
Abstract
Scaffolds are materials used for delivery of cells for regeneration of tissues. They support three-dimensional organization and improve cell survival. For the repair of small skeletal muscles, injections of small volumes of cells are attractive, and injectable scaffolds for delivery of cells offer a minimally invasive technique. In this study, we examined in vitro the cell instructive effects of three types of injectable scaffolds, fibrin, alginate, and poly(lactic-co-glycolic acid)-based microparticles on primary human myoblasts. The myoblast morphology and progression in the myogenic program differed, depending on the type of scaffold material. In alginate gel, the cells obtained a round morphology, they ceased to proliferate, and entered quiescence. In the fibrin gels, differentiation was promoted, and myotubes were observed within a few days in culture, while poly(lactic-co-glycolic acid)-based microparticles supported prolonged proliferation. Myoblasts released from the alginate and fibrin gels were studied, and cells released from these scaffolds had retained the ability to proliferate and differentiate. Thus, the study shows that human myogenic cells combined with injectable scaffold materials are guided into different states depending on the choice of scaffold. This opens for in vivo experiments, including testing of the significance of the cell state on regeneration potential of primary human myoblasts.
Introduction
Skeletal muscle tissue has a remarkable capacity to regenerate due to the potential of the myogenic stem cell, the satellite cell. Upon injury, quiescent muscle stem cells are activated, enter the myogenic program, and begin to proliferate. When the myoblasts later differentiate, they are able to fuse with already existing myofibers or fuse and form polynucleated myotubes that afterwards differentiate into mature myofibers, thereby restoring the muscle. 1 These characteristics make the satellite cell the primary candidate for use in muscle repair.
Strategies for stem cell therapy must deal with both type and size of muscle damage. Thus, small or diffuse lesions and volumetric loss should be handled differently. For small and diffuse lesions, cell delivery by injection has been employed and is clinically well accepted because of the minimal invasiveness of this technique.
Cultured myoblasts have been injected and studied for their regenerative potential in several types of lesions, reviewed by Ciecierska et al. 2 However, effective clinical procedures for repair of skeletal muscle tissue are not yet available because of low transplantation efficiencies, reviewed in Sicari et al. 3
In generalized muscular dystrophy, myoblast implantation by injection has been studied in animal models,4,5 as well as in human trials.6,7 While results in animal models were promising, most clinical trials turned out disappointing.
However, clinical trials on the treatment of incontinence by implantation of cells in the urethral sphincter wall have been more successful.8–12 Likewise, animal models for incontinence13–15 show that the continence improves following implantation of various cell types, but still histological examinations reveal a low recovery of implanted cells.
A major problem thus appears to be the survival in the tissues of injected myoblasts. 16 The injection procedure has been shown to be harmful, exposing the cells to mechanical forces 17 and oxidative stress. 18 Furthermore, the innate inflammatory response at the site of implantation19–21 has been considered to have adverse effects on implanted cells.
The use of injectable scaffolds to deliver myoblasts for regenerative medicine applications could be part of a solution. Such scaffolds can potentially reduce the shear force during injection 17 and possibly protect the cells against inflammatory host response, and thereby improve cell survival. 22 Further advantages in the use of injectable scaffolds could be the possibility to obtain a localized deposition of cells in the region of interest, and to obtain a local concentration of myoblasts in close proximity, permitting their subsequent fusion to form fibers. Also, injectable scaffolds have been shown to be effective for local delivery of bioactive components that support the implanted cells and optimize the micro-environment.
An additional parameter in optimizing implantation of myoblasts could be the selection of cells in specific states. In this study, we examined the cell responses induced by exposure to fibrin, alginate, and poly(lactic-co-glycolic acid) (PLGA): three Food and Drug Administration (FDA)-approved, injectable materials previously used for clinical purposes.23–25 We demonstrated in vitro how these scaffold materials differ in their regulation of the myogenic program.
Materials and methods
Isolation and propagation of human myoblasts
Human myoblasts were isolated from biopsies taken from young men (20–25 years). We established human primary myoblast cultures as previously described.26,27 In short, muscle biopsies were obtained from m. vastus lateralis, freed from connective tissue, minced, washed, and dissociated with 0.05% trypsin–ethylenediaminetetraacetic acid (EDTA; Invitrogen) for 3 × 30 min. Harvested mononuclear cells were pooled, and fetal bovine serum (FBS; Invitrogen) was added as protease inhibitor.
To obtain a myoblast-enriched cell culture, the cells were preplated in non-coated dishes to reduce the fraction of fibroblasts. We seeded isolated cells (maximum five passages) on flasks (NUNC) coated with extracellular matrix gel (from Engelbreth-Holm-Swarm mouse sarcoma; Sigma–Aldrich), and during every passage, the cells were preplated for 20–30 min before transfer to Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS and 1% penicillin and streptomycin (PS; Invitrogen).
In each experiment, we used cells from three individuals. The procedure was approved by The Regional Ethical Committee of Southern Denmark (S-20070079).
Cultivation of myoblasts in fibrin and alginate gels
Alginate
The alginate used in the study was LVG alginate (Novamatrix), a low-viscosity gel composed of a minimum 60% guluronate. A number of 6 × 105 primary human myoblasts were mixed with 600 µL of sterile 1% LVG alginate solution in phosphate-buffered saline (PBS) at room temperature. We released 25 µL alginate into a 100-mM CaCl2 solution for 60 s to produce polymerized alginate spheres.
Fibrin
A number of 6 × 105 primary human myoblasts were dissolved in 300 µL of human plasma. A volume of 300 µL of bovine thrombin (100 U/mL in isotonic NaCl; Biofac A/S) was added and incubated for 30 min at 37°C. The resulting gel was cut into six equal portions.
The alginate and fibrin gels were rinsed twice in sterile PBS and transferred to 24-well plates. Each gel was placed in a well containing 0.5 mL growth medium (GM) (DMEM with 2% Ultroser G (Pall Life Sciences), 2% FBS, and 1% PS) and incubated at 37°C and 5% CO2 in a humidified incubator. GM was renewed every second day.
After 7 days in GM, the medium was changed to differentiation medium (DM) (DMEM with 2% FBS, 1% PS, and 25 pmol insulin (Actrapid; Novo Nordisk)) to induce differentiation. 28
Gels incubated in GM at day 1 and day 4 and gels incubated in DM at day 14 were harvested for paraffin embedding for histology.
Cell retrieval from degraded alginate and fibrin gels
Each gel was placed in a well (24-well plate) containing a 12-mm circular glass coverslip (Menzel) at the bottom. Cells released passively during cultivation due to degradation of the gels were allowed to attach to the coverslip, and when growth was observed in all wells, medium was changed from GM to DM to induce differentiation.
Cell retrieval from dissolved alginate gels
To obtain fast and synchronous cell retrieval, sodium citrate buffer, acting as calcium chelator, was employed to dissolve the alginate gels. After 4 days of incubation in GM, each gel was dissolved in 100 µL isotonic solution of 50 mM trisodium citrate dihydrate (Sigma–Aldrich) and 104 mM NaCl (Sigma–Aldrich), pH 8.0 at 37°C. After 10 min, 900 µL of GM was added.
For the study of proliferation, cells retrieved from one gel were equally distributed to four wells in a 24-well plate, and cultured with coverslips in 0.5 mL GM for 1–4 days.
For the study of differentiation, cells from one gel were distributed to two wells containing 0.5 mL GM for 2 days, and then in 0.5 mL DM for 5 days.
Cultivation of myoblasts on PLGA-based microparticles
PLGA-based spherical microparticles, 10–100 µm in diameter, were used for the study (a gift from Coloplast A/S). For details on manufacturing, see Wen et al. 29
Before culturing, the microparticles were washed in 12% ethanol. A number of 10,000 primary human myoblasts were cultured with 1.5 mg microparticles in ultra-low attachment culture dishes (60 mm; Corning) for 7 days before changing to DM for another 7 days. The harvested microparticles were spun down to a pellet and embedded in paraffin.
Paraffin embedding procedures
Standard procedure for paraffin embedding results in dissolution of alginate, and PLGA microparticles and thus loss of cells. Consequently, to enable later sectioning of alginate gels and microparticle-cell aggregates from paraffin blocks, the harvested materials were infiltrated with plasma and thrombin for 30 min and then fixed in 4% neutral buffered formalin (NBF), followed by paraffin embedding. This initial fibrin embedding trapped the cells in their position in alginate and on particles, respectively.
Immunocytochemical staining
Immunocytochemistry was performed on either 3 µm sections from formalin fixed, paraffin embedded specimens or on cultured myoblasts on coverslips. Staining was performed on a Ventana BenchMark immunostainer (Roche) with the OptiView-DAB detection system. Before staining of cytological preparations, the coverslips were mounted on glass slides. The primary antibodies used were anti-CD56 (neural cell adhesion molecule (NCAM)):MRQ-42 (NCAM), rabbit monoclonal (Cell Marque), anti-myogenin: F5D, mouse monoclonal (Ventana Medical Systems), Ki67: CONFIRM anti-Ki-67 (30-9) rabbit monoclonal (Ventana Medical Systems), anti-MHCfast: Anti-Myosin (Skeletal, Fast) antibody, mouse monoclonal, and clone MY-32 (Sigma–Aldrich).
Phalloidin staining
Paraffin sections of alginate and fibrin gels were deparaffinized and incubated with phalloidin 1:40 conjugated to Alexa fluor 546 (Life Technologies) in Dako dilution buffer for 1 h at room temperature and dark conditions, then rinsed 3 × 10 min in Tris-buffered saline (TBS). The mounting medium (Vectashield) contained DAPI (4′,6-diamidino-2-phenylindole) to visualize nuclei.
Fixation and processing of microparticles and cells for electron microscopy
Transmission electron microscopy
Microparticles cultured with proliferating myoblasts were spun down to a pellet and resuspended in 2% glutaraldehyde in 0.04 M phosphate buffer, pH 7.4 for 60 min at 20°C. The cells were spun down again and washed in 0.1 M phosphate buffer for 10 min. The pellet was resuspended in 15% bovine serum albumin (BSA) for 60 min at 20°C, and then incubated in 2% glutaraldehyde over night at 4°C to solidify.
Following rinsing, the solidified material/pellet was post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer for 60 min at 4°C, dehydrated through a graded series of alcohol and acetone at 4°C–20°C, and infiltrated in Epon for 90 min at 20°C. Ultra-thin sections were cut from the Epon block on a Leica Ultracut UCT ultramicrotome (Rowako AB), contrasted with uranyl acetate and lead citrate, and examined and photographed in a precalibrated Philips EM 208 electron microscope and a Megaview III FW camera (both FEI Company).
Scanning electron microscopy
Microparticles cultured with proliferating myoblasts were dried, spread out on a metal specimen stub, and gold coated by sputter coating before imaging using a scanning electron microscope (Hitachi TM3000 Tabletop scanning electron microscope).
Quantitation
The Ki67 index was calculated from counting of cells on scanned images (NanoZoomer 2.0-HT digital Slide Scanner and NDP.view2 software; Hamamatsu) of the stained coverslips. The entire coverslip was scanned, and because the coverslip is round, the orientation in the images are random. From each culture, pictures (10× magnification, field: 1240 × 697 µm2), covering the entire horizontal diameter, were imported to ImageJ. 30 Using the Cell Counter plugin, the cells were counted in all imported pictures or in every second picture when the amount of cells was high. More than 200 cells were counted, except for one culture at 24 h where only 134 cells were found in the horizontal diameter zone.
Results
We cultured primary human myoblasts, isolated from young men, with three types of scaffolds to examine the influence on proliferation and differentiation of myoblasts of the different materials. As a model for testing, the cell instructive effect in the selected materials, we produced 25–50 µL portions of gel, each containing in the range of 25,000–50,000 cells.
In standard monolayer culture, the DM in 4 days is able to induce differentiation of confluent myoblasts and consequently cease most proliferation. 28 According to this, proteins expressed during myogenesis was examined with immunohistochemistry in proliferating cells at days 1 and 4, and in differentiating cells at days 5–7 after change to DM. Ki67, which is only expressed in proliferating cells, 31 was used as a marker for proliferation. We used myogenin and MHCfast as an early marker and a late marker of differentiation, respectively. NCAM is constitutively expressed by myoblasts and myotubes, and was used as a marker for myoblasts and immature fibers. The vast majority of the cells in both the proliferating and differentiating cultures were NCAM positive as seen in Figure 1(c) and (d).
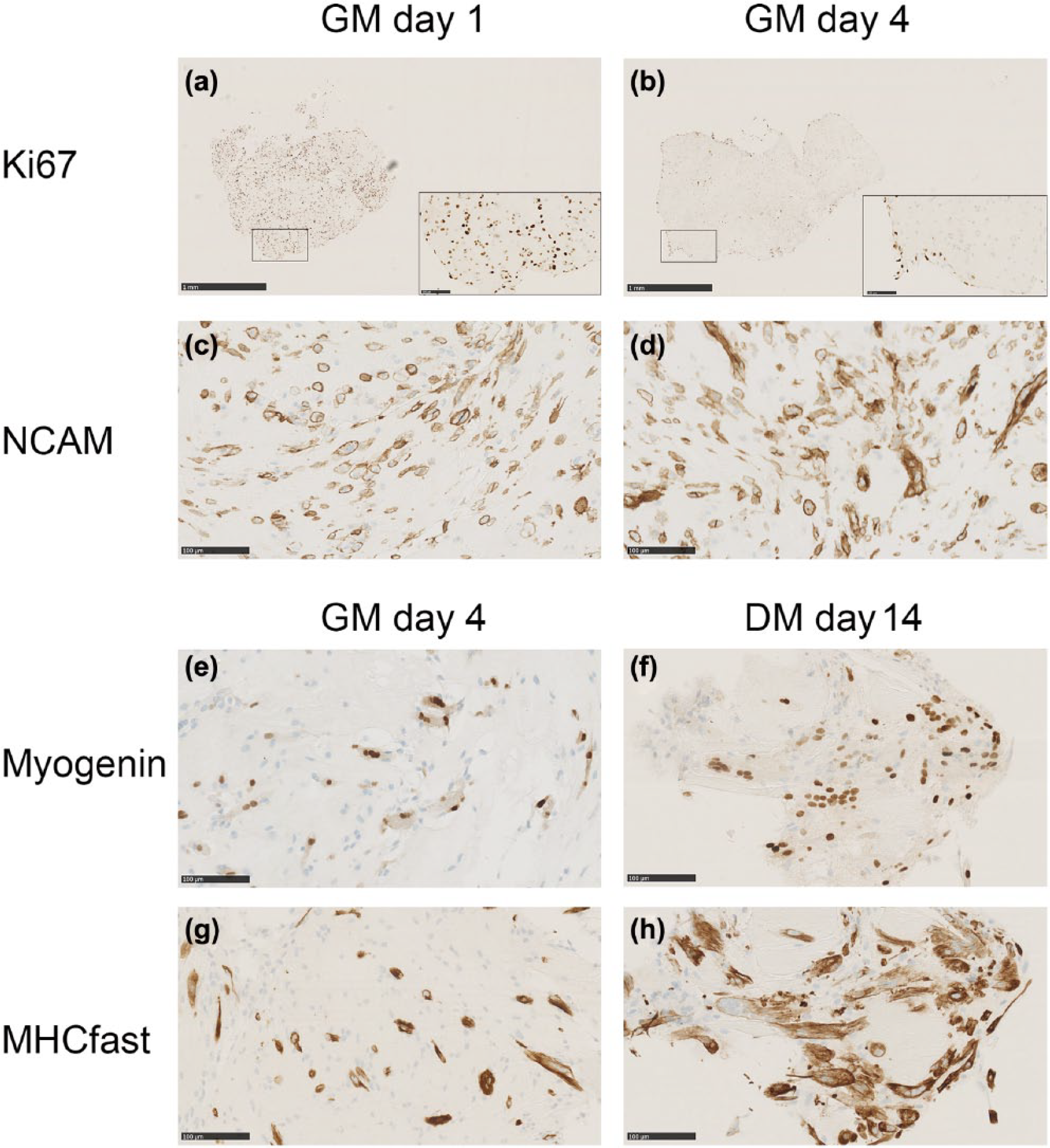
Proliferating human myoblasts embedded in fibrin gels, cultured in growth medium (GM) for 7 days and then in differentiation medium (DM) for another 7 days. The immunostaining for Ki67 at (a) day 1 shows a uniform distribution of Ki67-positive cells throughout the gel. (b) Day 4 Ki67-positive cells are mainly located at the surface. The vast majority of cells are positive for the myoblast marker NCAM at (c) day 1 and (d) day 4. At day 4 in GM, spontaneous formation of myotubes was detected, as well as expression of (e) myogenin and (g) MHCfast. More myotubes were detected in DM at (f, h) day 14 and with more nuclei per tube. During the experiment, the fibrin gels decreased in size and turned into more compact pellets. Scale bars for (a) and (b) represent 1 mm, and 100 µm in the insets. Scale bars for (c)–(h) represent 100 µm.
Fibrin gel
Proliferating myoblasts embedded in fibrin gel and cultured in GM appeared spindle shaped after 24 h, and the majority of the cells were proliferating and expressing Ki67 (Figure 1(a)). After 4 days, Ki67 was lost in the central part of the gels, but positive cells were still present in the periphery of the gel where the surface was exposed to the medium (Figure 1(b)). In contrast, myoblasts situated inside the fibrin gel at day 4 in GM had initiated a spontaneous differentiation, forming myotubes and expressing myogenin and MHCfast (Figure 1(e) and (g)). To optimize conditions for differentiation, the fibrin gels were then placed in DM for 7 days. Under these conditions, the differentiation was further enhanced (Figure 1(f) and (h)), but still a few superficial Ki67-positive cells were seen (not shown). In conclusion, embedding myoblasts in the fibrin gel induced fast differentiation. However, at the interface to culture medium, cells retained their capacity to proliferate.
Alginate gel
Myoblasts embedded in alginate gel quickly obtained a round shape and remained round without cell extensions at all time points examined (Figure 2(a)–(h)). After 1 day of culture in GM, the alginate embedded myoblasts still expressed Ki67 (Figure 2(a)), but after 4 days, they had exited cell cycle, and very few Ki67-positive cells were seen (Figure 2(b)). No signs of differentiation were found after 4 days in GM (Figure 2(c) and (e)), and even the standard procedure to induce differentiation, by changing to DM, did not produce multinucleated myotubes or induce expression of myogenin and MHCfast (Figure 2(d) and (f)). The phalloidin stain visualizing the actin cytoskeleton (Figure 2(g) and (h)) confirmed that the cells were rounded up and did not have extensions. Thus, the present formulation of LVG alginate hydrogel induced myoblasts to exit the cell cycle and enter quiescence. Moreover, alginate kept the myoblasts resistant to induction of differentiation by DM.

Proliferating primary human myoblast embedded in 1% LVG alginate gel spheres. The spheres were placed in growth medium (GM) for 7 days and then in differentiation medium (DM) for another 7 days. (a) At day 1, most myoblasts were positive for Ki67, (b) but at day 4, this expression had ceased. Neither did the cells express markers for differentiation, (c) MHCfast and (e) myogenin. (d, f) Exposure to DM did not induce expression of differentiation markers. Accordingly, the phalloidin staining showed a round morphology of the cells at both (g) day 4 in GM and (h) day 14 in DM. Scale bars for (a)–(f) = 100 µm, (g) = 200 µm, and (h) = 10 µm.
PLGA microparticles
When myoblasts were cultured with PLGA particles, ranging in diameters from 10 to 100 µm, a single myoblast could attach to the surface of several microparticles, hence aggregates of microparticles held together by myoblasts were formed (Figure 3(a) and (b)). Culturing myoblasts with microparticles promoted proliferation, seen as continued expression of the Ki67 at day 4 in GM (Figure 3(c)). Even when exposed to DM for 7 days, the amount of Ki67 expressing cells was prominent (Figure 3(d)). However, markers for differentiation were expressed by a few scattered myoblasts in GM day 4 (Figure 3(e) and (g)). When cultured in DM, a small population of mononuclear cells expressed myogenin, and a small number expressed MHCfast indicating some degree of differentiation, though multinucleated myotubes remained scanty (Figure 3(f) and (h)). This suggests that culturing myoblasts on PLGA microparticles favors proliferation and inhibits differentiation. The contact with PLGA even counteracted the induced differentiation.

Human myoblasts were cultured with microparticles in growth medium (GM) for 7 days before changing to differentiation medium (DM) for another 7 days. (a) Transmission electron microscopy (TEM) shows a myoblast joining together two microparticles. The myoblasts attach to the microparticles by growing into cavities in the particle surface (arrow). (b) Scanning electron microscopy (SEM) shows proliferating myoblasts adhering to and joining the particles. In the paraffin-embedded aggregates, the PLGA-based material melts during processing leaving empty gabs (*). (c) The majority of cells were Ki67 positive at day 4 in GM, and (d) even after changing to DM, Ki67 expression was seen in part of the cells. (e) In GM day 4, some myogenin-positive cells were seen and (f) culture in DM only slightly increased the expression. (f, inset) Mainly mononuclear cells were stained, although a few myofibers were detected. (g, h) The same pattern was observed in the MHCfast staining. Scale bars for (a) = 5 µm, (b) = 50 µm, and (c)–(h) = 100 µm.
Cells retrieved from alginate and fibrin gels
A prerequisite for successful integration of transplanted myoblasts is that they retain their ability to fuse and form new myofibers. We examined the ability of myoblasts released from alginate and fibrin to proliferate and differentiate outside the gels.
Cells were continuously released from the surface of the alginate and fibrin gels to the culture medium. We collected cells for 9 days by letting the released cells attach to a cover slip and grow in GM. The vast majority of released cells were still the NCAM-positive myogenic cells (Figure 4(a)–(d) and (g)). Cells retrieved from the gels in this way were able to both proliferate, and after induction by DM, differentiate into multinucleated myotubes (Figure 4(a)–(f)). The cells retrieved from alginate gels were observed to form larger fibers containing more nuclei than those retrieved from fibrin gels (Figure 4(c) and (d)).

Cells spontaneously retrieved from alginate and fibrin gels. Proliferating primary human myoblasts were embedded in alginate and fibrin gels and placed in growth medium (GM) for 9 days. During this period, the gels were partly dissolved resulting in cell release. In both (a, c) alginate and (b, d) fibrin, the majority of released cells were NCAM positive and (e, f) formation of MHCfast-positive myotubes could be found after induction of differentiation with differentiation medium (DM). For induced retrieval of cells from alginate gels, myoblasts embedded in alginate gels were placed in GM for 4 days and then the cells were retrieved by dissolving the gel by sodium citrate buffer. (g) Retrieved cells cultured in GM for 2 days followed by DM for 5 days showed strong NCAM positivity with myotube formation. In retrieved cells, cultured in GM for 1–4 days, (h) the Ki67 labeling index showed synchronous reactivation (n = 3). Scale bars for (a), (b), and (g) represent 500 µm and scale bars for (c) and (d) represent 100 µm.
In an additional experiment, we dissolved alginate gels by adding sodium citrate buffer, a calcium chelator. This resulted in a fast dissolution of the gel and retrieval of cells, which were later able to differentiate in DM (Figure 4(g)). When the cells were cultured in GM, we could demonstrate that about 20% of the cells within 24 h entered cell cycle and the expression of Ki67 rose between 24 and 72 h after release to around 90%, thus indicating that the cells were synchronously reactivated from quiescence (Figure 4(h)).
Discussion
In the present in vitro study, we demonstrated that the injectable scaffold materials, fibrin, LVG alginate, and PLGA-based microparticles, all influenced the cell state of primary human myoblasts; however, they had different cell instructive effects resulting in promotion of differentiation, quiescence, and proliferation, respectively (Table 1).
Cell instructive properties of injectable scaffold materials.

Moreover, we showed that the myoblasts during contact with or after retrieval from the three materials retained the capacity for proliferation and differentiation.
Concerning particularly alginate, it has been reported that also C2C12 mouse myoblasts survive embedding, while, in contrast, olfactory ensheathing cells did not. 32 The survival of cells embedded in alginate thus appears to be cell-type dependent.
The nature of lesions, that could be candidates for treatment with myoblast transplantation, includes both localized acute and chronic lesions with or without fibrosis, as well as atrophy and more diffused degeneration. Therefore, the option to choose different priming of the myogenic cells could be valuable in approaches to develop protocols for implantation of myogenic cells.
Fibrin is widely used clinically for surgery as a tissue sealant. It is produced by thrombolytic cleavage of fibrinogen. In vivo, it is rapidly degraded but the degradation rate of fibrin gel can be modulated by change in the concentration of fibrinogen. 33 The fibrin gel used in this study was prepared from human blood plasma. This can be favorable for clinical purposes because of the possibility to use autologous plasma for fibrin scaffolds. This concentration of fibrin, approximately 3 mg/mL has also previously been found preferable compared to higher concentrations. 33
Myoblasts have previously been cultured with fibrin in vitro, 34 and fibrin has been shown to improve the survival and integration of primary rat myoblasts injected into skeletal muscle tissue.33,35
In a study, primary human myoblast cultured in a fibrin gel increased the expression of differentiation-associated genes as compared to monolayer culture. 33 Our study confirms this finding, showing early formation of myotubes and upregulation of differentiation-associated proteins in myoblasts cultured in fibrin gel. This differentiation was obtained without the use of DM, thus induced by the conditions in the fibrin. While myoblasts embedded deeper inside the gel differentiated into myotubes, we, in addition, found a population of proliferating cells located on the surface of the gel. Moreover, myoblasts released from degrading fibrin gel were able to proliferate in monolayer culture, and they had retained their capability to fuse into myotubes. This indicates that the fibrin gels besides the postmitotic, differentiating cells harbored a population of cells with proliferative capacity. Distance to surface and number of cells are important parameters in models for estimating availability of nutrition and oxygen in scaffolds. 36 Moreover, the method to induce myoblast differentiation in vitro 28 includes a reduction in the serum content and the amount of insulin (or insulin-like growth factor (IGF)) in the medium. A reduced supply of nutrients and growth factors in the scaffold thus could be factors contributing to the observed differentiation.
The fibrin gel has myotubes located centrally, and proliferating myoblasts located at the surface. Consequently, the fibrin gel offers the possibility that proliferating myoblasts in the surface of the gel in vivo could promote the fusion of myotubes formed in the gel with existing myofibers.
Alginates are linear polysaccharides composed of varying proportions of mannuronic and guluronic acid. By addition of a polyvalent cation, such as calcium, the alginate will polymerize and form a gel. The ratio of guluronic acid to mannuronic acid affects mechanical strength and porosity of the gel. A study showed that primary murine myoblasts embedded in alginate gel have a round shape, and that they lack the ability to differentiate, while the ability to proliferate depended on the stiffness and degradability of the gel. 37 Alginate has also been suggested as an alternative to cryopreservation for the short-term storage of stem cells. 38
In our study, the myoblasts cultured in a 1% LVG alginate gel within 96 h had a rounded morphology and did not express proliferation or differentiation markers indicating that alginate gel induced quiescence of the myoblasts.
It has been shown that the stiffness of the substrate influences the cell state of myoblasts,37,39,40 and we have previously found that primary human myoblasts entered quiescence when cultured in a high-viscosity medium for 96 h. 27 Similarly, a 0.7% LVG alginate surface has been reported not to support attachment of C2C12 mouse myoblasts. 32 Lack of cell binding motifs in alginate has also been addressed in context of cell adhesion and morphology. Thus, RGD (arginine-glycine-aspartate-peptide)-modified alginates were reported to support attachment of C2C12 myoblasts in two-dimensional (2D) cultures, but did not have a clear effect on survival or growth pattern of the cells in three-dimensional (3D) cultures. 32 Cytoskeletal arrangement is an important factor for transmitting signals to the nucleus, and it has been shown that blocking intracellular adhesion–dependent signals mimics absence of adhesive contacts. Rounding up of the cells is also seen here and correlates with quiescence,41,42 indicating a molecular mechanisms behind alginate induced quiescence.
When the myoblasts were released from degrading alginate gel, their ability to fuse into myotubes was retained, and when compared to myoblast released from fibrin gel, they had an ability to form larger tubes with a higher number of nuclei. Moreover, it seems that more cells were spontaneously released from alginate than from fibrin gel. One reason for this could be that part of the cells released from the fibrin gel had already differentiated, while the cells in the alginate gel were still able to adhere and proliferate. When alginate gel after 4 days was quickly dissolved by calcium chelation, the retrieval of cells was much larger than what was observed from spontaneously degrading alginate gels in culture medium.
In vivo, an injury will result in a local, synchronous activation of myogenic stem cells. 1 The synchronous release and reactivation of myoblasts from quickly dissolved gels mimic the induction of regeneration after a lesion. Thus, in our in vitro study on activation of primary human myoblasts, we found that the cells synchronously progressed in the myogenic program. 27 Considering this, in a clinical setting, it could be advantageous to inject myoblasts in alginate, and later, when they have entered quiescence, to dissolve the gel with a calcium chelator.
In this study, we cultured primary human myoblast on PLGA microparticles with a diameter ranging from 10–100 µm. In a previous comparable study, primary human myoblasts were cultured on porous PLGA microspheres with a diameter ranging from 250–425 µm. 43 In both studies, the PLGA promoted continuous proliferation of myoblasts and prevented spontaneous differentiation. However, with our choice in particle size, cells are able to attach to more particles and thus form larger structures of cell-particle aggregates. This could be advantageous for subsequent muscle formation, having many myoblasts kept in close proximity by attachment to particles. A disadvantage of using microparticles for cell delivery could be that in contrast to hydrogels, the microparticles may offer less protection against a hostile environment in vivo. However, cells located in the interior of larger aggregates are likely to be shielded. In this connection, it can also be mentioned that transplanted myofibers with adherent myoblasts in mice have been shown to be successful. In that case, the myoblasts were also situated on a surface, here of the implanted fibers, and thus survived and proliferated extensively without a protective coverage. 44 The size of particles we chose in our study forms chains of cell-particle aggregates with dimensions resembling those of muscle fibers, which in vivo could be an advantage, simulating the implantation of fibers. During degradation, PLGA releases acidic monomers. These could be locally harmful, which calls for minimizing the amount of material.
In our in vitro cultured gels, the organization of myotubes was random. However, it has been shown that when a strain is applied to myoblasts cultured in fibrin gel, they align in the direction of the strain. 45 This has been further developed into bioengineered muscle constructs able to generate contractile force upon electrical stimulation.34,46 Similarly, implantation of fibrin containing material embedded with mesangioblasts underneath the skin upon the tibialis anterior muscle in mice created aligned myofibers in the construct. 47 This suggests that when integrated in muscle in vivo, the organization of muscle formed in implanted hydrogel scaffolds will be induced by the direction of tension. Similarly, the myoblasts in the PLGA-cell aggregates in vivo would be exposed to stretch. More generally, cells and scaffolds in vivo will be exposed to dynamic loading, and it has been reported that dynamic loading can influence both the formation of tissue and the degradation of scaffolds.48–50 The impact appears to be cell-type dependent, and besides the effect of stretch, little is known concerning myogenic cells. Regarding the impact on scaffolds, the degradation of polycaprolactone (PCL) and PLGA has been found to be faster when exposed to dynamic loading. The final rate of degradation in vivo, however, is determined by the combined influence of physical and biological conditions.
In this study, we show that cell state can be included as an element for the optimization of myoblast transplantation.
The degradability of the three injectable scaffold materials employed in this study can be controlled. In addition, the hydrogels can be formulated to obtain different stiffness. Cell concentration, degradation, and gel stiffness all affect cell state. This means that there are many options for directing the myoblast toward a specific cell state. As an element in the development of implantation procedures, some of the cell instructive effects of scaffolds can be studied in vitro. Performing immunochemistry on myoblasts cultured with scaffold materials in vitro, before using them for in vivo experiments, is an informative and simple first-step strategy to screen the effects on cell behavior, morphology, and distribution in the materials.
Footnotes
Acknowledgements
PLGA microparticles were a generous gift from Monica Gallego. Lene Feldskov and Tania Thedchanamoorthy kindly helped performing the SEM.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by Indo-Danish grant from the Government of India, Department of Biotechnology (BT/IN/Denmark/02/PDN/2011 DTD 26-05-11) and the Danish Council for Strategic Research (10-093757) and grants from the Novo Nordisk Foundation, and Odense University Hospital.