
Abstract
Background and aims:
Rotator cuff tendinopathy is common and is related to pain and dysfunction. However, the pathological mechanism of rotator cuff injury and shoulder pain is unclear. Objective: to investigate the pathological mechanism of rotator cuff injury and shoulder pain, and screen out the marker proteins related to rotator cuff injury by proteomics.
Methods:
Subacromial synovium specimens were collected from patients undergoing shoulder arthroscopic surgery. The experimental group were patients with rotator cuff repair surgery, and the control group were patients with habitual dislocation of the shoulder joint. Pathological examination was performed, and then followed by non-labeled quantitative proteomic detection. Finally, from analysis of the biological information of the samples, specific proteins related to rotator cuff injury and shoulder pain were deduced by functional analysis of differential proteins.
Results:
All the patients in experimental groups were representative. A large number of adipocytes and inflammatory cells were found in the pathological sections of the experimental group; the proteomics analysis screen identified 80 proteins with significant differences, and the analysis of protein function revealed that S100A11 (p = 0.011), PLIN4 (p = 0.017), HYOU1 (p = 0.002) and CLIC1 (p = 0.007) were closely related to oxidative stress and chronic inflammation.
Conclusion:
Rotator cuff injury is closely related to oxidative stress and chronic inflammatory response, and the results suggest that the expression of S100A11, PLIN4, HYOU1 and CLIC1 in the synovium of rotator cuff injury provides a new marker for the study of its pathological mechanism.
Introduction
Rotator cuff injury is a common cause of chronic shoulder pain; Milgrom et al. 1 conducted an epidemiological survey on adults aged 30 to 99 years with ultrasound, and found that rotator cuff tear significantly increased in patients over the age of 50, with over 50% of rotator cuff tears at the age of 70 and up to 80% at the age of 80. The above data indicate that rotator cuff injury increases with age. The clinical manifestations of rotator cuff injury include shoulder pain, dysfunction and muscle atrophy, which seriously affect limb function and quality of life. Early and effective diagnosis and treatment are of great significance for relieving shoulder pain and recovering shoulder function, preventing and reducing disability.
The etiology of chronic rotator cuff tear is multifactorial, with extrinsic and intrinsic factors.2–4 However, the above theories only explained rotator cuff injury from the aspects of pathological pathology and etiology, instead of molecular mechanism, and the prevention and treatment of rotator cuff injury is still limited. Therefore, understanding the molecular mechanism of rotator cuff injury is a scientific problem that needs to be solved, so as to prevent or control the progress of the disease, which has important clinical value.
Rotator cuff injury is not only a tendon problem, it is often accompanied by progressive and irreversible fat infiltration and involves adjacent muscles. 5 The pathological description of this intramuscular fat infiltration was first reported by Goutallier et al. 6 The muscle microstructures correspondingly change, such as myofibril lysis and degeneration, and are finally replaced by adipose tissue; the increased fat will accumulate outside and inside the muscle bundle. 7 Therefore, muscle atrophy and fat infiltration become two major complications of rotator cuff tendinopathy, especially chronic rotator cuff injury or huge rotator cuff tear, and seriously affect the postoperative recovery, resulting in rotator cuff re-tear.
Free fatty acids (FFA) are metabolites of fat, which can lead to increased production of highly reactive molecular oxygen cluster and nitrogen cluster, triggering oxidative stress reaction, and imbalance of redox reaction can cause tissue damage. These active molecules can directly oxidize and damage DNA, proteins and lipids, and also serve as functional molecular signals to activate a variety of stress-sensitive signaling pathways in cells.8–10 High concentrations of FFA can increase the production of reactive oxygen species (ROS) and activate stress-sensitive signaling pathways. 11 Studies have shown that circulating FFA is related to the occurrence and development of diseases such as metabolic syndrome, atherosclerosis, acute coronary syndrome and heart failure.12–14
Experiments have confirmed that reactive oxygen radicals can induce cell apoptosis under certain conditions. 15 In addition, other studies have found that long-term local high temperature and repeated ischemia/reperfusion in athletes’ tendons can produce an amount of reactive oxygen species, resulting in chronic tendinopathy. 16 The pathology of supraspinatus tendon in mice has been confirmed that the deficiency of superoxide dismutase can accelerate the degeneration of the rotator cuff, and the addition of oxidants such as H2O2 can also induce the programmed death of tendon fibroblasts.17,18 These studies suggest that rotator cuff injury may be closely related to oxidative stress-induced apoptosis. In this study, the subacromion synovium was analyzed by proteomics to screen out the marker proteins related to oxygen radicals for rotator cuff injury and shoulder pain.
Material and methods
Ethics approval
The study adhered to the Declaration of Helsinki and was approved by the National Regional Committee for Medical and Health Research Ethics, and registered with the Ethics Committee of Jinling Hospital (2019NZGKJ-006). Written informed consent was obtained from all participants prior to any study-related procedure.
Patients
Subacromial synovium was obtained from patients undergoing rotator cuff repair and labial lesion repair in Jinling Hospital from October 2019 to December 2019. A total of 30 patients were included in the study, and all met the criteria. Part of typical results are shown below. The experimental group included six patients with rotator cuff injury (supraspinatus) (three women and three men, mean age 61.9 years), and the control group included three patients with labial lesion of shoulder (two men and one woman, mean age 21.2 years). Inclusion criteria: the magnetic resonance imaging (MRI) evaluation of the rotator cuff injury in the experimental group was graded 3 and combined with persistent non-relief shoulder pain, excluding other basic diseases and history of shoulder trauma; patients in the control group had habitual dislocation of the shoulder joint due to labial lesion, excluding patients with neuropathy and congenital articular sac relaxation, and patients without basic diseases and rotator cuff injury.
Clinicopathologic examination
During shoulder arthroscopic surgery, the synovial tissue in the joint needs to be cleaned to expose the visual field. It was not difficult to find that the subacromial synovial tissue in the patients with rotator cuff injury presented hyperemia with extensive hyperplasia, while the patients with labial lesion showed no obvious synovial hyperemia (Figure 1). The subacromial synovium specimen was collected during operation. Hematoxylin-eosin (HE) staining was performed to observe the morphological changes of synovial cells, and proteomic analysis was also conducted.

(A) Preoperative oblique coronal and oblique sagittal images of rotator cuff injury in 61-year-old man; (B) preoperative oblique coronal and oblique sagittal images of labial lesion in 22-year-old man; magnetic resonance imaging showed joint effusion, subchondral signal changes, synovitis and supraspinatus fat infiltration in patients with rotator cuff injury. Arthroscopically, supraspinatus injury with extensive synovial hyperplasia and hyperemia can be seen in patients with rotator cuff injury (A versus B).
Experimental methods
Project process
The principle of the Maxquant algorithm adopted in this project, non-labeled quantitative method, is based on MS1, to calculate the integral of each peptide signal on liquid chromatography–mass spectrometry (LC-MS) chromatography and analyze the quantitative and significant differences of proteins in multiple groups of samples. The process of this project is divided into two parts: pre-experiment and formal experiment. The pre-experiment includes protein extraction, protein quantification, SDS-PAGE, protein digestion, LC-MS/MS analysis, database query, quality control and issuing a pre-experiment report. The formal experiment was performed on the basis of pre-experiments. The qualified samples in the preliminary experiment were tested by high resolution mass spectrometry to obtain the original data of mass spectrum.
Data analysis process
In the process of data analysis for label-free projects, database query and result evaluation of the original mass spectrum are usually performed first, and subsequent information analysis is performed on the project data that is qualified for quality control, including trusted peptides and protein identification and screening, quantitative analysis of protein and screening differentially expressed proteins, and then enriched annotations by gene ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, and clustering the functions of differential proteins to screen related gene proteins.
Instrument and analysis software
Experimental instruments
Easy nLC chromatography system (Thermo Fisher Scientific), Agilent 1260 infinity II HPLC system, low-temperature high-speed centrifuge (Eppendorf 5430R), electrophoresis system (BIO-RAD), ultrasonic breaker (Ningbo Scientz JY96-IIN), Votex oscillator GENIE Votex-2), Nano Drop (Thermo Fisher scientific ND2000), Q Exactive Plus Mass Spectrometer (Thermo Fisher Scientific), Multiskcan FC Microplate Reader (Thermo Fisher Scientific), Vacuum Centrifuge Concentrator (Huamei LNG-T98), MP Fastprep-24 homogenizer (MP Fastprep-24 5G), constant temperature incubator (Shanghai Jinghong GNP-9080), electronic balance (OHAUS AX324Z), compact constant temperature mixer (Da long HCM-100 pro).
Analyzing software
Perseus 1.3 (Max Planck Institute of Biochemistry, Martinsried, Germany), MaxQuant 1.5.5.1, R version 3.3.1.
Reagents and consumables
Urea (BIO-RAD 161-0731), SDS (Biotech SB0485-500g), Tris (Biotech T0826-500g), iodoacetamide (IAA, Sigma I1149-5G), C18 EmporeTM Solid Phase Extraction Disk (Sigma 66883-U, BCA) Quantitation Kit (Beyotime Biotechnology P0012), NH4HCO3 (Sigma A6141-25G), formic acid (Thermo Fisher Scientific, A117), acetonitrile (Merck 1000304008), C18 Cartridge (Waters WAT023590), trifluoroacetic acid (TFA, Sigma, T6508), SDS-PAGE protein loading buffer (Beyotime Biotechnology, P0015F), Lysing Matrix A (MP, 6910-100-99219), dithiothreitol (DTT; Sigma, 43819-5G), HCl (Sinopec, 10011018), BSA (Biotech, A0332), trypsin (Promega, V5117), 30kD ultrafiltration centrifuge tube (Sartorius, VN01H22), 0.22 μm ultrafiltration centrifuge tube (Corning Spin-X, 8160), Multiple Affinity Removal LC Column-Human 14/Mouse 3 (Agilent); C18 analysis column: Thermo Fisher Scientific, Acclaim PepMap RSLC 50 µm × 15 cm, nano viper, P/N164943; SDT lysate: 4% SDS, 100 mM Tris-HCl, pH 7.6; UA buffer: 8 M urea, 150 mM Tris-HCl, pH 8.5; EASY nLC mobile phase A: 0.1% FA; EASY nLC mobile phase B: 0.1% FA, 80% CAN.
Protein extraction
Homogenization + SDT lysis method: take the synovial tissue and add an appropriate amount of SDT lysate, transfer it to Lysing Matrix A tube, and apply MP homogenizer to homogenize and break (24 × 2, 6.0 M/S, 60 s, twice). After sonication, a boiling water bath was used for 10 min. Centrifuge at 14,000 g for 15 min, take the supernatant, filter through a 0.22 μm centrifuge tube, and collect the solution. BCA method was used for protein quantification. Samples were aliquoted and stored at −80°C.
Homogenate +SDT lysis method: the synovial tissue was added with an appropriate amount of SDT lysis fluid and transferred to a Lysing Matrix A tube. The homogenizer was used for homogenate crushing (24 × 2, 6.0 M/S, 60 s, twice). After sonication, boiling water bath for 10 min. After centrifugation at 14,000 g for 15 min, the supernatant was taken and filtered with a 0.22 centrifuge tube to collect the liquid. Protein quantification was carried out by BCA method, samples were packaged and stored at −80°C.
SDS-PAGE electrophoresis
Twenty micrograms of protein was added to each sample and 6× loading buffer was added. The samples were subjected to 12% SDS-PAGE electrophoresis (constant pressure 250 V, 40 min) in a boiling water bath for 5 min, and Coomassie-stained blue.
FASP enzymatic hydrolysis
Take 80 μg protein solution for each sample, add DTT to the final concentration of 100 mM, bath in boiling water for 5 min, and cool to room temperature. Add 200 μl of UA buffer and mix, transfer to a 30 kD ultrafiltration centrifuge tube, centrifuge at 12,500 g for 15 min, and discard the filtrate (repeat this step once). Add 100 μl of IAA buffer (100 mM IAA in UA), oscillate at 600 rev/min for 1 min, and react at room temperature for 30 min in the dark, and centrifuge at 12,500 g for 15 min. Add 100 μl of 40 mM NH4HCO3 solution and centrifuge at 12,500 g for 15 min. Repeat this step twice. Add 40 μl trypsin buffer (4 μg trypsin in 40 μl 40 mM NH4HCO3 solution), shake at 600 rev/min for 1 min, and leave at 37°C for 16–18 h. Replace the new collection tube, centrifuge at 12,500 g for 15 min; add 20 μl of 40 mM NH4HCO3 solution, centrifuge at 12,500 g for 15 min, and collect the filtrate. C18 Cartridge was used to desalinate the peptides. After the peptides were lyophilized, 40 μl of 0.1% formic acid solution was added and the peptides were quantified (OD280).
Mass spectrographic analysis
Each sample was separated using a nanoliter flow rate Easy nLC system. Buffer A is a 0.1% formic acid aqueous solution, and B is a 0.1% formic acid acetonitrile aqueous solution (80% acetonitrile). The column was equilibrated with 100% liquid A. The sample was separated from the autosampler onto an analytical column (Thermo Fisher Scientific, Acclaim PepMap RSLC 50 µm × 15 cm, nano viper, P/N164943), and the flow rate was 300 nl/min.
The samples were separated by chromatography and analyzed by Q Exactive Plus mass spectrometer. The analysis time was 60 min, the detection method was positive ion, the scanning range of the parent ion was 350–1800 m/z, the resolution of the mass spectrometry was 70,000, the AGC target was 3e6, and the maximum IT was 50 ms. The mass-to-charge ratio of peptides and fragments was collected according to the following methods: 10 fragment spectra (MS2 scan) were collected after full scan, MS2 Activation Type was HCD, isolation window was 2 m/z, secondary mass spectrometry resolution was 17,500, microscans was 1, secondary maximum IT was 45 ms, and normalized collision energy was 27 eV.
Data analysis
Mass spectrum raw file processing
Maxquant is a leading qualitative algorithm for proteomics and has gradually become one of the standard solutions in recent years. The use of the label-free algorithm in Maxquant for non-labeled quantitative calculation of proteomics data has also become one of the important applications of this algorithm. 19
Protein quantitative analysis parameters
In this project, NCBInr, a comprehensive protein database, was selected to carry out qualitative analysis on mass spectrometry data. MaxQuant software (version 1.5.5.1) was used for database search, and the LFQ (label free quantitation) algorithm was used for quantitative analysis.
Method of bioinformatics analysis
GO functional annotation
The GO annotation process of target protein sets by Blast2 GO can be roughly summarized into four steps: sequence alignment (Blast), GO item extraction (Mapping), GO annotation and annotation augmentation. First, the sequence alignment tool NCBI BLAST+ (ncbi-blast-2.3.0+) was used on the Linux server to compare the target protein set with the appropriate protein sequence database, and the first 10 alignment sequences satisfying e-value ⩽1e−3 were retained for subsequent analysis. Second, Blast2 GO Command Line was used to extract the GO items associated with the bit-score sequence with the highest bit-score in the target protein set and Blast retention results (download address: www.geneontology.org). In the annotation process, Blast2GO command line annotates the GO items extracted in the Mapping process to the target protein sequence by comprehensively considering the similarity of the target protein sequence and the alignment sequence, the reliability of the GO item source, and the structure of the GO directed acyclic graph. After completing the annotation, to further improve annotation efficiency, we can search the conservative motif matching the target protein in EBI database through InterProScan3, and annotate the motif related functional information to the target protein sequence. It also runs ANNEX to further supplement the annotation information and to establish links between the different GO categories to improve the accuracy of the annotation.
KEGG path annotation
In the KEGG database, KO (KEGG Orthology) is a classification system of genes and their products. Lineal homologous genes with similar functions on the same pathway and their products are grouped together and assigned the same KO (or K) label. KOALA (KEGG Orthology And Links Annotation) software was used to annotate the target protein set. By comparing the KEGG GENES database, the target protein sequence was KO classified, and the pathway information involved in the target protein sequence was automatically obtained based on KO classification.
GO annotation and KEGG annotation enrichment analysis
In the enrichment analysis of GO annotation or KEGG pathway annotation on the target protein set, Fisher’s exact test was used to compare the distribution of each GO classification or KEGG pathway in the target protein set and the total protein set, so as to evaluate the significance level of protein enrichment of a GO term or KEGG pathway.
Protein cluster analysis
In the clustering analysis, first, the quantitative information of the target protein set is normalized. Second, two dimensions (distance algorithm: Euclid, connection method: Average linkage) were classified by matplotib software, and a hierarchical cluster heat map was generated.
Results
Pathological changes of subacromion synovium
All the specimens were from the subacromial synovium. Compared with the patients with labial lesion, the patients with rotator cuff injury showed obvious hyperplasia, hyperemia and inflammatory changes in the synovium under the microscope. Pathological examination revealed obvious cell proliferation, disordered arrangement of fiber cells, accompanied by a large number of new blood vessels, and the presence of chronic inflammatory cells and fat cells (Figure 2).

Synovial histopathological changes in patients with labial lesion (A) and rotator cuff injury (B). The subacromial synovium and morphological structure in the control group were normal, and the fibroblasts were arranged orderly. However, the synovial structure of the experimental group was changed, showing a large number of collagen fibers, fat cells and collagen deposition with disordered arrangement; meanwhile, amounts of inflammatory cells, such as macrophages, lymphocytes and neutrophils, could be seen in the pathological sections (hematoxylin-eosin staining, 40×).
Results of label-free experiment
Quality of the specimen
The quality of samples collected meets the test requirements, the electrophoresis bands are clear, and the total amount meets the requirements of two or more experiments. The quantitative results of protein extraction are shown in Table 1, and the SDS-PAGE results are shown in Figure 3.
Results of protein extraction and quantification.

A represents experimental group, B represents control group.

SDS-PAGE test results.
Protein clustering
The synovial peptide data of shoulder joint were analyzed by cluster analysis, and the data were grouped and classified based on similarity. The results of clustering and grouping show that the similarity of data patterns within the group is higher, while the similarity of data patterns between the groups is lower. The clustering results of the samples can test the rationality of the selected target proteins, that is, whether the changes in the expression of target proteins represent the significant influence of biological treatment on the samples; it can help us distinguish protein subsets with different expression patterns from protein sets. Proteins with similar expression patterns may have similar functions or participate in the same biological pathway, or be in adjacent regulatory positions in the pathway (Figure 4).

Cluster analysis results (A versus B).
Mass spectrometry identification and quantitative result evaluation
The mass spectrometry data in this experiment were collected from a Q Exactive Plus high-resolution mass spectrometer, which can obtain high-quality MS1 and MS2 spectra. Then, Andromeda was used to analyze the MS spectrum data, and finally the score of each MS2 spectrum was obtained. The Andromeda score of MS2 is ideal, and about 70% of the peptide score above 60 points. In the qualitative analysis of each set of labeled free data, peptide FDR ⩽0.01 and protein FDR ⩽0.01 are taken as the screening criteria to obtain the distribution of excellent peptide score, which further indicates the high quality of MS experimental data.
The following data are shown in Figures 5–10: distribution of peptide ion score (Figure 5), relative molecular mass distribution of proteins (Figure 6), peptide sequence length distribution (Figure 7), distribution of the number of identified peptides (Figure 8), distribution of protein abundance ratio (Figure 9) and fusiform diagram (Figure 10).

Peptide ion score distribution chart. The abscissa is the peptide score; the ordinate number of peptides corresponding to the column in the graph; the secondary ordinate corresponds to the cumulative curve in the figure, which represents the cumulative percentage of peptides that are not higher than the corresponding ion score.

Distribution diagram of the molecular weight of the identified protein; the abscissa is the relative molecular mass of the identified protein; the histogram of the number of proteins corresponding to the main ordinate indicates the number of proteins with the corresponding relative molecular mass; the secondary ordinate corresponds to the cumulative curve in the figure, which represents the cumulative percentage of proteins with no higher than the corresponding relative molecular mass.

Distribution of peptide sequence length. The abscissa is the number of amino acids of the identified peptide sequence. The ordinate is the number (percentage) of identified peptides.

Distribution diagram of the number of identified peptides.

Distribution of protein abundance ratio; the abscissa is the difference multiple (logarithmic transformation with base 2); the ordinate is the number of identified proteins.

Fusiform diagram; two factors, fold change of protein expression and label free quantitation (LFQ) intensity, were used to make a shuttle pattern. The p values obtained from each data point according to the t test algorithm were identified by different colors: blue represents p > 0.05, red represents 0.01 < p < 0.05, yellow represents 0.001 < p < 0.01, and green represents p < 0.001. The abscissa is the difference multiple (logarithmic transformation with base 2); the ordinate is the sum of the peptide intensity values (logarithmic transformation with base 10).
Qualitative results of proteins
In the analysis of the significant difference in quantitative results, first of all, screen at least half of the repeated experimental data in the sample group for non-null values for statistical analysis (i.e. more than three non-null values in group A and more than two non-null values in group B), which conform to the expression differences ratio greater than two times (up and down) and p < 0.05 (t test) screening standard protein as differentially expressed proteins. A total of 80 proteins with significant differences were detected in synovial specimens, including 54 up-regulated proteins and 26 down-regulated proteins (Tables 2 and 3).
Statistics of protein identification results.

Database: name of database species selected for use; peptides: total number of peptides identified; protein groups: total number of proteins identified.
Differences in protein amounts between the experimental group and the control group.

GO functional annotation
The massive data generated by proteomics through gel electrophoresis, mass spectrometry, et cetera represent all the processes and changes in the organism. It is the main task of proteomic biological information to find the changes of organisms and the source and mechanism of these changes from these huge and complex experimental data. GO is a standardized classification system, providing a set of dynamic updates of standardized vocabulary, and from three aspects describes the biology of gene and gene product attributes: biological processes, molecular function and cellular components. The results of GO functional annotation of synovial samples showed that, compared with the control group, the differential proteins were mainly involved in cell metabolism, biological regulation, cell proliferation and biological adhesion, while their molecular functions were concentrated in antioxidant activity, molecular regulation, integration and transduction activities (Figure 11).

Level 2 statistics of gene ontology annotation results (A versus B).
GO enrichment analysis of differentially expressed proteins
Target proteins can be categorized by GO annotation in terms of biological processes involved, molecular functions, and cellular components. Although the proportion of each classification can reflect the degree of influence of biological treatment on each classification in the experimental design to a certain extent, it is not accurate to evaluate the significance of each classification based solely on this proportion, and the distribution of each classification in the overall protein set needs to be considered at the same time.
In this study, the GO annotation significant enrichment analysis was performed by using Fisher’s exact test to evaluate the significance level of GO term protein enrichment (Figure 12).
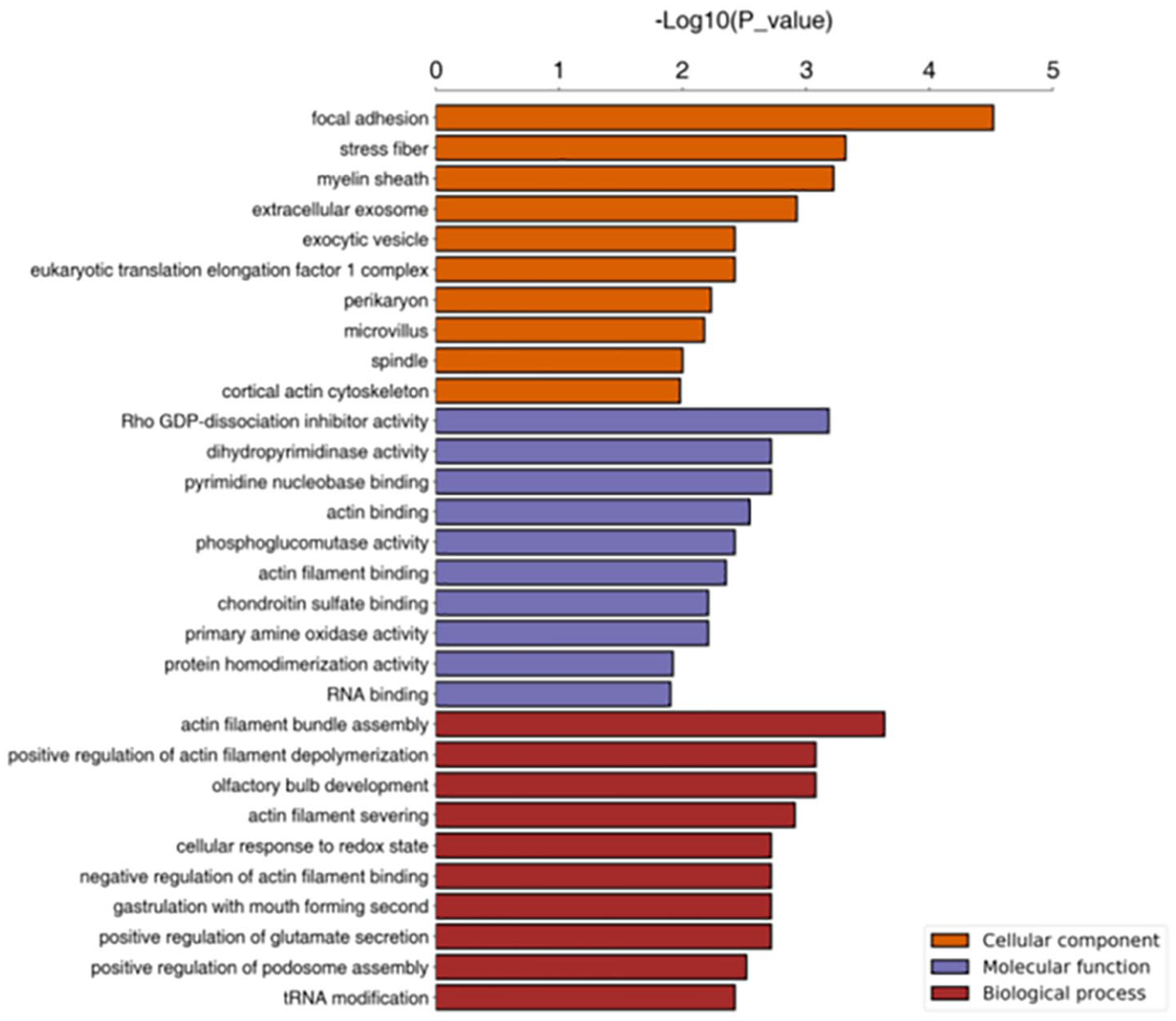
Significantly enriched gene ontology term statistics (A versus B) (top 10).
KEGG pathway annotation
Proteins in living organisms do not perform their functions independently; different proteins coordinate with each other to complete a series of biochemical reactions to perform their biological functions. Therefore, pathway analysis can provide a more systematic and comprehensive understanding of cell biological processes, traits or disease pathogenesis and drug action (Figure 13).

KEGG signaling pathway annotation (A versus B).
Enrichment analysis of differential protein KEGG pathway
The KEGG pathway enrichment analysis method is similar to GO enrichment analysis, that is, the KEGG pathway is taken as the unit and all qualitative proteins are taken as the background. Fisher’s exact test is used to analyze and calculate the significance level of protein enrichment of each pathway, so as to determine the metabolic and signal transduction pathways significantly affected (Figure 14).

KEGG pathway statistics with significant enrichment (A versus B) (top 10).
Results of proteomics analysis
Proteomics results suggest that there are 80 significantly different proteins in the experimental group and the control group. According to the GO molecular function, these proteins are involved in processes such as cell metabolism, biological regulation, cell proliferation and biological adhesion, and their molecular function is focused on the antioxidant activity, molecular regulation, integration and transduction activity. The purpose of this study is to investigate the pathological mechanism of rotator cuff injury and pain. At the same time, our research found that S100A11, PLIN4, HYOU1 and CLIC1 are significantly up-regulated in the rotator cuff injury synovium. These proteins are closely related to oxidative stress and chronic inflammation (Tables 4).
Results of proteomics analysis.

Discussion
The main results of this study showed that S100A11, PLIN4, HYOU1 and CLIC1 were significantly up-regulated in synovium of the rotator cuff injury. In addition, the synovial membrane presented hyperemic inflammatory changes under arthroscopy, and pathological sections also observed obvious inflammatory cells. It suggests that rotator cuff injury and shoulder pain are related to oxidative stress and chronic inflammation.
The upregulated expression of S100A11
S100A11 (also known as S100C or calgizzarin) is a member of the large calcium-binding S100 protein family, which is involved in specific biological processes such as endocytosis and extracellular secretion, enzyme activity regulation, cell growth, apoptosis and low-degree inflammation. 20 S100A11 has different expression levels in the nucleus, cytoplasm and peripheral tissues. Studies have shown that S100A11 can enhance the release of pro-inflammatory cytokines by peripheral blood monocytes and synovial fibroblasts.21–23 Compared with normal synovial tissue, S100A11 protein in the patients with rotator cuff injury was significantly up-regulated (p < 0.05), and the mass spectrometry data covered 82.9% of the predicted amino acid sequence. The results showed that rotator cuff injury was closely related to chronic inflammation and apoptosis.
HYOU1 and CLIC1 expression upregulated
The HYOU1 protein is called oxygen regulatory protein 150 (ORP150). Under hypoxia conditions, the endoplasmic reticulum can accumulate ORP150, to protect cells from hypoxia, while inhibiting ORP150 expression can accelerate cell apoptosis. 24 Moreover, HYOU1 protein has a fairly extensive target gene profile, including nearly 100 target genes related to hypoxia adaptation and inflammation development.25,26 The results showed that HYOU1 protein in the synovial tissue of patients with rotator cuff injury was significantly up-regulated (p < 0.01), and mass spectrometry data covered 36.2% of the predicted amino acid sequence. CLIC1, as a transmembrane protein, activation depends on the activation of NADPH oxidase produced by the oxidation of ROS. 27 Not only is ROS generation necessary, but also activation by oxidants and NADPH oxidase activities. 28 The HYOU1 protein was significantly up-regulated in the synovial tissue (p < 0.01), which means that there is an oxidative stress response in the damaged rotator cuff.
The upregulated expression of PLIN4
PLIN4 is involved in the formation of lipid droplets and plays an important role in regulating lipid metabolism, usually located at or near the myometrium. 29 Lipid metabolism is important for muscle contraction, and the number of lipid droplets (LDs) in tissue depends on lipid utilization and the activity of enzymes and co-activators required to synthesize and degrade LDs.30,31 The expression of PLIN4 is mostly limited to adipocytes, brain, skeletal muscle, et cetera, 32 and its expression in the synovium of rotator cuff injury is significantly up-regulated compared with normal (p < 0.05), suggesting that a large amount of lipid is produced in the synovium of shoulder joint in patients with rotator cuff injury, which is closely related to fat infiltration. Its expression in synovium of patients with rotator cuff injury was significantly up-regulated compared with the control group (p < 0.05), which suggests that PLIN4 is closely related to fat infiltration.
In this study, for the first time, we performed proteomic analysis of the subacromial synovium, combined with MRI and pathological studies, screened out proteins related to rotator cuff injury, and clarified the molecular pathological mechanism of rotator cuff injury. The current research on rotator cuff injuries focuses on the surgical repair and efficacy of rotator cuff injuries, and analysis of postoperative complications, but no etiology research has been performed on rotator cuff injuries. In recent years, with the development of muscle physiology, it has been found that rotator cuff injuries are usually accompanied by various muscle changes, including atrophy, fibrosis and fatty infiltration, which can have a significant impact on the mechanical and biological properties of tendons.33,34 The degree of fat infiltration is also closely related to postoperative rehabilitation and re-tear injury, and also has a significant impact on muscle atrophy and contractility.
Studies have shown that adipocytes can reduce the expression of contractile and structural proteins, promote muscle fiber atrophy and disrupt muscle regeneration. 35 Chemokines such as fatty acids, adipokines, cytokines and IL-1b58 produced by adipokines can promote inflammatory responses and induce oxidative stress, thereby reducing the viability of muscle fibers. 36 In addition, high free fatty acids lead to production of a large number of ROS and oxidative stress. 37 Hypoxia significantly up-regulates the expression of fatty acid synthase (FAS) gene, which is phosphorylated by Akt and activates hypoxia inducible factor 1 (HIF1), and significantly up-regulates the main transcription regulator of FAS gene, sterol regulatory element-binding protein-1. 38 HIF-1 is a nuclear protein with transcriptional activity and has a fairly broad target gene spectrum, including nearly 100 target genes related to hypoxia adaptation, inflammation development and tumor growth. When combined with target genes, the body produces a series of reactions through transcription and post-transcriptional regulation. 39 Some reactions often bring pathological damage to the body although they have adaptive compensation. Under hypoxic conditions, the level of FAS protein also significantly increased, leading to increased fatty acid synthesis. Studies have shown that endoplasmic reticulum HYOU1 can accumulate under hypoxia to protect cells from hypoxia interference, while inhibiting the expression of HYOU1 protein can accelerate cell apoptosis. 40 Our study found that the expression of HYOU1 in the subacromial synovial of patients with rotator cuff injury was up-regulated. The up-regulation of HYOU1 also indicates hypoxia around the diseased tissue, and the synthesis of fatty acid enzymes will be intensified. Meanwhile, the physiological function of CLIC1 is not only necessary for ROS production, but also can be activated by oxidant and NADPH oxidase activity. 27 Therefore, the up-regulation of CLIC1 will increase the amount of ROS and aggravate the oxidative stress response of the tissue around the rotator cuff, leading to the apoptosis of tendon cells. Oxidative stress can cause extensive destruction of lipids, proteins and DNA, may alter neuronal function and cause cell death. Studies have pointed out that the ROS in microglia is produced by NADPH oxidase have a direct impact on surrounding cells and are required as a signal for microglial proliferation through regulation of TNF production and promotion of further signaling cascades. 41 The inhibition of CLIC1 can reduce microglial proliferation and TNF production, which means CLIC1 regulation of ROS as a potential signaling mechanism controlling these functions. Therefore, rotator cuff injury is closely related to apoptosis induced by oxidative stress.
Perilipin is a protein that covers the surface of fat particles (LDs). LDs were considered to be inert fat depots composed of neutral lipid cores, containing triacylglycerin and/or cholesterol esters encased in phospholipid monolayer; aliphatic granules are now shown to be highly dynamic and actively involved in the physiological activities of cellular lipid accumulation, storage and metabolism. 42 Many proteins have been shown to be associated with LDs and change under different physiological conditions, interacting with cytoplasmic proteins and other organelles to control intracellular lipid balance. Studies on PLIN4 in skeletal muscles have found that PLIN4 mRNA expression is related to fatty acid metabolism, and the two main pathways are propionic acid and fatty acid metabolism. 43 Our previous studies have also confirmed that fatty acid metabolism is closely related to oxidative stress, which can lead to red blood cell damage.11,37 PLIN4 is located near the plasma membrane/SS region, and research has found that the expression of PLIN4 mRNA is significantly reduced in long-term training athletes. The decrease in PLIN4 mRNA in skeletal muscle is related to the decrease in the number and volume of LDs in the SS region, and it is positively related to the genes involved in the de novo synthesis of phospholipids and the concentrations of phospholipids PE and PC.32,44 In proteomics studies, PLIN4 expression was also up-regulated in the subacromion synovium of patients with rotator cuff injury, which further verified the existence of oxidative stress. At the same time, studies have also pointed out that fat infiltration is closely related to muscle atrophy and loss of contractility.5,45 However, when the tendon is severed, muscles show significant fat infiltration, rather than further atrophy. The experimental group were all chronic degenerative rotator cuff injuries, characterized by obvious shoulder pain, weakness and limited mobility, MRI showed significant fatty infiltration of the supraspinatus muscle. The expression of PLIN4 is up-regulated in the synovium around the injured tendon, indicating that PLIN4 plays an important role in muscle contraction and pain of shoulder joint, and the channel mechanism of its action needs further study.
S100A11 participates in the regulation of various biological functions, and plays an important role in regulating cell growth, apoptosis, inflammatory response and cytoskeletal construction.20,21 S100A11 has been shown to regulate the stability of cell cycle regulators and cell cycle-dependent kinase inhibitors (1p21CIP1/WAF1) in human keratinocytes. 46 The study by Cerezo et al. 23 found that increased expression of S100A11 was found in articular cartilage of patients with rheumatoid osteoarthritis (RA). Histopathologic features of RA encompass infiltration by macrophages and T cells, synovial lining hyperplasia, neoangiogenesis, pannus formation and destruction of cartilage and bone. S100A11 expression and release from chondrocytes were induced by the pro-inflammatory cytokines interleukin-1β and tumor necrosis factor-α and the chemokine CXCL8.22,47 Studies have found that S100A11 is up-regulated in synovial from the knee joint of RA patients, especially in inflammatory infiltrated synovial lining. S100A11 in synovial of knee can enhance the secretion of pro-inflammatory cytokines, thereby forming a positive feedback loop in RA. 48 This suggests that local upregulation of S100A11 protein may reflect inflammatory processes and immune responses in patients with rotator cuff injury.
Conclusion
Our study reveals that pathological mechanism of rotator cuff injury and shoulder pain is closely related to oxidative stress and chronic inflammation. Meanwhile, the differential expressions of S100A11, PLIN4, HYOU1 and CLIC1 in the synovium of rotator cuff injury provide a new way to study the molecular pathological mechanism of rotator cuff injury. The limitation of this study is that the sample size is small. Meanwhile, the parallel analysis of different grades of rotator cuff injuries can make the research results more comprehensive and reliable. Whether there are more meaningful results of this has to be studied further.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by National Natural Science Foundation of China (81772318) and Natural Science Foundation of Jiangsu Province (BE2017723 and QNRC2016916).
Informed consent
This study was approved by Ethics Committee of Jinling Hospital (2020DZGZRZX-006). We confirm that all patients gave written informed consent.