffolds were pre-wetted in Dulbecco’s phosphate-buffered saline (DPBS, Sigma-Aldrich) prior to seeding. Excess saline was removed prior to the addition of 30 μL of fibrinogen (15 mg/mL in DPBS) containing 4 × 105 hLEC or hBEC with or without 2 × 105 hvSMC and 6 μL of thrombin (25 U/mL) by gentle scaffold compression which when released allowed ‘suction’ of cells/fibrin into the pores. Following uptake of the cells, scaffolds were held at room temperature for 5 min before being transferred to a 24-well plate containing 2 mL growth media (EGM2-MV or EGM2-MV + SMC growth Medium 2) and cultured for 1, 3 or 7 days. Media was replaced every other day. For co-cultures, EGM2-MV and SMC culture media were used at a ratio of 2:1.
Ratio of endothelial cells:vascular smooth muscle cells
Our choice of hEC:hvSMC ratio was dictated by a number of factors. We had previously used this ratio in a hiPSC EC ± hvSMC study in the same scaffold material. 4 Other in vitro engineering studies report quite divergent EC:support cell ratios (5:1 to 1:3 blood EC:support cells; 3:2 to 1:3 lymphatic EC: support cells).1,2,8,9,33,34
The EC:pericyte ratio in the literature for human blood capillary networks appears to vary depending on the tissue site, and may depend on the degree of tightness of inter-endothelial cell junctions and the level of microvascular blood pressure. 35 Ratios vary dramatically from 1:1 in the retina to 100:1 in striated muscle. 35 For skin blood capillaries pericyte coverage has been reported to be less than 80%. 36 Human skin lymphatic capillaries do not have a support cell coverage, 37 yet most tissue engineering studies use support cells for the formation of lymphatic capillaries.
Given that: (1) Physiologically there are generally more endothelial cells than vascular support cells in human capillary networks, (2) Perivascular cells often overgrow cultures, and this had to be avoided, and (3) Blood ECs and lymphatic ECs were assembled under identical conditions, so a direct comparison could be made between the two endothelial cell types in in vitro and in vivo conditions. We therefore chose an EC:hvSMC ratio in a mid-position (2:1).
Immunohistochemistry of seeded scaffolds following in vitro culture
Following in vitro culture, scaffolds were fixed in 4% PFA at 4°C overnight prior to processing and paraffin embedding. Cross sections (5 μm thickness) were subjected to immunolabelling with mouse anti-human CD31 antibody (clone JC70A, 1:100, Dako), mouse anti-human podoplanin (clone D2-40, 1:100, Dako) or mouse anti-human α smooth muscle actin (α-SMA) antibody (clone 1A4, 1:500, Dako) to label hBECs, hLECs and hvSMCs, respectively. Primary and secondary antibodies were diluted in antibody diluent (Dako) and washes used PBS-T. For hLEC/hBEC immunolabelling, dewaxed sections were incubated with human CD31, or podoplanin-specific antibody overnight at 4°C, following antigen retrieval (30 min, 10 mM citric acid buffer, pH 6.0, 95°C), quenching (3% (v/v) hydrogen peroxide, 5 min) and protein blocking (Dako, 10 min) steps. For immunostaining of hvSMCs, α-SMA antibody was applied overnight at 4°C. Sections were then incubated with polyclonal rabbit anti-mouse immunoglobulin biotinylated conjugate (30 min, 1:200, Dako). Specific antibody binding was detected using Vectastain Elite ABC kit (30 min, Vector Laboratories, Burlingame, USA) for hCD31 and podoplanin, or Streptavidin-HRP conjugate (1:400, Dako) for α-SMA, and DAB Substrate Chromogen System (Dako) according to manufacturer’s instructions. Sections were counterstained with haematoxylin and coverslips mounted with Entellan (Merck, NJ, USA).
Immunofluorescence of frozen sections following in vitro culture
All steps were completed at room temperature unless otherwise specified. Scaffolds were fixed for 4 h in 10% neutral buffered formalin (Trajan Scientific, Australia), washed and submerged in Tissue-Tek OCT (optimal cutting temperature) compound (Sakura Finetek, Tokyo, Japan) for 16 h. Samples in OCT were transferred to cryomolds and frozen prior to sectioning. For immunofluorescence, 6 micron thick sections were washed and placed in antigen retrieval solution (10 mM citrate buffer, pH 6.0) at 95°C for 20 min followed by permeabilization buffer (0.2% (v/v) Triton X-100 in PBS) for 10 min then 10% (v/v) goat serum blocking solution for 30 min. Sections were overlaid with primary antibody (rabbit polyclonal CD31 antibody, 1:50 Thermo Fisher Scientific), mouse monoclonal podoplanin antibody (clone D2-40, 1:100, Dako), mouse monoclonal smooth muscle actin (clone 1A4, 1:100, Dako) diluted in 5% (v/v) goat serum, overnight at 4°C. Secondary goat anti-rabbit IgG-AlexaFluor594 and anti-mouse IgG-AlexaFluor488 conjugates (5 mg/mL, Life Technologies) diluted in 5% (v/v) goat serum were applied for 60 min prior to incubation with DAPI (1 mg/mL, Sigma) for 10 min. Sections were washed and coverslips mounted with fluorescence mounting medium (Dako).
Immunostaining and microscopy of whole mounts
Immunostaining of wholemounts
Scaffolds prepared with hBEC ± hvSMC, or hLEC ± hvSMC (N = 1–2 scaffolds per cell seeding protocol, per time point) were infiltrated with 4 × 105 hBECs or hLECs ± 2 × 105 hvSMC in human fibrin and cultured in vitro for 48–96 h. Scaffolds were then fixed with PFA at 4°C, and subjected to permeabilization for 30 min in 0.5% (v/v) Triton X-100/PBS with gentle agitation, followed by Ultravision Protein Block (Thermo Fisher Scientific, KS, USA) for 15 min. Scaffolds were then submerged in mouse anti-human CD31-specific antibody (clone JC70A, 1:40, Dako) for 2 h with agitation. hBEC scaffolds were washed (PBS-T) and incubated with secondary antibody (goat anti-mouse IgG AlexaFluor555 conjugate, 1:200, Life Technologies) for 2 h followed by DAPI (0.5 µg/mL) for 30 min. hLEC whole mount scaffolds were permeabilized for 90 min, blocked with Ultravision Protein Block for 90 min, and the primary antibody (mouse anti-human podoplanin, 1:20) applied overnight with SM22 (1:50, Abcam, Cambridge, United Kingdom), overnight. hLEC scaffolds were then washed (PBS-T) and incubated with secondary antibodies, goat anti-mouse AlexaFluor555 and goat anti-rabbit AlexaFluor488, were also applied overnight followed by DAPI for 90 min. Washed scaffolds were then infiltrated sequentially by 25%, 50% and 75% sucrose for 1 h each and mounted in 75% sucrose containing 0.5% n-propyl gallate (Sigma Aldrich) in an imaging chamber (CoverWell™ imaging chambers, Grace-Bio Labs, Bend, OR. USA) comprised of a silicone ring sealed to a square 20 mm glass coverslip.
Confocal and Thunder microscopy of wholemounts
Microscopy was performed using a Nikon A1R confocal microscope (Tokyo, Japan), or a Leica Thunder microscope (Wetzlar, Germany). On the confocal, image stacks were acquired with a 20x or 40x (oil) lens with 512 × 512 resolution, and 561 and 635 nm lasers. Z-step size was 1.2 μm for the 20x acquisition and 0.5 μm for the 40x acquisition. On the Thunder system, stacks were acquired with a 20x or 63x (oil) lens and with 0.5 or 0.21 µm z-steps, respectively. All stacks are presented as either maximum intensity projections or volume views.
In vivo studies
Surgical implantation
Scaffolds seeded with hLEC ± hvSMC or hBEC ± hvSMC were cultured in vitro for 1 h, 1 day or 3 days, prior to implantation under the dorsal skin in nude adult male rats (CBH-rnu/Arc). Under sterile conditions and general anaesthesia and analgesia, a midline incision was made on the rat back and the skin on both sides lifted up, and two scaffolds on each side spaced, at 2 cm apart, were sutured to the overlying skin (Figure 1). (Each scaffold/rat had a different cell seeding protocol or period of pre-culture from the other three scaffolds implanted). The midline incision was closed and, 7 or 14 days later, implanted scaffolds were excised with skin attached and fixed overnight at 4°C in PFA.
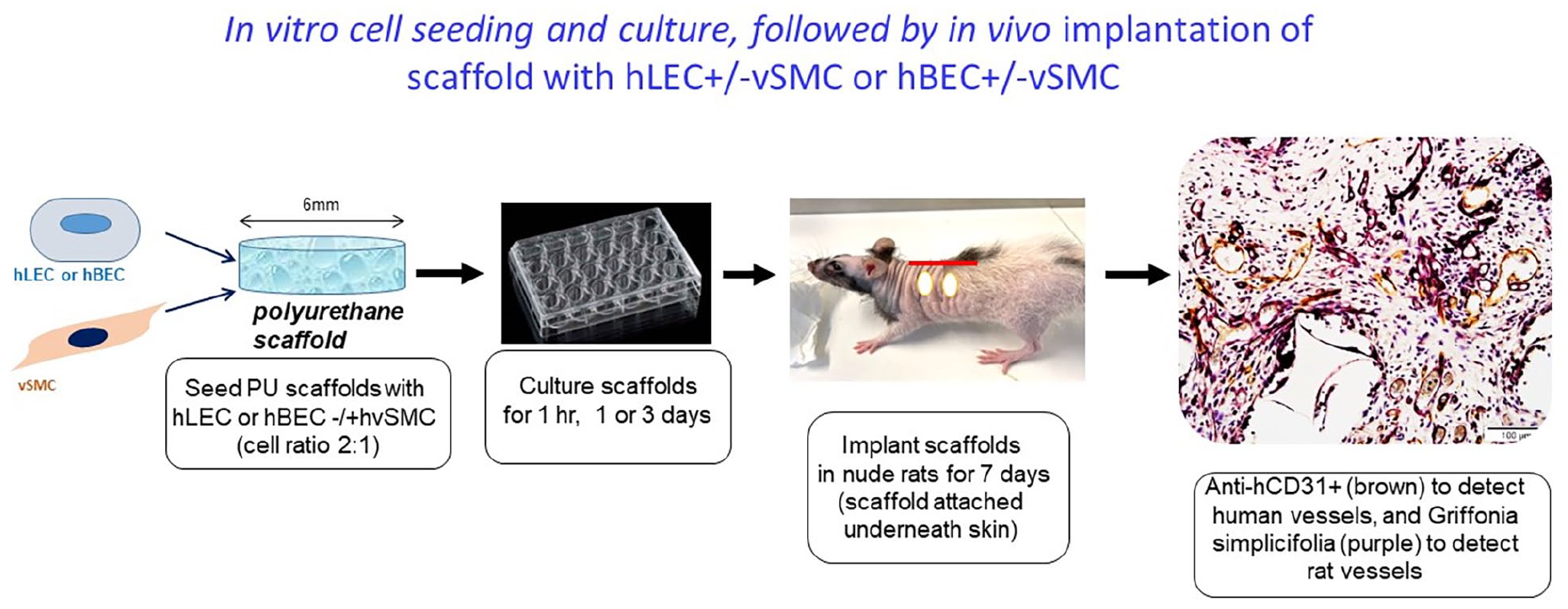
Diagram illustrating the 3D porous scaffold cell seeding steps in vitro, followed by in vivo transplantation.
Immunohistochemical analysis of in vivo scaffolds
Paraffin embedded scaffold cross sections were stained routinely with either hCD31 or podoplanin antibodies (as described previously). Paraffin embedded scaffold cross sections were also doubled labelled with human-specific CD31 antibody and Griffonia simplicifolia lectin-biotin conjugate to label human and rat host vessels, respectively. Briefly, Proteinase K (Dako) was applied to dewaxed sections for 6 min followed by 3% (v/v) hydrogen peroxide quench and protein blocking (Dako) steps. Human-specific CD31 antibody (clone JC70A, 1:100, Dako) was applied for 120 min, followed by rabbit anti-mouse immunoglobulin-biotin conjugate (1:200, Dako) for 30 min, then Vectastain Elite ABC kit (30 min, Vector Laboratories) and DAB Substrate Chromogen System, for 5 min. Sections were washed and overlaid with Griffonia simplicifolia lectin-biotin conjugate (1:300, Vector Laboratories) for 30 min, followed by detection with Streptavidin-HRP conjugate (30 min, 1:400, Dako) and Vector VIP substrate kit (5 min, Vector Laboratories). Slides were counterstained with haematoxylin and coverslips mounted with Entellan (Merck).
Immunolabelling of hvSMC in implanted scaffolds was performed using human-specific KU80 antibody (Abcam) and α-SMA antibody (clone 1A4, 1:500, Dako) as described in Kong et al. 4
Rat lymphatic vessels were stained with podoplanin HG-19 antibody (1:800, Sigma-Aldrich) for 60 min followed by goat anti-rabbit immunoglobulin-HRP conjugate (1:200, Vector Laboratories) for 30 min prior to DAB Substrate Chromogen System for 5 min.
Host (rat) CD68-positive macrophages were labelled using mouse anti-rat CD68 antibody (MCA341R, 1:400, BioRad, CA, USA) following antigen retrieval with Proteinase K for 6 min, 3% (v/v) hydrogen peroxide quench and blocking (UltraVision, Thermo Fisher Scientific) steps. CD68 antibody was applied overnight at 4℃, followed by rabbit anti-mouse immunoglobulin-biotin conjugate (1:200, Dako) for 30 min, Vectastain Elite ABC kit for 30 min and DAB Substrate Chromogen for 5 min.
In addition, four nude rats had abdominal skin tissue samples taken at scaffold harvest, processed for histology and embedded in paraffin. Five micrometer sections were cut and subjected to Griffonia simplicifolia lectin-biotin staining for rat skin blood vessel per cent vascular volume (PVV) morphometric counting (as described below).
FITC dextran perfusion and immunofluorescence of cryosections
Anaesthetized rats just prior to scaffold harvest (N = 3 rats including scaffolds with all cell seeding protocols) were intravenously injected with FITC-dextran conjugate (MW 2 × 106) comprising 5 mg of lysine fixable FITC-dextran (Life Technologies) and 5 mg of FITC-dextran (Sigma-Aldrich) in 1 mL of 0.9% saline, administered 10 min prior to excision of implanted scaffolds. Scaffolds with skin attached were fixed in 4% PFA for 24 h at 4°C, followed by infiltration with 30% (w/v) sucrose at 4°C for 24 h. Scaffolds were then embedded and frozen in OCT media (Sakura Finetek, Tokyo, Japan). Ten micrometer thick cryosections were permeabilized (0.2% TritonX-100, 5 min), blocked (10% (v/v) goat serum, 30 min) and immunostained with either human-specific CD31 (clone JC70A, 1:100, Dako) or human podoplanin antibody (clone D2-40, 1:100, Dako) at 4°C overnight, followed by secondary anti-mouse IgG-AlexaFluor594 conjugate (1:200) and DAPI (10 ng/mL), for 60 min. Coverslips were mounted with fluorescence mounting medium (Dako).
Microscopy
Brightfield and fluorescence images of stained cells and frozen sections of FITC dextran infused tissues were acquired on an Olympus (Tokyo, Japan) BX61 upright microscope using analySIS software with 4x, 10x, 20x or 40x objectives.
Fluorescence microscopy of frozen sections for CD31/podoplanin and CD31/alphaSMactin were taken on an Olympus IX71 microscope.
Morphometric analysis
Analysis was performed using software and parameters previously described, 4 on cross sections of labelled human CD31 antibody cultured scaffolds; and subdermal scaffolds labelled with human CD31 antibody and Griffonia simplicifolia-lectin to visualize human and rat host vessels, respectively. All morphometric counting was conducted with the observer blinded to the identity of the tissue specimens. Briefly, PVV and density were determined using Stereo Investigator V11 software (MBF Bioscience, VT, USA) using a sampling fraction of 25% for each cross section assessed and counting grids (200 μm × 200 μm) containing grid points at 40 μm intervals. The PVV was calculated as the percentage of grid points falling over human CD31, or lectin-positive vessels (including vessel lumen and wall) over the total number of points positioned over tissue and fibrin matrix within the scaffold pores while vessel density was expressed as number of vessel profiles/mm2 in the fibrin.
Image J measurements of vessel diameter, cross sectional area and perimeter in vivo
Vessel diameter, perimeter and cross-sectional area were measured using brightfield images of human CD31-immunolabelled sections acquired on an Olympus BX61 microscope, using Image J software. For hLEC, sections from three scaffolds per group were analysed and for hBEC, 2–5 scaffolds/group were analysed. For each section, at least five fields with resolution of 4080 × 3072 pixels were acquired using a 20x objective. The total number of vessels measured for each section ranged from 26 to 58.
For vessel diameter measurement, a straight line was drawn across the widest section of the vessel. Length of line was measured in microns. Vessel perimeter and cross-sectional area were measured by manually drawing a line around the inner wall of the vessel using the freehand tool. Perimeter and area were measured in microns and square microns, respectively.
Image J measurements of alive and regressing vessel diameter in vitro
Vessel diameter measurements using Image J (as described above) were taken from in vitro hLEC ± hvSMC and hBEC ± hvSMC 3 and 7 day cultured scaffolds immuno-stained for hCD31. Alive complete vessel profiles were defined as having an intact endothelial lining positively stained with hCD31 and there was little cellular debris in the vessel lumen. Vessel diameter measurements from regressing vessels (which may also immuno-stain positive for hCD31) where the lumen was full of cellular debris and the endothelial cells lining the vessel wall were not intact were also taken. For all in vitro measurements the smallest vessel diameter was measured. Vessel measurements were taken of 3–5 alive and 3–5 regressing vessels per scaffold and N = 2–3 scaffolds per cell seeding scaffold type.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical significance of morphometric counts was determined using two-way ANOVA on Graph Pad Prism version 9, with a subsequent Tukey post hoc test to determine statistical significance between individual groups. A p value of less than 0.05 was considered statistically significant.
Results
Immuno-fluorescent staining of plated endothelial cells
All endothelial cell subtypes were positive for CD31 (Figure 2). Only Cascade microvascular EC and hDLEC were positive for podoplanin (D2-40), confirming that both were lymphatic endothelial cells (Figure 2), with the classic cell marker signature of CD31+/D2-40+ (termed hLEC throughout the text), whilst Lonza microvascular EC were universally CD31+/D2-40− and were therefore blood microvascular endothelial cells, termed hBEC, throughout the text.

Plated cell types: human cardiac microvascular endothelial cells (hCMEC, PromoCell), hBEC (Lonza HMVEC-dBLAD), hLEC (Cascade HMVECad), and human dermal lymphatic endothelial cells (hDLEC, PromoCell) immuno-stained for hCD31 (left hand panel) podoplanin (middle panel). DAPI: blue nuclei. Corresponding bright field images of CD31 and podoplanin immuno-stained cells appear to the right of each immuno-stained image. The right panel indicates a negative control with DAPI (blue nuclei) staining. Scale bar = 50 µm in each image.
Formation of hLEC ± hvSMC and hBEC ± hvSMC capillary networks in vitro
Appearance over 7 days in vitro, IHC labelling
hLEC ± hvSMC at 1 day in scaffold sections showed complete filling of scaffold pores with the human fibrin evenly distributed throughout all pores, and which supported scattered small groupings of hCD31-positive cells either clumped and without a lumen, or with a narrow central lumen. hLEC (hCD31+) at 3 days demonstrated mostly small vessel cross sections, whilst hLEC + hvSMC were hCD31-positive and wider and irregular in shape. At 7 days, vessel numbers were reduced when hLEC were cultured alone, but vessel numbers were maintained in hLEC + hvSMC co-cultures (Figure 3).

Vertical sections through porous scaffolds at 1, 3 and 7 days in vitro seeded with four cell type combinations used in the study: hLEC, hLEC + hvSMC, hBEC and hBEC + hvSMC. Scale bar = 200 µm in every image.
At day 1 hCD31-positive hBEC ± hvSMC were seen evenly distributed in fibrin in all pores as small circular structures with a discernable lumen. By day 3 hBEC, regardless of whether they were co-cultured with hvSMC, became larger and irregular in shape, and remained hCD31-positive (Figure 3). At 7 days the numbers of hBEC ± hvSMC capillaries were noticeably reduced, indicating vessel regression between 3 and 7 days both with and without hvSMC.
hLEC maintained their hCD31+/podoplanin+ profile when assembled as capillaries in scaffolds in vitro (Figure 4(a)–(d)), and hBEC maintained their hCD31+/podoplanin− profile when forming vessel structures in in vitro 3D culture (Figure 4(e)–(h)).

(a–h) hCD31 (red), podoplanin (D2-40, green) and DAPI (blue nuclei) fluorescent staining of frozen sections of 3 day in vitro hLEC and hBEC scaffolds. (a)–(d) are hLEC capillaries in scaffolds with (d) demonstrating merging of staining in hLEC capillaries, (e)–(h) are hBEC capillaries in scaffolds. Note hLEC capillaries are hCD31+/podoplanin+, whilst hBEC capillaries are hCD31+/podoplanin−. Scale bars = 20 µm. (i–p): hCD31, alpha smooth muscle actin (SMA), and DAPI (blue nuclei) fluorescent staining of 3 day frozen sections of hLEC + hvSMC and hBEC + hvSMC scaffold cultures. (i)–(l) are hLEC seeded scaffolds, and (m)–(p) are hBEC seeded scaffolds. Both hLEC and hBEC capillaries (hCD31+, red) show attachment of αSMactin+ (green) hvSMCs and merged staining in (l) and (p). Scale bars = 20 µm.
Attachment of hvSMC to hLEC and hBEC capillaries
In both hLEC + hvSMC and hBEC + hvSMC co-cultures, alpha smooth muscle actin-positive hvSMC were observed attaching to the abluminal surface of hCD31-positive capillaries occasionally at 1 day and more frequently at 3 days in culture (Figure 4(i)–(p)). Seeded hvSMC at 1 day appeared as single cells either not attaching to hLEC or hBEC structures or occasionally loosely attached to the abluminal vessel surface.
Whole mounts
Whole mounts of hLEC (as observed in confocal and Thunder microscopy) did not reveal interconnected lymphatic capillaries at 2 days. hLEC cultured alone demonstrated numerous small groups of bunched cells, some with elongated extensions although a central lumen was generally not obvious in confocal or thunder microscope observations (Figure 5(a) and (b)). However, in hLEC + hvSMC co-cultures at 2 days, occasional larger vessels with lumens occurred (Figure 5(c)), and these large vessels had smooth muscle cells clearly attached abluminally (Figure 5(d)). At 3 days, hLEC showed some interconnection of vessels with lumens (Figure 5(e)).
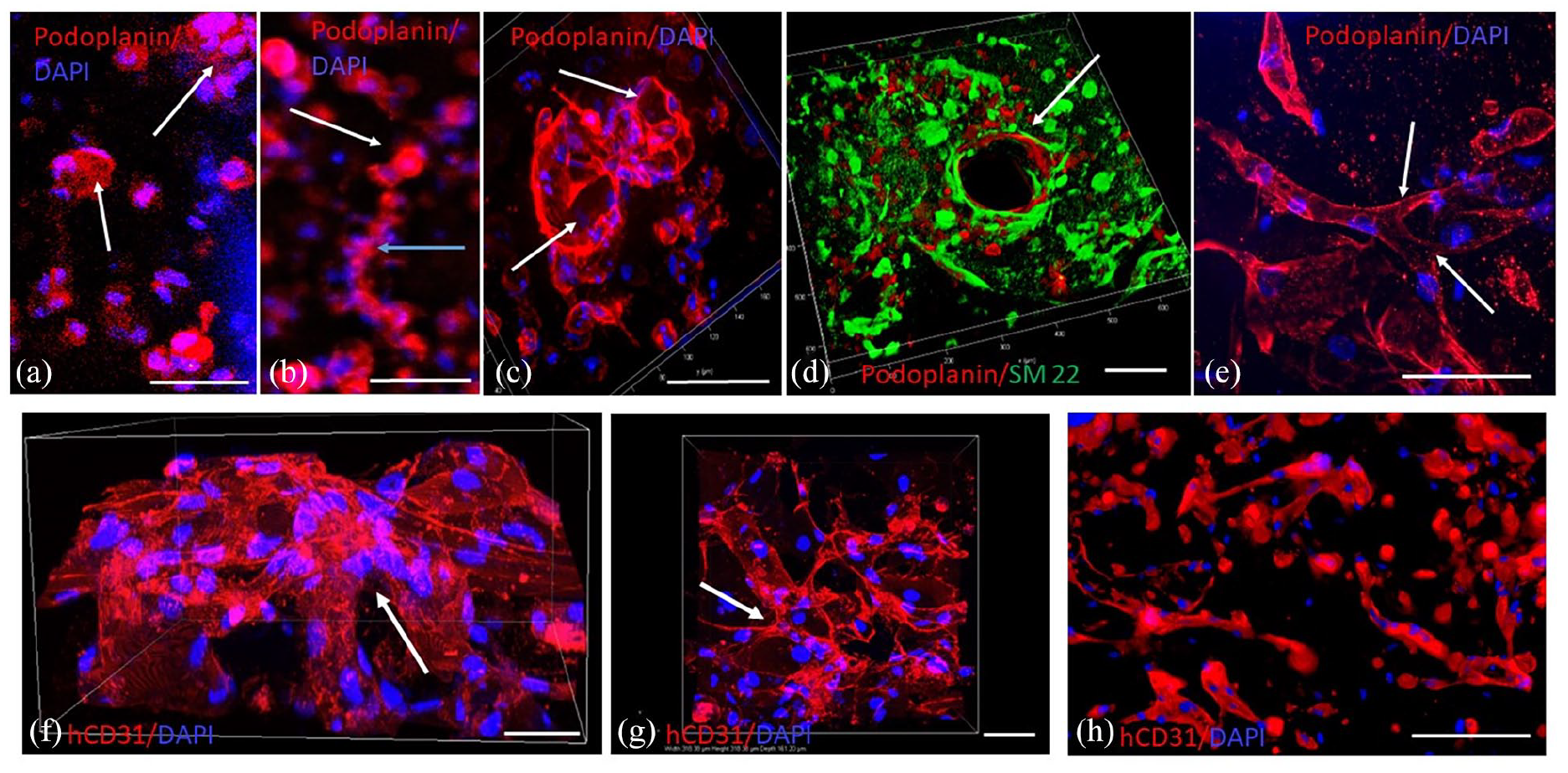
Confocal and Thunder microscope images of the early stages of hLEC and hBEC vessel formation in whole mounts (a) hLEC (-hvSMC) capillaries at 2 days in vitro appearing as balls (clusters) of podoplanin+ cells (red) with little evidence of lumen formation. Blue: DAPI stained nuclei. Nikon confocal, 20x objective, maximum intensity projection, 50 µm range, 1.2 µm z-step. Scale bar = 50 µm. (b) hLEC (-hvSMC) capillaries at 2 days in vitro appearing not only as balls of cells (white arrow), but also as elongated projections without apparent lumens (blue arrow). Blue: DAPI stained nuclei. The image is a single optical section, taken with 10x objective on the Thunder microscope. Scale bar = 50 µm. (c) hLEC + hvSMC capillaries at 2 days in vitro. Some hLEC vessels, podoplanin+ (red, arrow) appear larger than in (a) and lumen formation is evident. Blue: DAPI stained nuclei. Leica Thunder, 63x objective, volume view, 43 µm range, 0.21 µm z-step. Scale bar = 50 µm. (d) LEC (podoplanin+, red) + hvSMCs (SM22+, green) at 2 days in vitro. hvSMC are wrapping around a large central hLEC capillary (arrow). Leica Thunder, 20x objective, volume view, 58 µm range, 0.5 µm z-step. Scale bar = 100 µm. (e) hLEC (podoplanin+, red) capillaries at 3 days in vitro. Arrows indicate branching of interconnected hLEC capillary network. Blue: DAPI stained nuclei. Scale bar = 50 µm. Leica Thunder, 63x objective, maximum intensity projection, 49 µm range, 0.21 µm z-step. Scale bar = 50 µm. (f, g) hBEC capillaries (hCD31+ red), (blue: DAPI stained nuclei) at 2 days in vitro. The hBEC capillary network demonstrates (arrows) a branching interconnected network. Nikon confocal, 40x objective, volume view, 161 µm range, z step 0.2 µm. Scale bars = 50 µm (both in (f) and (g)). (h) hBEC capillaries (hCD31+ red) (blue: DAPI stained nuclei) at 4 days in vitro. Leica Thunder, 63x objective, volume view, 40 µm range, 0.21 µm z-step. Scale bar = 100 µm.
Whole mounts of hBEC at 2 days in vitro indicated a more mature appearance with interconnected capillaries with lumens forming within scaffold pores (Figure 5(f) and (g)). hBEC at 4 days showed limited interconnection of capillaries (Figure 5(h)), suggesting vessel regression was occurring.
hlec and hBEC seeded scaffolds in vivo
Transplantation of seeded scaffolds from in vitro to in vivo was achieved easily as the scaffolds were easy to pick up and were in no way damaged by the handling and suturing involved.
The majority of scaffolds were assessed after 7 days in vivo, with a smaller number going onto 2 weeks in vivo. At 7 days the scaffolds were clearly seen positioned between the base of the dermis and the underlying skeletal muscle of the rat back. At this time human vessels (hCD31+) were visualized scattered in the pores of the scaffold, largely surrounded by fibrin matrix. At the periphery of the scaffold, rat (host) connective tissue containing rat blood vessels could be seen infiltrating the pores of the scaffold (Figure 6(a)). At 2 weeks the infiltration of the scaffold pores by rat tissue was complete and the fibrin matrix that had filled the pores was no longer present (Supplemental Figure 1(c) and (d)).
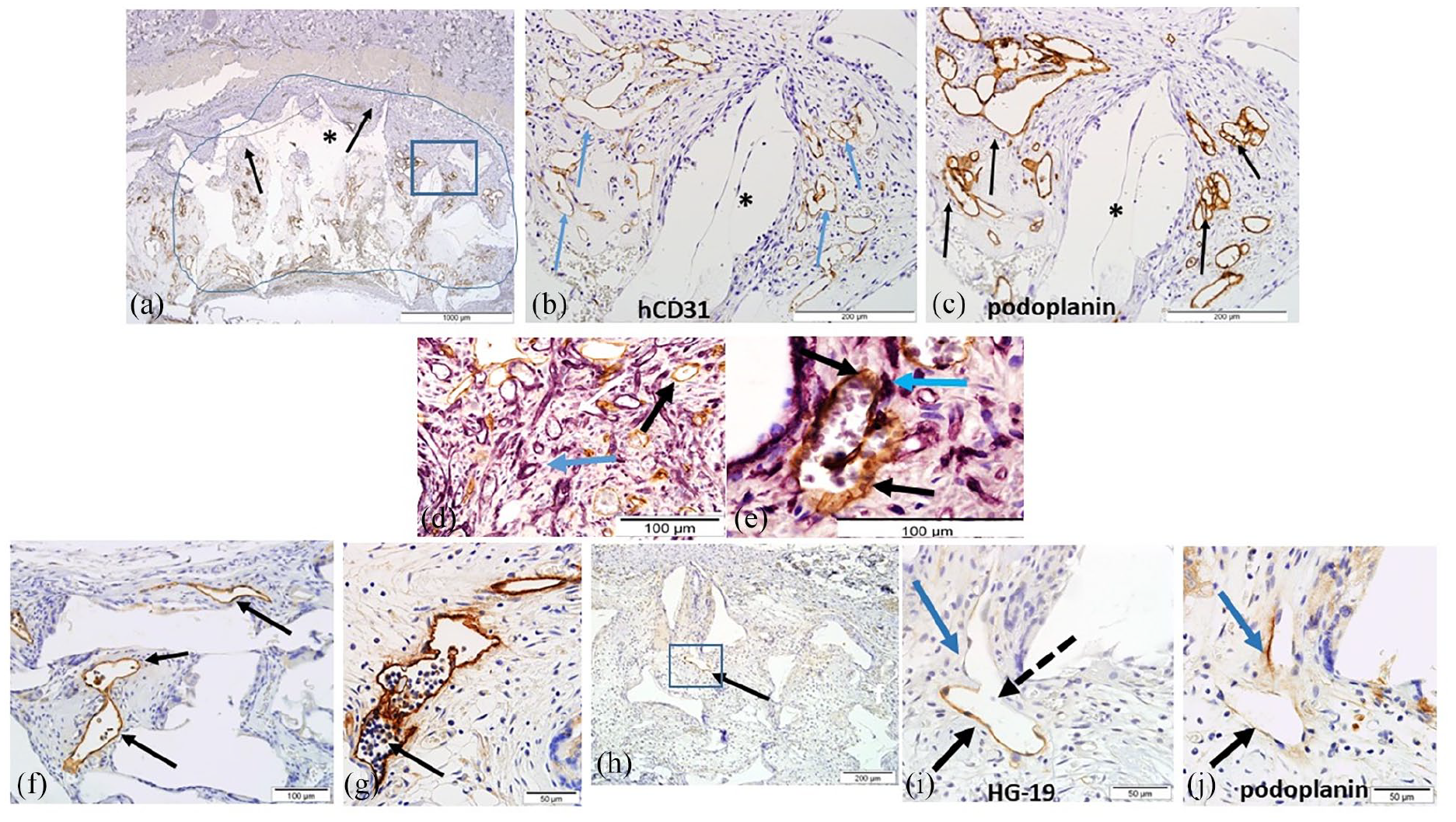
hLEC seeded scaffolds in vivo. (a) Low magnification image of section through an entire hLEC seeded scaffold (demarcated by a blue line, with the skin above) at 7 days in vivo. Area within the blue rectangle appears at higher magnification in (b) and (c) and includes numerous hLEC vessels positive for hCD31/podoplanin. Black arrows at the edge of the scaffold indicate rat tissue infiltration into the pores of the scaffold. * indicates scaffold material (appears white). Scale bar = 1000 µm. (b) Area within the blue rectangle in (a) showing numerous hLEC capillaries (arrows) immunostained positive for hCD31. The hLEC capillaries are surrounded by rat infiltrating connective tissue. * scaffold material. Scale bar = 200 µm. (c) Area within the blue rectangle of (a) and adjacent section to (b) with the same hLEC capillaries strongly immunostained with podoplanin (arrows). * indicates scaffold material. Scale bar = 200 µm. (d) Intermingling of hLEC capillaries (stained brown with hCD31, black arrow) and rat tissue in vivo at 7 days. Rat vessels stain purple (blue arrow) with Griffonia simplicifolia lectin. Scale bar = 100 µm. (e) Mosaic vessel in an hLEC seeded scaffold at 7 days in vivo. The black arrow (lower right) indicates a hCD31+ hLEC vessel (brown) joined to a mosaic vessel that has hCD31+ ECs (brown, indicated by black arrow at top) and rat mesenchymal support cells (purple, indicated by blue arrow) wrapping around its abluminal surface. Rat blood is seen in the vessel lumen. Scale bar = 100 µm. (f) Numerous podoplanin+ hLEC capillaries (arrows) in a scaffold in vivo at 14 days. Some human lymphatics show white blood cell only trafficking in their lumens. Scale bar = 100 µm. (g) hLEC + hvSMC seeded scaffold at 14 days in vivo demonstrating lymphatic function. Podoplanin+ human lymphatic vessel (arrow) exclusively trafficking rat white blood cells within its lumen. Scale bar = 50 µm. (h) Low magnification image of hLEC + hvSMC seeded scaffold at 14 days in vivo. Arrow indicates HG19+ rat lymphatic positioned centrally in the scaffold. Scale bar = 200 µm. (i) The area in the blue box in (h) demonstrating the rat lymphatic vessel (HG19+, black arrow) positioned centrally in a hLEC + hvSMC seeded scaffold at 14 days in vivo. The thick dotted black arrow indicates a branch point from the rat lymphatic. The blue arrow indicates the branch which is not HG19+. Scale bar = 50 µm. (j) Adjacent tissue section to (i) where the original HG19+ rat lymphatic is seen (black arrow) and the blue arrow indicates a human podoplanin+ lymphatic vessel, indicating inosculation between rat and human lymphatics within the scaffold. Scale bar = 50 µm.
hLEC ± hvSMC in vivo at 7 days
At 7 days in vivo hLEC capillaries had survived and numerous irregular capillaries labelled strongly with podoplanin antibody, but weaker hCD31 labelling (Figure 6(a)–(c)) was evident. hLEC capillaries were either surrounded by fibrin matrix, or where rat connective tissue infiltration had occurred intermingled in close association with rat capillaries (Figure 6(d)) and rarely a hybrid of rat and human lymphatic vessel could be identified (Figure 6(e)). Although in FITC dextran infused animals, FITC was routinely observed in rat vessels within the scaffold, it was difficult to identify any FITC dextran within hLEC vessels. However, rat blood cells (mostly erythrocytes) were seen in the lumen of some hLEC vessels indicating that inosculation with rat blood vessels did occasionally occur. Sporadic alpha-smooth muscle actin+/Ku80+ (human nuclear positive) hvSMC were seen abluminally attached to a hLEC capillary (Figure 8(a) and (b)) suggesting that some co-cultured hvSMC maintained their attachment to hLEC vessels in vivo.
hLEC ± hvSMC seeded scaffolds at 14 days
In hLEC ± hvSMCs seeded scaffolds at 14 days in vivo, hCD31 and podoplanin stained vessels were frequently identified. This suggests prolonged survival for hLEC in vivo. In addition, hLEC vessels at 2 weeks demonstrated lymphatic function as numerous hLEC vessels displayed a lumen full of white blood cells, without evidence of red blood cells (Figure 6(f) and (g)). Additionally, staining of hLEC seeded scaffolds at 2 weeks with the rat specific lymphatic marker HG-19 revealed rat lymphatics at the perimeter of hLEC seeded scaffolds, and on two occasions rat lymphatics had penetrated to the central scaffold (Figure 6(h)). Evidence of inosculation (functional joining) of rat lymphatics (HG-19+) with human podoplanin+ hLEC vessels within the central scaffold was observed (Figure 6(i) and (j)). (Rat lymphatics were not observed within any scaffolds regardless of cell seeding type at 7 days in vivo.)
hBEC ± hvSMC in vivo at 7 and 14 days
At 7 days in vivo hBEC capillaries were evident throughout the scaffold, some in fibrin only areas, others, in scaffold areas that were invaded by rat connective tissue (Figure 7(a)–(d)). In areas invaded by rat tissue, rat and human capillaries intermingled very closely and appeared to make contact with one another suggesting inosculation between the two blood vessel systems (Figure 7(e)). When scaffolds containing hBEC + hvSMC were implanted (Figure 7(c) and (d)), at 7 days the human capillaries were more robust in appearance and of wider diameter than hBEC-hvSMC implanted scaffolds (Figure 7(a) and (b)) and appeared to have rat blood (erythrocytes) in their lumens, although rat blood was also observed in hBEC-hvSMC vessels.

hBEC seeded scaffolds in vivo. (a) Low magnification image of hBEC seeded scaffold (demarcated by the blue line) in the skin at 7 days in vivo. Black arrow indicates hBEC immuno-stained positive for hCD31. Blue arrows indicate rat connective tissue infiltration into the scaffold pores from the rat skin above the scaffold. * indicates scaffold material (white). Scale bar = 1000 µm. (b) Higher magnification image of hCD31+ hBEC capillaries (arrows) at 7 days in vivo. The grey material around the hBEC capillaries is human fibrin. Scale bar = 200 µm. (c) Low magnification image of hBEC + hvSMC seeded scaffold (demarcated by the blue line) in a skin wound at 7 days in vivo. Black arrows indicate human capillaries immuno-stained positive with hCD31. These human vessels appear more numerous than those in (a). Blue arrows indicate rat connective tissue infiltration into the scaffold pores from the rat skin above the scaffold. * indicates scaffold material (white). Scale bar = 1000 µm. (d) Higher magnification image of hCD31+ hBEC + hvSMC capillaries at 7 days in vivo. The grey material around the capillaries is human fibrin. The human capillaries (arrows) appear larger than in (b) and contain rat blood. * indicates scaffold material. Scale bar = 200 µm. (e) Intermingling of human hCD31+ hBEC capillaries (immuno-stained brown, black dashed arrow) with rat capillaries (stained with Griffonia simplicifolia lectin (purple), blue dashed arrow). The vessels appear to contact each other at the thick black arrow. Scale bar = 50 µm. (f, g) Adjacent tissue sections through a hBEC + hvSMC seeded scaffold at 7 days in vivo. (f) is immuno-stained for hCD31, numerous positive human capillaries are seen (brown, arrows). In (g) the same capillaries as (f) are immuno-stained for podoplanin, but are negative (arrows). Scale bars = 100 µm. (h) FITC dextran tail vein infusion of nude rat at 7 days. hCD31+ hBEC capillaries (red, white dashed arrows) contain FITC dextran (green, white solid arrows) indicating inosculation between human and rat capillaries in the scaffold. Scale bar = 50 µm.
Immunohistochemical labelling of hBEC capillaries ± hvSMC demonstrated all hBEC capillaries in vivo maintained their hCD31+/podoplanin− cell marker profile (Figure 7(f) and (g)). FITC infusion via the tail vein demonstrated FITC dextran within hBEC ± hvSMC scaffold capillaries (Figure 7(h)) proving that the human capillaries had inosculated with the host (rat) vasculature. There was also evidence from triple labelling experiments that hBEC capillaries co-cultured with hvSMC maintained this smooth muscle cell coverage in vivo (Figure 8(c) and (d)), with alpha smooth muscle actin+/Ku80+ hvSMCs seen attached abluminally to hCD31+ hBEC capillaries.

hvSMC attachment to hLEC and hBEC capillaries in vivo (7 days). (a, b) hLEC capillaries in (a) double labelling with Ku80-specific antibody (anti-human nuclear, red) and alpha SM actin (brown), arrow indicates double labelled positive hvSMC attached to a hLEC capillary. In (b) the same capillary in an adjacent section to (a) is immunostained with hCD31 and positive (arrow) indicating it is a hLEC capillary. Scale bars = 50 µm. (c, d) hBEC capillaries in (c) double labelling with Ku80-specific antibody (anti-human nuclear, red) and alpha SM actin (brown), arrow indicates double labelled positive hvSMC attached to a hBEC capillary. In (d) the same capillary in an adjacent section to (c) is positive for hCD31 (arrow), demonstrating it is a hBEC capillary. Scale bars = 100 µm.
In hBEC ± hvSMC seeded scaffolds at 14 days in vivo, no human vessels were identified on hCD31 labelling.
Inflammatory response to the polyurethane scaffold
The polyurethane scaffold maintained its integrity over 2 weeks in vivo implantation. Although nude rats are immunocompromised they are still able to mount a macrophage inflammatory response to the foreign material of the polyurethane scaffold. In all in vivo implanted scaffolds at 7 and 14 days and regardless of the cell types implanted in the scaffold a CD68+ macrophage response was seen at both time points. CD68+ macrophages attached as a complete lining around the scaffold material and in direct contact with the rat connective tissue that had invaded the internal surface of the scaffold pores (Supplemental Figure 1(a)–(d)). In areas with little rat connective tissue (day 7 only) very few macrophages attached to the scaffold material around the pores (Supplemental Figure 1(a)).
Morphometric analysis
Two morphometric parameters were measured in vitro and in vivo for hLEC ± hvSMC and hBEC ± hvSMC – percent vascular volume (PVV) and vessel density (number of vessel profiles/mm2).
General trends hLEC ± hvSMC and hBEC ± hvSMC vessels in vitro
Human LEC and hBEC were cultured for 1, 3 or 7 days ± hvSMC. Cell seeding densities were identical for hLEC and hBEC.
hLEC vessels (cultured alone) in vitro displayed lower values of PVV (2.54% ± 0.55%) and vessel density (23.45 ± 2.73 vessels/mm2) at day 1 than hBEC cultured alone (PVV: 10.71 ± 4.06; BV density: 60.44 ± 16.74). hLEC values declined slightly over time to day 7 (Figure 9(a) and (b)). However, co-culture of hvSMC with hLEC greatly increased both PVV values and vessel density mean values at all time points in vitro (Figure 9(a) and (b)), and overall the addition of hvSMC significantly increased PVV (p = 0.0083) and vessel density (p = 0.0039). Time in culture overall exerted no influence on PVV or vessel density for hLEC (Figure 9(a) and (b)).

Morphometric assessment of hLEC and hBEC capillary networks. (a) hLEC capillary percent vascular volume (PVV) in scaffolds to 7 days in vitro. Overall pre-culture time (1, 3 or 7 days) made no significant difference to PVV. The co-culture of hvSMC with hLEC overall significantly increased the PVV (p = 0.0083). (N = 3–6 scaffolds/group). (b) hLEC capillary vessel density (vessels/mm2) in scaffolds to 7 days in vitro. Overall pre-culture time (1, 3 or 7 days) made no significant difference to hLEC vessel density. The co-culture of hvSMC with hLEC overall significantly increased the vessel density (p = 0.0039) (N = 3–5 scaffolds/group). (c) hBEC capillary percent vascular volume (PVV) in scaffolds to 7 days in vitro. Overall pre-culture time (1, 3 or 7 days), and co-culture with hvSMC made no significant differcence to PVV (N = 3–4 scaffolds/group). (d) hBEC capillary vessel density (vessels/mm2) in scaffolds to 7 days in vitro. Overall pre-culture time (1, 3 or 7 days) made a significant difference to BEC vessel density (p = 0.0019). The co-culture of hvSMC with hBEC overall made no significant difference. Individual group comparisons indicated by significance bars on the graph: *p < 0.05, ***p < 0.005 (N = 3–4 scaffolds/group). (e) hLEC capillary percent vascular volume (PVV) in scaffolds in vivo at 7 days. Overall pre-culture time (1, 3 or 7 days) made no significant difference to PVV. The co-culture of hvSMC with hLEC overall also made no significant difference (N = 4–5 scaffolds/group). (f) hLEC capillary vessel density (vessels/mm2) in scaffolds at 7 days in vivo. Overall pre-culture time (1, 3 or 7 days) made no significant difference to hLEC vessel density, whilst the co-culture of hvSMC with hLEC was significant overall (p = 0.0314) (N = 4–5 scaffolds/group). (g) hBEC capillary percent vascular volume (PVV) in scaffolds in vivo at 7 days. Overall pre-culture time (1, 3 or 7 days) made no significant difference to PVV. The co-culture of hvSMC with hBEC overall was significant (p = 0.0087) (N = 4–5 scaffolds/group). (h) hBEC capillary vessel density (vessels/mm2) in scaffolds at 7 days in vivo. Overall pre-culture time (1, 3 or 7 days) made no significant difference to hBEC vessel density. The co-culture of hvSMC with hBEC overall significantly increased the vessel density (p = 0.0035) (N = 4–5 scaffolds/group).
There was a general trend for hBEC vessels in vitro to demonstrate declining PVV and vessel density over time in culture. hBEC (cultured alone) parameters were highest at 1 day post-seeding (PVV: 10.71% ± 4.06%, vessel density: 60.44 ± 16.74 vessel/mm2) and declined at day 3 and 7 in culture. The co-culture of hvSMCs with hBEC made little difference to either the PVV or vessel density, and did not significantly affect either parameter. The factor of culture time was not significant overall for PVV, but the overall decline in vessel numbers/mm2 was significant over time (p = 0.0019) (Figure 9(c) and (d)).
Statistical comparison of hLEC ± hvSMC versus hBEC ± hvSMC in vitro
When directly comparing the PVV and vessel density data of hLEC versus hBEC cultured alone in vitro over 1, 3 and 7 days (Supplemental Figure 2(a), (b)), there were overall significant differences related to cell type (hLEC vs hBEC) in PVV (p = 0.0029) and vessel density (p = 0.0191) with larger values particularly at 1 day recorded for hBEC. Time in culture had no significant influence for PVV overall, but significantly influenced vessel density (p = 0.0007) with decreasing values in both cell types from one to 7 days.
When hBEC + hvSMC versus hLEC + hvSMC scaffolds in vitro were compared neither culture time nor cell type influenced the PVV, or vessel density (Supplemental Figures 2(c) and (d)).
The overall significant differences when hBEC and hLEC are cultured alone indicate that the two cell types have different capabilities to form vessels in vitro, and hBEC do slightly better than hLEC in culture, largely through higher 1 day in vitro PVV and BV density values. These differences are not apparent when hvSMC are co-cultured with hLEC and hBEC.
Diameter of alive and regressing vessels in vitro
The diameter of alive and regressing vessels in vitro in hLEC ± hvSMC and hBEC ± hvSMC scaffolds was measured in 3 and 7 days in vitro scaffolds. Overall there was a significant effect of alive v’s regressing vessels on vessel diameter for 3 day hLEC ± hvSMC scaffolds (p = 0.0299) and 3 day hBEC ± hvSMC (p = 0.0092). There was no overall effect at 7 days for hLEC ± hvSMC or hBEC ± hvSMC, and coculture with vSMC had no effect in either cell seeding regime or at either time point (Supplemental Figure 3).
hLEC and hBEC morphometric analysis in vivo
Scaffolds were pre-cultured prior to in vivo implantation. The pre-culture times were 1 h to establish if a minimal pre-culturing period could be successful in vivo, and 1 or 3 days pre-culture. All scaffolds were implanted sub-dermally in the rat back and harvested 7 days later. In each case scaffolds contained hLEC ± hvSMC or hBEC ± hvSMC at the same cell seeding density. The PVV and vessel density was determined for each cell combination and compared between hLEC versus hBEC after 7 days in vivo.
General trends hLEC ± hvSMC and hBEC ± hvSMC in vivo
After 7 days in vivo for hLECs cultured alone, the PVV was similar regardless of pre-culture time, and highest in the 1-h pre-culture group (4.86% ± 1.71%). The same trend applied to the vessel density analysis which was similar for all groups, and highest in the 3 days pre-culture group (20.83 ± 7.91 vessels/mm2) (Figure 9(e) and (f)). The co-culture of hLEC with hvSMC increased all PVV and vessel density values in vivo (Figure 9(e) and (f)), with the highest PVV in the 1-h pre-culture group (5.57 ± 2.30), and highest vessel density in the 1 day pre-culture group (64.87 ± 18.85 vessels/mm2). Overall co-culture with hvSMC was significant for the vessel density (p = 0.0314) but not significantly different overall for the PVV analysis. Pre-culture time made no difference to hLEC ± hvSMC PVV or vessel density in vivo.
At 2 weeks in vivo the PVV of hLEC and hLEC + vSMC was 0.63% ± 0.39% (n = 4) and 0.69% ± 0.51% (n = 4), respectively. The vessel density of hLEC and hLEC + vSMC was 4.49 ± 2.93 vessels/mm2 and 11.05 ± 10.65 vessels/mm2, respectively.
At 7 days in vivo scaffolds seeded with hBEC alone and pre-cultured for 1 h, 1 day and 3 days prior to in vivo implantation displayed very similar in vivo PVV and vessel density, and no trend was observed amongst these groups. However, when the scaffolds were co-cultured in vitro with hvSMC there was a significant and consistent increase in in vivo PVV and vessel density with the highest PVV in the 3 day pre-culture group (2.35% ± 0.65%) and the highest vessel density also in this 3 day pre-culture group (31.75 ± 8.96 vessels/mm2) (Figure 9(g) and (h)). Two-way ANOVA analysis indicated that overall pre-culture time had no influence on PVV or vessel density, but overall the co-culture of hvSMC with hBEC significantly increased PVV (p = 0.0087) and BV density (p = 0.0035) in vivo (Figure 9(g) and (h)). At 14 days in vivo no hBEC vessels were observed.
Statistical comparison of hLEC ± hvSMC versus hBEC ± hvSMC in vivo
When directly comparing the in vivo PVV and vessel density data of hLEC versus hBEC pre-cultured alone, cell type (hLEC vs hBEC) overall recorded significant differences in PVV (p = 0.0069) and vessel density (p = 0.0156), with hLEC consistently recording higher values than hBEC in vivo. Pre-culture time overall was not significant for either PVV or BV density (Supplemental Figure 4(a) and (b)). When hvSMC were co-cultured with hLEC or hBEC (hLEC + hvSMC vs hBEC + hvSMC) there was a significant overall difference between hLSEC + hvSMC and hBEC + hvSMC for PVV (p = 0.0276), but no overall difference for vessel density. hLEC + hvSMC tended to have higher values than hBEC + hvSMC, particularly for PVV values (Supplemental Figures 4(c) and (d)). Pre-culture time had no overall effect on PVV or vessel density.
These in vivo comparisons confirm that pre-culture time has no significant effect on PVV or vessel density for hLEC or hBEC, but the cell types significantly influenced PVV and BV density with hLEC showing significantly increased PVV and BV density over hBEC especially when the cells were cultured alone. The addition of hvSMC particularly improved the vessel volume and density in vivo of hBEC.
Statistical comparison of in vitro versus in vivo parameters for hLEC and hBEC
hLEC ± hvSMC seeded scaffolds showed no overall statistical difference between in vitro and in vivo values of PVV and vessel density, nor did the pre-culture time overall exert any significant difference (Supplemental Figure 5(a)–(d)).
hBEC (without hvSMC) showed a distinct decline in PVV and BV density between in vitro and in vivo values. Overall this was significant for PVV (p = 0.0003) (the time in pre-culture was not significant). Vessel density for hBEC was also overall significantly different for in vitro versus in vivo (p = 0.0003). In addition the pre-culture time overall was significant (p = 0.0074). (Supplemental Figure 6(a) and (b)).
hBEC + hvSMC also showed overall a significant difference between in vitro and in vivo PVV values (p = 0.0416) with some decline from in vitro to in vivo, but there was no overall difference between pre-culture times. Vessel density did not change significantly between in vitro and in vivo for hBEC + hvSMC, nor was the pre-culture time overall significantly different (Supplemental Figure 6(c) and (d)).
In summary hLEC capillary formation was unaffected by transplantation in vivo whilst hBEC particularly when cultured without hvSMC showed a significant decline in PVV and BV density in vivo compared to in vitro, indicating significant hBEC capillary loss after transplantation. The co-culture of hvSMC with hBEC diminished this difference between in vitro and in vivo values – with the PVV overall statistical difference being just significant (p = 0.0416), whilst the difference in vessel density between in vitro and in vivo was not significant overall.
Individual hLEC and hBEC vessel parameters: diameter, perimeter and cross sectional area in vivo
Individual in vivo vessel parameters – vessel diameter, perimeter length and cross sectional area were measured using Image J on scaffolds implanted with hLEC ± hvSMC (N = 3 scaffolds/group analysed), or hBEC ± hvSMC (N = 4–5 scaffolds/group analysed, except for 1 h pre-culture hBEC, where N = 2 scaffolds were analysed).
hLEC vessels: Diameter, perimeter, and cross sectional area in vivo
For all hLEC ± hvSMC vessel parameters, a universal trend demonstrated that the 1 h pre-culture period produced the highest vessel diameter, perimeter and cross sectional area for all in vivo scaffold groups (Figure 10(a)–(c)).

Individual vessel morphometric parameters: diameter, perimeter and cross sectional area. (a) hLEC vessel diameter at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0011). The co-culture of hvSMC with hLEC overall was not significant (N = 3 scaffolds/group). (b) hLEC vessel perimeter at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0007). The co-culture of hvSMC with hLEC overall was not significant (N = 3 scaffolds/group). Individual group differences are indicated by a horizontal line above the columns. *p < 0.05. (c) hLEC vessel cross sectional area at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0007). The co-culture of hvSMC with hLEC overall had no effect (not significant) on cross sectional area. Individual group differences are indicated by horizontal lines above the columns. *p < 0.05. (N = 3 scaffolds/group). (d) hBEC vessel diameter at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0290). The co-culture of hvSMC with hBEC overall was also significant (p = 0.0177). (N = 2–5 scaffolds/group). (e) hBEC vessel perimeter at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0232). The co-culture of hvSMC with hBEC overall also had a significant effect (p = 0.0218) (N = 2–5 scaffolds/group). (f) hBEC vessel cross sectional area at 7 days in vivo. Overall pre-culture time had a significant effect (p = 0.0451). The co-culture of hvSMC with hBEC overall had no effect (not significant) on cross sectional area (N = 2–5 scaffolds/group).
The co-culture of hvSMC with hLEC did not alter overall individual vessel diameter, perimeter or cross sectional area. However, the overall effect of pre-culture time was significant for hLEC ± hvSMC vessel diameter (p = 0.0011), perimeter (p = 0.0007) and cross sectional area (p = 0.0007).
hBEC vessels: Diameter, perimeter, and cross sectional area in vivo
The measurements of individual hBEC vessels demonstrated a completely different trend than those shown for hLEC vessels. hBEC vessels in vivo, showed overall that increasing pre-culture time significantly increased vessel diameter (p = 0.0290), vessel perimeter length (p = 0.0232) and vessel cross sectional area (p = 0.0451). Overall, the additional co-culture of hvSMC also significantly increased hBEC vessel diameter (p = 0.0177), and hBEC vessel perimeter (p = 0.0218), but not cross sectional area (Figure 10(d)–(f)).
The mean values of vessel parameters for hLEC groups were always higher than equivalent hBEC cell seeding groups, suggesting than hLEC vessels in vivo were universally larger than hBEC vessels in vivo. This was also seen in immunohistochemical staining of hLEC compared to hBEC vessels in vivo (Figure 6(b) and (c) compared to Figure 7(b) and (d); all figures taken at identical magnifications).
Statistical comparison of hLEC and hBEC vessel diameter, perimeter and cross sectional area
Comparing the individual vessel parameters when the two main cells types hLEC and hBEC were pre-cultured alone it was very clear that overall vessel diameter (p < 0.0001), vessel cross sectional area (p < 0.0001) and vessel perimeter (p <0.0001) were all significantly different, with hLEC consistently forming larger individual vessels than hBEC in vivo. Pre-culture time was overall generally not significant except for cross sectional area (p = 0.0313) (Supplemental Figure 7(a)–(c)). When hLEC or hBEC were co-cultured with hvSMC again overall cell type was significantly different for vessel diameter (p < 0.0001), vessel perimeter (p < 0.0001) and vessel cross sectional area (p < 0.0001), with hLEC + hvSMC consistently having larger values in vivo than hBEC + hvSMC. In vitro pre-culture time was generally not significant overall except for vessel cross sectional area (p = 0.0158) (Supplemental Figures 7(d)–(f)).
Rat blood vessel and connective tissue invasion into scaffolds
Invasion of rat blood vessels and connective tissue into scaffolds was clearly evident in hLEC and hBEC seeded scaffolds at 7 days in vivo, and although the degree of rat tissue infiltration was variable, at 7 days host tissue had not invaded all the scaffold pore volume (Figures 6(a), 7(a) and (b)). At 2 weeks in vivo the invasion of rat blood vessels and connective tissue was complete into the pores which were filled with host tissue (Supplemental Figure 1(c) and (d)).
There was a general tendency for scaffolds seeded with hBEC + hvSMC to have a lower rat vessel PVV within the scaffold than in hBEC-hvSMC scaffolds (this was overall significant p = 0.0249) (Supplemental Figure 8(b)).
There was a similar (but not overall significant) trend for the hLEC + hvSMC to have a lower rat PVV than hLEC-hvSMC seeded scaffolds, except for the 3 days pre-culture group (Supplemental Figure 8(a)). Both hLEC − hvSMC and hLEC + hvSMC groups with 1 h pre-culture had much lower levels of rat PVV (there was a significant overall difference due to pre-culture time of p = 0.0168).
In vivo scaffolds seeded with fibrin alone demonstrated a rat vessel PVV in the 4%–6% range (in hLEC seeded scaffolds rat PVV (mean ± SEM) was 5.618% ± 1.926%; whilst in hBEC seeded scaffolds mean rat PVV was 4.097% ± 0.658%) (Supplemental Figure 8(a) and (b)). These are higher values than normal nude rat skin PVV (1.665% ± 0.192%) (N = 4) (Supplemental Figure 8(c)).
Discussion
This study successfully assembled primary human lymphatic endothelial cells into a lymphatic vessel network in a porous scaffold in vitro, and demonstrated the significant positive influence of co-culture with human vascular smooth muscle cells in expanding in vitro lymphatic network formation. The study also successfully transplanted lymphatic networks in vivo with no evidence of early regression of the transplanted hLEC capillary network, whilst transplanted blood endothelial capillaries (hBEC) exhibited early vessel regression. hLEC vessels demonstrated connection to rat (host) lymphatics at 2 weeks, and functioned as lymphatics within scaffolds in vivo as evidenced by the trafficking of white blood cells exclusively within their lumen, whilst hBEC capillaries inosculated with rat blood capillaries by 1 week. In comparing this engineered human lymphatic capillary network in vivo to an identically assembled human blood capillary network, it was evident, both with and without hvSMC co-culture, that the lymphatic network showed significantly increased individual vessel size, and vessel network PVV and vessel density in vivo (Figures 9 and 10; Supplemental Figures 4 and 7).
In vitro an interconnected lymphatic capillary network formed in human fibrin taking at least 3 days in the porous scaffold, demonstrated in whole mounts. Additional growth factors were not necessary for lymphatic network formation. In alternative models mesenchymal support cells8,30 or mesenchymal stem cells 9 have been considered essential to form human lymphatic networks in vitro. However, in this scaffold/fibrin model we were able to assemble lymphatic capillaries without mesenchymally-derived support cells, although the addition of hvSMC did significantly expand the lymphatic network formed in vitro. The fibrin support matrix infiltrated within the scaffold pores confirmed in this and other studies 29 is highly suitable for lymphatic capillary assembly.
In vitro blood capillary network formation occurs more rapidly than lymphatic network assembly in this scaffold model, with hBEC interconnected capillaries apparent in 2 day whole mounts, and hBEC initially forming a significantly larger vascular network (PVV and vessel density values) at 1 day in vitro than hLEC. The relative slowness of hLEC vessel network formation compared to hBEC parallels skin wound lymphatic angiogenesis and infiltration, which lags behind blood capillary infiltration of the granulation tissue by several days.38,39
Given that human skin lymphatic capillaries do not have a mural cell component 37 it was unexpected that co-culture of hvSMC with hLEC accelerated lymphatic capillary formation, as larger more mature vessels were observed at 2 days if hvSMC were co-cultured with hLEC in whole mounts. Similarly, coculture with hvSMC also enlarged the overall hLEC network as observed in PVV and vessel density analyses in vitro. Previous studies have also confirmed that mesenchymal cells (of various types) enhance lymphatic capillary formation. Cell types known to have this function have included fibroblasts, 8 adipose-derived stromal cells, 31 dermal pulp stem cells 9 and in this study human vascular smooth muscle cells. The later were observed attaching abluminally to lymphatic vessels in vitro and sporadically in vivo. Despite the absence of mural cells around human lymphatic capillaries, it could be postulated that during development of lymphatic capillaries local mesenchymal cell attachment may occur transiently or exert influence through paracrine secretions. 30 Conversely, hBEC did not demonstrate any increase in PVV or vessel density in vitro when co-cultured with hvSMC. However, the presence of hvSMC significantly increased hBEC’s PVV and BV density in vivo. Similar increased blood capillary growth and maturation has been previously demonstrated for human blood endothelial cells co-cultured with mesenchymal cell types.2,40
The overall significant differences between hLEC and hBEC when cultured without hvSMC (PVV and vessel density) indicate that the two endothelial cell types have different capabilities to form vessel networks in vitro, and hBEC do better than hLEC initially. However, hBEC vessels regress over time in culture to a greater extent than hLEC. These differences between hLEC and hBEC were reversed when hvSMC were co-cultured with each cell type, as hLEC co-cultured with hvSMC significantly increases PVV and vessel density, but had no effect in vitro on hBEC (Figures 3 and 9).
The co-culture of hvSMC with hBEC did significantly increase both PVV and vessel density compared to hBEC-vSMC in vivo (Figure 9). hBEC-hvSMC transplanted in vivo demonstrated a significant decline in PVV and vessel density compared to in vitro values. This decline was reduced when hBEC + vSMC were transplanted with only the PVV being marginally significantly different overall from in vitro values (Supplemental Figure 6). Conversely hLEC were quite resistant to vessel network regression when transplanted in vivo. Both hLEC and hLEC + hvSMC groups, showed little decline in PVV or vessel density at 7 days post transplantation (Supplemental Figure 5). This suggests that hLEC can survive the harsher in vivo hypoxic wound conditions 41 far better than hBEC. The maintenance of hLEC vascular volume and vessel density in the hypoxic wound indicates the possibility that hLEC have upregulated expression of pro-survival genes, which promote greater survival in hypoxic environments than hBEC are capable of.
The robust survival of hLEC capillaries was further confirmed by their medium term 2-week in vivo survival (although diminished compared to 1 week values), whilst hBEC capillaries demonstrated no survival at 2 weeks in vivo. In addition, there was evidence of hLEC capillary inosculation (functional joining) with rat lymphatics (HG-19+) within the scaffold at 2 weeks (Figure 6), and their appearance at this time indicated hLEC capillaries were functioning as lymphatics with numerous leucocytes being conveyed in their lumen, without evidence of red blood cells (Figure 6). This demonstrates the attainment, at least in part, of mature human lymphatic functioning of transplanted hLEC capillaries with demonstrated leucocyte trafficking.12,13
In vivo hBEC vessels demonstrated inosculation with rat blood vessels at 7 days, with evidence of tail vein injected FITC dextran within hBEC capillaries in vivo (Figure 7). However, the study was unable to demonstrate similar FITC dextran infusion in hLEC capillaries at 7 days in vivo despite that fact that FITC dextran was clearly visible in rat blood vessels within the scaffold. However, occasional hLEC capillaries containing rat blood erythrocytes were observed at 7 days. This suggests that capillary inosculation with rat blood vessels is less frequent in hLEC than hBEC capillary networks at 7 days, but still does occasionally occur.
Both hLEC and hBEC vessels maintained distinct cell marker characteristics (hLEC: CD31+/podoplanin+ and hBEC: CD31+/podoplanin−) from 2D culture to 3D in vitro culture and similarly when vessel networks were transplanted in vivo (Figures 2, 3, 6 and 7). In addition the morphometric characteristics of individual blood and lymphatic vessels were distinctly different, with hLEC in vivo showing a consistent significant increase in diameter, perimeter length and cross sectional area over hBEC vessels with and without hvSMC (Figure 10; Supplemental Figure 7). This characteristic vessel size difference reasserts that both endothelial cell types formed vessels characteristic of their cell lineage, in cell marker profile and vessel structural morphology, which was maintained under in vivo perfusion conditions.
Individual vessel parameters for hBEC and hLEC vessels in vivo demonstrated distinctly opposite trends in relation to pre-culture time and the influence of co-culture with hvSMC. hBEC capillaries displayed significant increases in vessel diameter, vessel perimeter and cross-sectional area with increased pre-culture time, thus the largest individual vessel parameters were after 3 days in culture prior to in vivo transplantation. Additionally the co-culture of hvSMC with hBEC significantly increased individual vessel diameter and perimeter (Figure 10). hLEC vessels in vivo demonstrated opposite trends. Co-culturing hLECs with hvSMC made no difference to individual hLEC vessel parameters in vivo, but pre-culture time was significant overall for all parameters, with 1 h pre-culture consistently producing the largest hLEC diameter, perimeter length and cross sectional area (Figure 10).
It should also be noted that these individual vessel morphometric parameters were significantly and consistently larger for hLEC than hBEC. hLEC diameter was on average in the 35–50 µm range, with 50 µm diameter vessels forming in vivo with only 1 h of pre-culture. This hLEC vessel size is within the range reported for skin lymphatics.12,42 Valves were not observed in hLEC vessels in vivo, similar to human skin lymphatic capillaries. 43 The individual vessel diameter of hBEC capillaries was 12–25 µm, and this is in the diameter range of skin blood capillaries.36,44
This study examined hBEC and hLEC capillaries to 2 weeks in vivo to assess early-mid-term survival and donor capillary function when transplanted in vivo. Similar in vivo time points have been used in comparable studies (Landau et al. 9 7 days in vivo, and Marino et al. 8 15 days in vivo). The literature suggests that donor autologous vessels generally do not survive longer than 2–3 weeks. In transferred autologous skin grafts, graft vessel blood perfusion is evident from 48 to 72 h, 45 graft blood vessel regression is observed from day 3 to 21, 46 with replacement of graft vessels by host wound bed ECs moving along the existing graft vessel channels. 45 Blood capillary invasion into the graft is largely complete at 3 weeks post-transplantation, with 60% replacement in central graft positions 46 and greater replacement peripherally. Our results do not differ greatly from this pattern with loss of hBEC scaffold capillaries more rapid than lymphatics. The latter to our knowledge have not been examined in a similar autologous skin flap model. Inosculation of hBEC capillaries and host capillaries occurred by 7 days in our model (Figure 7(e) and (h)), and inosculation of scaffold hLEC capillaries with host rat lymphatics was slower but evident at 2 weeks in vivo (Figure 6(h)–(j)) with some evidence of mosaic donor hLEC capillaries and rat cell components also evident (Figure 6(e)).
In vitro pre-culture time had no overall significant influence on vessel network size as measured by PVV or vessel density for any cell type or cell combination implanted in vivo. At 1 h post-seeding in vitro, small individual clumps of cells have formed, without lumens or any vessel structure. 4 Yet this minimal period of pre-culture ultimately formed the largest human lymphatic vessels in vivo, and 1 h pre-culture hLEC seeded scaffolds were also able to form lymphatic vessel networks with equivalent network size in terms of PVV and vessel density as longer pre-culture periods. Therefore, 1 h pre-culture would be adequate for all future hLEC implantations. In vivo, co-culture with hvSMC significantly increased hBEC’s PVV and vessel density in vivo, and hLEC’s vessel density in vivo, and co-culture with hvSMC would be recommended for all future implantations of primary blood and lymphatic microvascular cells.
The porous polyurethane scaffold (NovoSorb) used in this study has been used by our group previously to form human induced pluripotent stem cell-derived endothelial cell capillaries in vitro with subsequent transplantation in vivo into a mouse model. 4 Other scaffolds with wide interconnected pores have been used to serve a similar purpose, including poly(lactide-co-glycolide) (PLGA) and poly(L-lactide) (PLLA) scaffolds (pore size 212–600 μm). 9 Other wide-pored scaffolds such as polyester-ether hydrogel scaffolds 47 and used by our group as an adipose tissue carrier48 could also be employed for lymphatic capillary assembly. It should be noted however that the Landau et al. 9 study and our study used fibrin to support the cells within the scaffold pores. Fibrin is a naturally occurring element of all skin wound healing where angiogenesis and lymphangiogenesis occur. Fibrin was a vital factor in creating a supportive 3D environment for lymphatic capillary assembly in our study and has been successfully used by others.29,31 Other materials used for lymphatic formation for in vitro assembly include Celgro (Orthocell) a type 1 collagen material, 9 hydrogels such as collagen 18 or fibroblast cell layers. 30
NovoSorb has been used in vivo as a dermal replacement both experimentally and clinically. Vascularized connective tissue infiltrates NovoSorb pores and some weeks later a split skin graft can be placed over the scaffold to heal the wound. 49 NovoSorb has also been used to provide a large vascularized wound base for a delayed application of engineered skin in a pig model. 50 Recently it has been used as a dermal substitute in human applications for reconstruction of complex wounds.51–53 In these reports the wide interconnected pore size permits ingrowth of connective tissue and blood vessels over several weeks providing a well vascularized bed for second stage application of split thickness skin grafts. The degradation of a 2 mm thick polyurethane scaffold in vivo takes 12–18 months. 51 NovoSorb’s continued use clinically is likely to expand.
Conversely, NovoSorb’s purpose in our study was as a cell carrier for which it has a number of advantages. The highly porous polyurethane provided space for the assembly of the lymphatic and blood capillary networks as its elastic properties enabled a ‘manual suction method’ to be used to seed the cells in human fibrin into all scaffold pores in vitro, resulting in complete pore filling. The scaffold’s large interconnected pores (300–600 µm) allowed the 3D vessel network to assemble in an interconnected manner, and it was sturdy enough to enable surgical transplantation into a subdermal environment without damage to the capillaries. The scaffold maintained its structural integrity up to 2 weeks in vivo, and permitted host connective tissue and vessel ingrowth from the recipient site permitting inosculation between host and transplanted capillary networks. However, the scaffold’s major disadvantage is the degree of macrophage inflammation it attracts in vivo. This inflammation may be diminished by specific anti-inflammatory coatings 54 yet to be trialled for NovoSorb.
The athymic nude rat used in this study has a marked reduction in T lymphocyte production and function, 55 but retains macrophages and is therefore able to mount an inflammatory response to NovoSorb’s ‘foreign’ material. This inflammatory response may have been detrimental to human blood and lymphatic capillary survival. In creating the skin wound some inflammation would occur, but we believe that the scaffold material has promoted an increased response (as also observed in a SCID mouse model 4 ). We have noted that other synthetic scaffold materials cause a significant inflammatory response in non-immunosuppressed rats56,48 as opposed to biologically derived scaffolds/matrices where the inflammatory response (even if the scaffold/matrix is derived from a different species) is less in the sub-dermal environment. 57 However it should be noted that despite this inflammatory reaction NovoSorb has been implanted in humans for many months without detrimental outcomes, 51 and the inflammatory response is likely to subside over time.
Conclusion
The primary human lymphatic networks generated in this study were surprisingly resilient to in vivo transplantation. The maintenance of extensive and functional interconnected lymphatic networks in vivo indicates that with further optimization in vitro assembly of hLEC networks could be utilized as a regenerative treatment in conditions where lymphatic drainage is impaired as occurs in lymphoedema, radiation injury, cancer resection sites and areas of significant or deep skin wounds. In addition, they hold possibilities for further in vitro studies involving lymphatic disease modelling or drug studies involving lymphatic vessel responses.
Supplemental Material
sj-docx-1-tej-10.1177_20417314221140979 – Supplemental material for Engineering transplantable human lymphatic and blood capillary networks in a porous scaffold
Supplemental material, sj-docx-1-tej-10.1177_20417314221140979 for Engineering transplantable human lymphatic and blood capillary networks in a porous scaffold by Anne M Kong, Shiang Y Lim, Jason A Palmer, Amanda Rixon, Yi-Wen Gerrand, Kiryu K Yap, Wayne A Morrison and Geraldine M Mitchell in Journal of Tissue Engineering
Footnotes
Acknowledgements
The authors acknowledge Priya Sivakumaran who assisted with supplying the human vascular smooth muscle cells. The authors are also indebted to the Experimental Medical and Surgical Unit (the late Sue Mc Kay, Anna Deftereos and Liliana Pepe) at St Vincent’s Hospital Melbourne.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by grants from the Research Endowment Fund, St Vincent’s Hospital, Melbourne, Australia; the Australian Catholic University; the Stafford Fox Foundation, Australia; and the Victorian State Government’s Department of Innovation, Industry and Regional Development’s Operational Infrastructure Support Program. KKY was supported by scholarships from the Australian Government Research Training Program, the National Health & Medical Research Council; Australia and New Zealand Hepatic, Pancreatic, and Biliary Association; and St Vincent’s Institute Foundation.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.