
Abstract
Interleukin-2 receptor alpha (IL2RA) defect (OMIM- # 606367) is an immune disease where affected patients are vulnerable to developing recurrent microbial infections in addition to lymphadenopathy and dermatological manifestations. This condition is known to be caused by pathogenic variants in the IL2RA gene, which are inherited in an autosomal recessive fashion. In this case report, we present a patient with IL2RA defect from Saudi Arabia who presented with chronic diarrhea, poor weight gain, mild villous atrophy, malnutrition, hepatomegaly, nonspecific inflammation, and an eczematous skin rash. His genetic analysis revealed a novel, homozygous, and likely pathogenic variant, that is, c.504 C>A (Cys168Ter), located in the exon 4of the IL2RA gene, which was inherited from his parents in an autosomal recessive mode of inheritance. This variant produces a 272-amino-acid shorter IL2RA protein chain, which most likely becomes degraded in the cytosol. Thus, we assume that the c.504 C>A is a null allele that abolishes the synthesis of IL2RA, malforms the IL-2 receptor complex, and eventually causes immunodeficiency manifestations. To our knowledge, this is the first time a person with IL2RA defect has shown signs of granulomatous hepatitis on a liver biopsy.
Introduction
Interleukin receptor 2 alpha (IL2RA) defect (OMIM- # 606367) is an autosomal recessive disorder of immune system dysregulation caused by homozygous or compound heterozygous mutations in the IL2RA gene, located on chromosome 10p15. Hyperproliferation of CD8+ T cells occurs owing to defects in T-cell regulation, weak antibody response, and increased production of cytokines, as demonstrated by immunology investigations. 1 This condition usually presents in infancy with recurrent infections, lymphadenopathy, autoimmune enteropathy, and eczematous dermatitis. 1 Here we describe a case with an IL2RA mutation that had elevated liver enzymes with a liver biopsy showing granulomatous hepatitis.
Case report
A 12-year-old Saudi Arabian boy of consanguineous parents first presented in 2009 at the age of 7 months with chronic diarrhea, poor weight gain, malnutrition, and an eczematous skin rash. Anti-tissue transglutaminase IgA was slightly elevated [39.3 units, upper limit of normal (ULN) = 0–20] and mucosal biopsy of the duodenum showed mild villous atrophy and nonspecific inflammation [Figure 1(c)]. The patient failed to gain weight normally following a trial of a gluten-free diet, despite partial improvement of diarrhea on lactose-free and amino acid formulas. Skin manifestations were progressive, involving the whole skin surface with areas of localized dermatitis and scaling [Figure 1(a) and (b)]. Skin biopsy revealed mild hyperkeratosis with foci of lymphocytic exocytosis and interstitial chronic inflammatory infiltrate, mainly consisting of lymphocytes, with perivascular and peri-appendiceal aggregation seen mainly in the superficial and deep layers of the dermis [Figure 1(d)]. There was one episode of urinary tract infection with Klebsiella pneumoniae organism, which was treated with antibiotics. Renal ultrasonography showed bilateral miniscule renal stones that were detected on serial imaging studies.
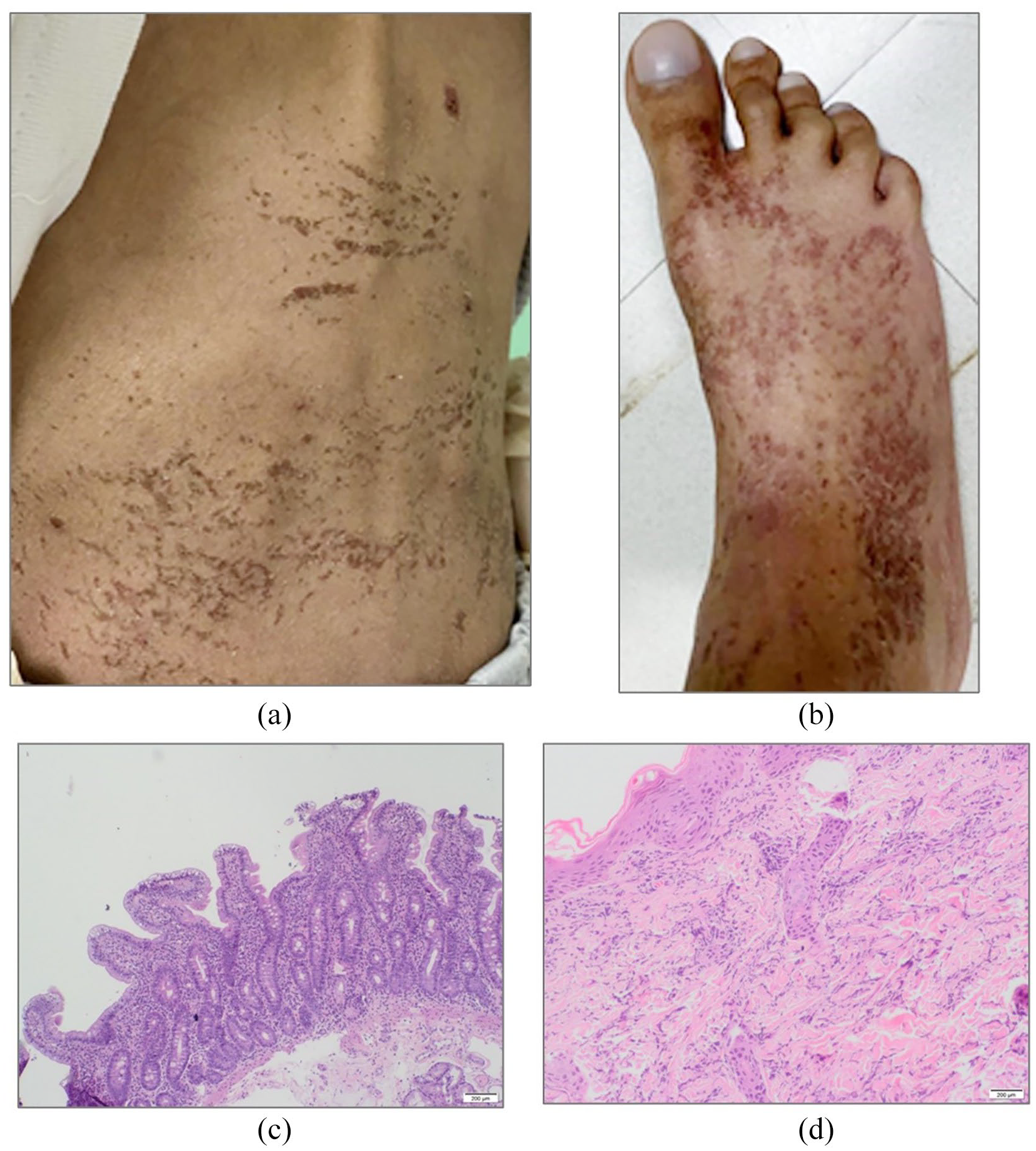
(a) and (b) Area of localized scaly dermatitis and clubbing. (c) Histological findings in the duodenum. Low power view revealing moderate villous shortening and intraepithelial lymphocytosis. (Hematoxylin & Eosin stain, 5×). (d) Dermatitis. Skin biopsy showed superficial and deep moderate mononuclear infiltrate (Hematoxylin & Eosin stain, 10×).
At the age of 2 years, hepatomegaly with a moderate increase in liver enzymes was found (γ-glutamyl transferase = 211 U/l, aspartate amino transferase = 132 U/l, alanine amino transferase = 226 U/l, alkaline phosphatase = 382 U/l, bilirubin = 9 umol/l). Percutaneous liver biopsy was performed, which showed expansion of the portal tracts and distension with chronic inflammatory cells, mainly lymphocytes and occasionally eosinophils, along with an ill-defined granuloma surrounded by dense inflammatory cells (Figure 2). Screening for autoimmune markers, including anti-smooth muscle antibody (SMA), anti-nuclear antibody (ANA), and anti-liver kidney microsomal antibody-1 (LKM-1), was negative as were the immunoglobulin (Ig) levels [IgG 19.5 g/l (5.4–16.1 g/l), IgA 3.87 g/l (0.8–2.8 g/l), IgM 1.35 g/l (0.5–1.9), and IgE 77 kU/l (0–195 kU/l)]. Liver enzymes declined with time, although liver enlargement continued.

Granulomatous hepatitis. Some of the portal tracts contain ill-defined noncaseating granulomas formed of epithelioid histiocytes that are surrounded by dense mononuclear infiltrate (Hematoxylin & Eosin stain, 20×).
At 4 years of age, following adenotonsillectomy, the boy developed mild splenomegaly and generalized lymphadenopathy of the cervical, axillary, and inguinal lymph nodes, accompanied by digital clubbing. Test results for tuberculosis (tuberculin skin test and chest X-ray), sarcoidosis, and parasitic infections were negative.
At 9 years of age, owing to persistent diarrhea, failure to grow, and a high fecal calprotectin level (6200 mg/kg), upper gastrointestinal endoscopy and colonoscopy were performed. Pancolitis was found during colonoscopy and normal upper gastrointestinal endoscopy. Chronic active inflammation was reported in the biopsy specimens, taken from the esophagus, stomach, duodenum, and colonic mucosa, with marked lymphocytic infiltration. In consideration of a possible diagnosis of IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, a screening for possible endocrinopathies was performed, but the thyroid function tests with thyroid antibodies were negative, and there were normal fasting blood sugar and hemoglobin A1c levels. There was no evidence of autoimmune neutropenia, anemia, or thrombocytopenia.
The patient continued to have alternating diarrhea and normal bowel motions. Anthropometric measures for his age were always under the third percentile for weight, height, and body mass index. Unfortunately, the patient passed away aged 10 years after developing septic shock.
Written informed consent was obtained from the parents for genetic testing and for reporting the case. Under HIPAA (Health Insurance Portability and Accountability Act), ethical approval is not needed for case reports.
Family pedigree and molecular studies
Figure 3 shows the family pedigree of the patient being studied. The whole exome sequencing of the proband was performed on the Illumina, Hiseq2000 sequencing platform at 30× coverage. The full details of DNA purification, library preparation, sequencing, data filtration, and variation identification strategies we adopted are outlined in our recent publications.2–4 From the exome data of the proband, we observed a novel, homozygous, and likely pathogenic c.504 C>A variant located in the exon 4 region of the IL2RA gene, resulting in the substitution of a cysteine codon at amino acid position 168 with a termination codon in the IL2RA protein. Sanger sequencing of the proband and of both parents has confirmed the autosomal recessive mode of inheritance of this variant. This variant is absent from the GnomAD, 1000 Genomes Project, Saudi Human Genome Program, and DALIA databases. The functional characterization of the IL2RA variant was performed by various systems biology methods. First, the 3D model of IL2RA variant protein was constructed using the ab-initio method as described. 5 Then, RNAfold webserver was used to estimate the effect of the IL2RA, c.504 C>A variant on the corresponding RNA structure.6,7 Findings from mRNA secondary structure of IL2RA predictions have indicated no significant changes in the thermodynamic stability of the mRNA folding pattern due to c.504 C>A variant (Figure 4). Finally, the NCBI conserved domain database was used to map the IL2RA, c.504 C>A variant on the functional domains of the protein molecule. 8 From the results of functional domain analysis, we identified that the c.504 C>A variant falls into a canonical sushi-like functional domain (complement control protein, CCP), spanning between the 125th and 184th amino acids, and it most likely produces a premature protein (104 amino acids) that is shorter than the native IL2RA protein molecule (272 amino acids).

A multigeneration family pedigree showing the autosomal recessive mode of inheritance of IL2RA defect in the patient.

Bioinformatic analysis of the human IL2RA. Images (a) and (b) show the RNA secondary structure predictions for wild-type and mutant mRNAs (c.504 C>A) in the form of a mountain plot (MP) representation of minimum free energy (MFE), thermodynamic ensemble (pf), and the centroid structure predictions, respectively. The color gradient in the scale of 0 to 2 represents the MFE calculations of nucleotide base pairing. The MP representation shows the secondary structures in a height versus position in which the helices are represented in slopes, loops in plateaus, and hairpin loops in the peaks. The bottom graph represents the entropy of predicted RNA structure in which higher entropy means the RNA structure has lower stability. Image (c) shows that the c.504 C>A variant falls in the complement control protein (CCP domain). Also, the location of Cys168Ter is shown in the 3D model.
Discussion
Human IL-2 receptor α chain deficiency is an immune dysregulation disorder involving dysfunctional interleukin-2 (IL-2) receptors and occurs owing to mutations in the α chain (CD25). 9 The IL-2 binds to IL-2R (IL-2 receptor), which is basically composed of three subunits, namely, α (CD25), β (CD122), and γ (CD132). The β and γ chains are predominantly located on T lymphocytes, whereas α chain is mainly expressed on activated mature T lymphocytes during early thymocyte development stages. 10 All three subunits (α, β, and γ chains) are required for a functional IL-2 receptor. 1 The CD25 is constantly expressed by CD4+CD25+FOXP3+regulatory T cells, which allows them to be the initial responders to IL-2 in an immune response. 11 It also enhances IL-2 signaling by stimulating the FOXP3 transcription. 12 Single nucleotide polymorphism in IL-2A has been associated with multiple autoimmune disorders, such as type 1 diabetes. 13 Upon antigenic stimulation, CD25 is essential for the expansion of effector T cells in response to IL-2. 1 Goudy et al. 1 have suggested that IL-2 consumption by FOXP3+ Tregs is an important step in maintaining immune system homeostasis. The absence of CD25 on the surface of FOXP3+ Tregs, however, helps CD8+ T cells grow and causes autoimmune reactions in CD25-deficient patients.
Tregs is divided into thymus-derived (tTregs) and induced Tregs (iTregs). The tTregs develop in the thymus, but iTregs differentiate from naïve T-cell precursors peripherally. The generation in the thymus depends on the presentation of thymic self-peptide/MHC activation, which activates the precursors to differentiate into functional antigen-specific suppressive regulatory T cells. This process needs the activation of a number of Treg-specific enhancers, CD25, Ctla4, Eos, and Helios, which become gradually activated at the precursor stage before FoxP3 expression. The differentiation of peripheral iTregs, however, most likely occurs in response to nonself antigens, for example, commensal microbiota, food, and allergens, and depends on the signaling strength and abundance of the antigen, giving harmony of immune tolerance. 10 Treg generation is largely dependent on the presence of IL-2, required for homeostatic maintenance for their thymic development. 11 IL-2 is produced mainly by CD4+ T cells and cannot be synthesized by Treg cells. IL-2 has also been shown to be crucial for the development and maintenance of Treg cells. IL-2 also directly affects suppressive function of Tregs. 12
One study has found that mice with IL-2 or IL-2 receptor deficiency display an enlargement of peripheral lymphoid organs, including lymph nodes and spleen, impaired activation-induced cell death, and autoimmune disorders, which have been attributed to a decreased generation of Tregs. 13
IL2RA, along with two other receptors, IL2RB and IL2RG, forms a complex cell–surface receptor for the pleiotropic cytokine IL-2. When IL-2 binds to the receptor, the Jak3 (tyrosine kinase) and STAT5 (signal transducer) 7 proteins are activated. 14 The IL2RA structure is characterized by D1 and D2 domains linked by linker peptides (T65 to H103), and the second domain has a 54-amino-acid-residue-long peptide, which connects it to the cell membrane (G165 to E217). It is thought that deleterious genetic mutations can eliminate or considerably reduce gene function. This could be true in the present case, since the c.504 C>A nonsense mutation introduces a protein termination codon at the 168th codon and truncates the IL2RA 104 amino acids shorter than the original IL2RA. The mRNAs with protein truncating code (PTC) often undergo nonsense-mediated decay 15 or generate premature proteins, which will eventually undergo a ubiquitin-mediated protein degradation pathway. 16 Thus, we assume that the c.504 C>A is a null allele that abolishes the synthesis of IL2RA and malforms the IL2 receptor complex. Because IL-2R is not available on the surface of resting T and B cells, but expressed by aberrant T cells in patients with leukemia, autoimmune conditions, or organ transplant rejection, it is considered to be an ideal target for therapeutic modulation.17,18 In recent years, some molecular studies have shown that the effect of nonsense mutations can be mitigated in the LDLR gene by using translation inhibitory molecules, such as aminoglycoside antibiotics, which were shown to be effective in partial restoration of full-size proteins encoded by genes with premature stop mutations. 15 A recent study has shown that pathogenic mutations identified in the IL2RA gene (CD25) in siblings presenting monogenic autoimmunity can be corrected in their affected primary T cells using the CRISPR-Cas9 genome-targeting system. 19 Whether a similar strategy could be useful to treat patients harboring IL2RA mutations, however, has not yet been established.
A few case reports have been published regarding IL2RA defect patients.1,9,20,21 Sharfe et al. describe a novel case of an IL2RA-deficient patient who suffered from chronic infections and severe autoimmunity, resembling immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome, caused by mutations in the FOXP3 gene. This IPEX-like patient possessed a translation frameshift mutation in the IL2RA gene, abating its expression. Similarly, a second report 21 describes a patient with a different frame shift mutation in the IL2RA gene, leading to a CD25 null phenotype with comparable clinical manifestations. A third report by Goudy et al. involved a patient with the development of progressive manifestations of both autoimmunity, such as enteropathy, erythroderma, and severe alopecia, and immunodeficiency with chronic cytomegalovirus (CMV) infection. A case study reported by Bezrodink et al. involved a girl with CD25 mutation-associated chronic inflammatory lung disease (follicular bronchiolitis with lymphocytic hyperplasia). 20
To our knowledge, the present case report is the first instance in the medical literature where an immunodeficiency-41 patient has shown positive evidence of developing granulomatous hepatitis on liver biopsy. The patient developed hepatitis and had extensive examination that excluded autoimmune and infectious causes, including CMV.
A liver biopsy revealed portal tract expansion and distension with chronic inflammatory cells, primarily lymphocytes and occasionally eosinophils, as well as an ill-defined granuloma surrounded by dense inflammatory cells. The examination rules out other causes of granulomatous liver disease, including sarcoidosis, tuberculosis, Crohn’s disease, and fungal infections. Liver enzymes, however, improved gradually with no specific treatment, although there was ongoing hepatomegaly. No further elevation of liver enzymes was found. A recent study has provided preclinical evidence that CRISPR-Cas9-based genome targeting has the potential to correct the pathogenic IL2RA variant in primary T cells from a monogenic autoimmune disease family. 19 Thus, we presume that the IL2RA variant identified in our patient can also be targeted by nonviral genome-targeting methods in primary T cells in vitro or in vivo in order to pave the way for future personalized medicine in patients suffering with the IL2RA defect.