
Abstract
Plaque psoriasis is one of the most common autoimmune skin diseases and is characterized by erythematous, scaly plaques. Many highly effective, targeted therapies have been developed as a result of an improved understanding of the pathogenesis of psoriasis. Using agents that target the central interleukin (IL)-23/IL-17 immune axis, this once difficult-to-treat disease is now among the most effectively treated autoimmune diseases with major clinical improvements possible in around 90% of patients. In this article, we outline the immune mechanisms responsible for the development of psoriasis and provide an overview of the novel IL-23 antagonists being used to manage this chronic skin disease.
Psoriasis is one of the most common autoinflammatory diseases in humans and affects approximately 3% of the US population. 1 This condition is characterized by inflammatory skin lesions that typically develop variably but can affect the entire skin surface. Psoriasis is also considered a systemic inflammatory condition due to its strong association with comorbidities that include psoriatic arthritis, obesity, metabolic syndrome, chronic kidney disease, stroke, and cardiovascular disease. 2 Peak disease onset is in adolescence or early adulthood and the disease course is chronic, with rare spontaneous improvement, thus creating the need to treat patients over a lifetime. 3 Over the last several decades, our understanding of the immune events that drive the development of psoriatic disease has significantly expanded. Because the pathogenic underpinnings of psoriasis are well known, targeted immune therapies have been developed and can effectively control psoriasis in 80–90% of patients. Full disease clearance is now seen in nearly half of patients treated with these selective antagonists, as described in further detail below.
Pathogenic activities of the IL-23/IL-17 axis in psoriasis
The immunopathogenesis of psoriasis is due to a complex interaction between multiple immune cell populations, including lymphocytes, dendritic cells (DCs), and keratinocytes. The dysregulation of the immune system in the skin promotes the proliferation of epidermal keratinocytes, resulting in the distinctive thickened, scaly plaques seen in psoriasis. The abnormal proliferation of keratinocytes is sustained by the release of various cytokines from pathogenic T lymphocytes (e.g. IL-17) and DCs infiltrating the skin. Laboratory studies have identified the IL-23/IL-17 axis as the primary signaling pathway leading to characteristic molecular, cellular, and structural changes in psoriatic skin.4, 5
In the initiation phase of psoriatic skin lesions (Figure 1), unknown environmental stimuli or loss of tolerance (e.g. autoantigen activation) lead to THE production of tumor necrosis factor (TNF) and the subsequent activation of dermal DCs.5–7 Increased production of IL-23 from dermal DCs activates distinct subsets of IL-17-producing cells (T17), including CD4+ helper T cells (Th17), cytotoxic CD8+ T cells (Tc17), innate lymphoid cells (ILC3), and γδ T cells.8–10 In the presence of IL-23, secreted mainly from inflammatory dermal DCs (TIP-DCs), T17 cells increase in number and produce large amounts of IL-17, which drive the upregulation of many psoriasis-related genes produced by epidermal keratinocytes (Figure 1).11,12 The clonal expansion of T17 cells and increased levels of IL-17 produce a feedforward inflammatory response and drive keratinocyte hyperproliferation.5,13,14 In addition to dermal DCs, epidermal keratinocytes and dermal macrophages are also found to produce IL-23 and may contribute to the pathogenesis and maintenance of psoriatic lesions.15,16
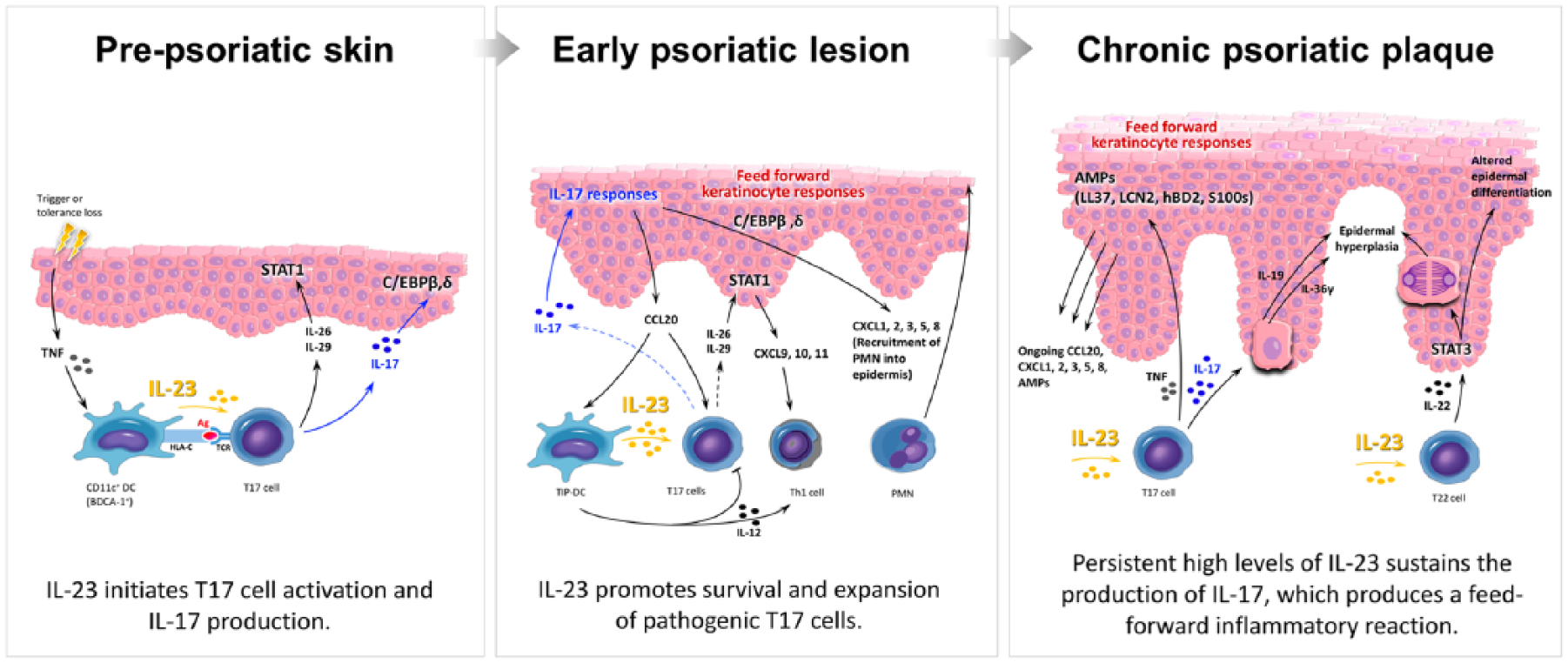
Updated pathogenic model of psoriasis. In prepsoriatic skin (left panel), stimuli initiate the pathogenesis of psoriasis. Increased amounts of tumor necrosis factor (TNF) and interferon mediate activation of interleukin (IL)-23-producing dermal dendritic cells (DCs) that trigger IL-17-producing (T17) cell clonal expansion. Activated T17 cells produce substantial amounts of IL-17, IL-26, IL-29, and TNF, leading to a ‘feedforward’ inflammatory cytokine cascade cytokine cascade in epidermal keratinocytes and the subsequent development of the early psoriatic lesion (middle panel). CCL20 secreted by activated keratinocytes recruits inflammatory dermal DCs (TIP-DCs) and T17 cells which perpetuate inflammation. IL-22, together with IL-17-induced IL-19 and IL-36 produced by keratinocytes, contribute to epidermal hyperplasia and altered terminal differentiation, which lead to the characteristic thickening of thickening of the skin seen in patients with chronic psoriasis (right panel). Without treatment, the ongoing ‘feedforward’ inflammatory cascade maintains a chronic psoriatic plaque by a self-amplifying, positive-feedback loop involving DCs, T lymphocytes, and keratinocytes.
TNF also contributes to the hyperproliferation of the keratinocyte through the activation of psoriasis-related genes and by acting synergistically with IL-17. 17 IL-17 further promotes a feedforward inflammatory response in keratinocytes by triggering increased chemokine production (e.g. CCL20) and transcriptional regulatory factor expression (e.g. C/EBPβ, δ), that lead to the recruitment of various immune cells into lesional skin, including neutrophils which are often found in the upper layers of the epidermis.18,19 IL-26 or IL-29 produced by T17 cells also activates STAT1 in keratinocytes and upregulates expression of CXCL9, CXCL10, and CXCL11. 29 CXCL9, CXCL10, CXCL11, and IL-12 secreted by dermal DCs promote the recruitment of Th1 lymphocytes, which can be found in well developed psoriatic plaques.
The feedforward keratinocyte responses seen in psoriatic skin are self-amplifying and sustain the pathogenic immune infiltrate leading to the development of mature psoriatic plaques (Figure 1). This IL-17-induced feedforward response also contributes to the increased production of keratinocyte-derived antimicrobial proteins keratinocyte-derived antimicrobial proteins (e.g. S100s, LCN2, hBD2, LL37),5,20 which may serve as psoriasis autoantigens and further promote disease progression. In response to DC-derived IL-23 stimulation, Th17 and Th22 cells induce the production of IL-19, IL-36, and IL-22 in lesional psoriatic skin.13,21–23 IL-36 is expressed in the skin, lung and gut, and has been shown to induce cellular inflammation through the activation of nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK). Following specific inflammatory stimuli in the skin (e.g. trauma or infection), IL-36 can activate DC subsets and promote the differentiation of IL-17-producing T cells through the increased production of keratinocyte-derived cytokines or chemokines.24,25 IL-36 expression in the skin is further upregulated by increased levels of IL-17 and acts cooperatively with this cytokine to promote epidermal proliferation and disrupt the normal differentiation of keratinocytes.26,27 Similarly, IL-19 is expressed in the skin and is strongly regulated by IL-17. In response to IL-17, keratinocytes produce increased amounts of IL-19 and IL-22, which stimulate epidermal hyperproliferation and the upregulation of proinflammatory signals, including antimicrobial peptides.28,29 In this way, IL-17 regulates the production of IL-36, IL-19, and IL-22, which in turn promote epidermal proliferation and the clonal proliferation of IL-17-producing lymphocytes, thus creating a feedforward loop of inflammation in psoriatic skin. The hyperproliferation and aberrant differentiation of keratinocytes lead to psoriasiform epidermal thickening (acanthosis), loss of the epidermal granular layer, and retention of nuclei in the stratum corneum (parakeratosis), several characteristic histopathological features of psoriasis.
In summary, the IL-23/T17 signaling pathway is central to the immunopathogenesis of psoriasis by stimulating the proliferation of IL-17-producing lymphocytes and sustaining the inflammatory milieu found in psoriatic plaques. In response to IL-23, these T17 cells create an immune environment that promote the hyperproliferation of keratinocytes and leads to the recruitment of other immune cell populations. Therefore, this feedforward mechanism and positive cytokine feedback loop represents the fundamental immune process responsible for the histologic features of psoriasis and its chronic disease course. Autoantigens that drive T-cell activation in psoriasis are also positively regulated by IL-17. 30 The critical importance of the IL-23/T17 axis in psoriasis is underscored by the high efficacy and rapid onset of action seen with the recent approval of multiple IL-17 and IL-23 antagonists.
Selective blockade of IL-23
The identification of IL-23 as the master regulator of IL-17-producing T lymphocytes led to the development of selective antagonists of IL-23 through the targeting of its p19 subunit. The primary effect of IL-23 blockade in psoriatic disease is to inhibit the differentiation and survival of pathogenic T17 cell populations in the skin. In July 2017, the US Food and Drug Administration approved guselkumab as the first IL-23p19 antagonist for the treatment of plaque psoriasis. Several other similar medications are currently in clinical trials, including tildrakizumab (MK-3222), risankizumab (BI 655066), mirikizumab (LY3074828), and LY2525623. Here, we provide an overview (Table 1) of the published clinical trial data for these agents in psoriasis.
Overview of clinical trial testing for current IL-23p19 antagonists.

FDA, US Food and Drug Administration.
Guselkumab
Guselkumab (also known as CNTO 1959) is a fully human immunoglobulin G1 (IgG1) λ monoclonal antibody that binds the p19 subunit of IL-23, thereby blocking the downstream signaling effects of IL-23. The high efficacy of IL-23 blockade in psoriasis was demonstrated in early proof-of-concept testing and phase I clinical trials. Since that time, results from three pivotal phase III clinical trials have been published for guselkumab: VOYAGE 1, 31 VOYAGE 2, 32 and NAVIGATE. 33 Guselkumab is approved for the treatment of moderate to severe plaque psoriasis and is administered subcutaneously at a dose of 100 mg at weeks 0 and 4, then every 8 weeks thereafter.
The VOYAGE 1 and VOYAGE 2 trials were randomized, double-blind studies evaluating the efficacy of guselkumab compared with adalimumab and placebo for the treatment of plaque psoriasis. In the VOYAGE 1 trial, patients with moderate to severe plaque psoriasis were randomized to 48 weeks of treatment with guselkumab (100 mg at weeks 0 and 4, then every 8 weeks) or adalimumab (80 mg at week 0, 40 mg at week 1, then 40 mg every 2 weeks), whereas patients in the control arm received placebo for 16 weeks before receiving guselkumab. The coprimary end points were the proportion of patients achieving a 90% improvement in their Psoriasis Area and Severity Index score (PASI90) and Investigator Global Assessment (IGA) of 0/1 (clear/minimal disease). At week 16, 85% (guselkumab) and 66% (adalimumab) of patients achieved an IGA 0/1 compared with only 7% of those subjects in the placebo arm. A total of 73% of patients in the guselkumab group achieved a PASI90 compared with only 50% and 3% in the adalimumab and placebo arms, respectively. The high efficacy of guselkumab is also underscored by the nearly 40% of guselkumab-treated subjects achieving a PASI100 compared with only 17% in the adalimumab arm. The safety profile of guselkumab was similar to adalimumab and the placebo groups with injections site reactions and nonserious infections (e.g. upper respiratory infection and nasopharyngitis) being the most commonly reported adverse events.
In the VOYAGE 2 trial, subjects were randomized to guselkumab (100 mg at weeks 0 and 4, then every 8 weeks), adalimumab (80 mg at week 0, 40 mg at week 1, then 40 mg every 2 weeks), or placebo. Those subjects randomized to the treatment arms (guselkumab and adalimumab) were treated for 28 weeks, whereas the control group received placebo for 16 weeks before receiving guselkumab until week 28. All groups then underwent a randomized withdrawal and retreatment period between weeks 28 and 72. As in the VOYAGE 1 trial, the coprimary end points were the proportion of patients achieving a PASI90 and IGA 0/1 at week 16. The proportion of patients at week 16 that achieved an IGA 0/1, PASI90, and PASI100 in the VOYAGE 2 trial is nearly identical to the VOYAGE 1 trial, and no new safety issues or concerns were noted. The unique design of the VOYAGE 2 trial, however, revealed several important treatment insights. First, in patient who were deemed nonresponders to 28 weeks of adalimumab treatment, subsequent treatment with guselkumab resulted in approximately two-thirds of patients achieving a PASI90. Second, patients treated with guselkumab maintained their higher clinical responses between weeks 24 and 48 compared with the clinical responses observed in the adalimumab group. The exact mechanisms underlying these two observations are not yet fully understood, but are likely due to the predominant role of IL-23 versus other cytokines on the regulation of IL-17-producing cells and other psoriasis-related cytokines (e.g. IL-22 and TNF).
In a third randomized, double-blind, 60-week, phase III clinical trial (NAVIGATE), the efficacy of guselkumab (100 mg at weeks 0 and 4, then every 8 weeks) for the treatment of moderate to severe plaque psoriasis was compared with ustekinumab (45 or 90 mg weight-based dosing at weeks 0, 4, and then every 12 weeks). 33 The trial design included an open-label run-in treatment period with ustekinumab for 16 weeks, followed by continued treatment with ustekinumab for those patients achieving an IGA of 0/1 or randomization of patients to guselkumab versus ustekinumab for those achieving an IGA of 2 or higher. The primary end point was the number of visits to achieve an IGA 0/1 at week 16 and at least a two-grade improvement (relative to week 16) from week 28 to week 40. At week 28, 31% of patients in the guselkumab group compared with only 14% of those in the ustekinumab treatment arm achieved an IGA 0/1 and at least a two-grade improvement relative to week 16. Approximately half of patients in the guselkumab treatment group also achieved a PASI90 at week 28 compared with 23% for ustekinumab. This study provides additional evidence for the clinical utility of guselkumab in patients with plaque psoriasis with inadequate responses to highly effective biologics such as ustekinumab or adalimumab. No new safety concerns for guselkumab were noted in this study.
Guselkumab is not currently approved for the treatment of psoriatic arthritis, though phase II clinical trials have been completed and phase III trials are currently underway. Similarly, phase II and III trials evaluating the efficacy of guselkumab for the treatment of palmoplantar pustulosis and erythrodermic or pustular psoriasis are ongoing. Of note, it will be important to see the long-term treatment and safety data for guselkumab in patients with psoriasis as current trial results suggest a possible protective effect of IL-23 blockade for the development of inflammatory bowel disease. This is of particular interest given the potential worsening or risk of developing inflammatory bowel disease while undergoing treatment with IL-17 inhibitors. Further research and clinical studies evaluating the biological relationship between IL-23, IL-17, and inflammatory bowel disease are necessary.
Tildrakizumab
Tildrakizumab (also known as MK-3222) is an IgG1 κ humanized monoclonal antibody against the p19 subunit of IL-23. Tildrakizumab is not currently approved for the treatment of plaque psoriasis or psoriatic arthritis. To date, two phase III clinical trials evaluating the efficacy of tildrakizumab have been reported: reSURFACE 1 and 2. 34
The reSURFACE 1 and 2 trials were three-part, double-blind, randomized, placebo-controlled studies evaluating the efficacy of guselkumab for the treatment of moderate to severe plaque psoriasis. In the reSURFACE 1 trial, subjects were randomized to tildrakizumab (100 mg or 200 mg) given at week 0, 4, and then every 12 weeks until week 28. At week 12 (part 2), patients in the placebo group were rerandomized to receive tildrakizumab (100 mg or 200 mg) until week 28. In part 3, subjects received tildrakizumab or placebo until week 64 (reSURFACE 1) or 52 (reSURFACE 2). The coprimary endpoints in this study were the proportion of patients achieving a PASI75 and PGA 0/1 with at least a two-grade reduction from baseline at week 12.
In part 1, approximately two-thirds of patients in the tildrakizumab treatment groups achieved a PASI75 at week 12 compared with only 6% and 48% in the placebo and etanercept arms, respectively; the proportion for subjects achieving a PASI90 was 35% compared with only 3% and 21% in the placebo and etanercept groups, respectively. Slightly less than 60% of patients achieved a PGA 0/1 compared with 7% in the placebo and 48% in the etanercept group. In part 2 (week 28), the proportion of patients achieving a PASI75 exceeded 80% whereas approximately 70% achieved a PGA 0/1. The reported PASI90 at week 28 was between 50% and 60% for tildrakizumab-treated patients. No major differences were observed between the 100 mg and 200 mg tildrakizumab groups throughout the reSURFACE trials.
Nasopharyngitis and upper respiratory tract infections were the most common adverse events observed with tildrakizumab. There was a very low (<1%) occurrence of serious adverse events such as malignancy, injection site reactions, and drug-related hypersensitivity reactions. No deaths or major cardiovascular adverse events were reported.
Risankizumab, mirikizumab, and LY2525623
Several other novel agents that directly target IL-23 are being developed and are in clinical testing. These other agents include risankizumab, mirikizumab, and LY2525623. To date, these agents are in the early or middle stages of clinical trial testing and do not have published phase III results.
Risankizumab (also known as BI 655066) is a high-affinity, humanized IgG1 monoclonal antibody against the p19 subunit of IL-23. In an early proof-of-concept, 24-week study in 39 patients with plaque psoriasis, 35 patients received single-dose intravenous (n = 18) or subcutaneous (n = 13) risankizumab compared with placebo. By week 12, nearly 90% of subjects in this study achieved a PASI75 compared with 0% of patients in the placebo group. In a 48-week, head-to-head phase II clinical trial published last year, 36 patients were randomized to either risankizumab (a single 18 mg dose at week 0, or a 90 or 180 mg dose at weeks 0, 4, and 16) versus ustekinumab (45 mg or 90 mg based on weight at weeks 0, 4, and 16). For the pooled 45 mg and 90 mg risankizumab treatment groups, 77% of patients reportedly achieved a PASI90 at week 12 compared with 40% for the ustekinumab group. Interestingly, a single 18 mg dose of risankizumab was similar in efficacy compared with the standard induction dosing for ustekinumab. In recent press releases for unpublished results from four phase III clinical trials evaluating the efficacy of risankizumab for plaque psoriasis, around 75% of patients with moderate to severe plaque psoriasis achieved a PASI90 at week 16 compared with less than half of patients in the ustekinumab or adalimumab treatment groups.37,38 The safety profile and tolerability of risankizumab were similar to other IL-23p19 antagonists but need to be further assessed.
Mirikizumab (also known as LY3074828) and LY2525623 are two additional, novel IL-23 antagonists in the early phases of clinical testing. Mirikizumab is currently being evaluated for efficacy in both psoriasis and Crohn’s disease, though the results from phase I studies have not yet been published and phase II studies are ongoing. LY2525623 testing has also progressed to phase II testing in psoriasis, but no published results are available. Of note, ClinicalTrials.gov indicates that phase II testing in psoriasis for LY2525623 was terminated for nonsafety reasons.
Conclusion and future directions
Advances in our understanding of the immunopathogenesis of plaque psoriasis have directly led to the development of selective IL-17 and IL-23 antagonists. The high efficacy of these novel agents has raised the bar for the primary study end points used in clinical trials (e.g. increasing use of PASI90 and PASI100), and these medications are shaping the treatment guidelines for psoriatic disease. While newer biologics in psoriasis have proven effective for the treatment of cutaneous psoriasis, psoriatic arthritis has proven to be much more resistant to therapy. To date, no biomarkers for psoriatic arthritis have been identified, and we are not yet able to predict which therapies these patients will be responsive to. Additional studies investigating the molecular mechanisms that drive the development of joint disease are needed and have the potential to identify new, potential therapeutic targets. Whether early, aggressive treatment of cutaneous psoriasis can reduce or prevent the subsequent development of psoriatic arthritis is not known.
The management of psoriasis-associated comorbidities represents another important area for future investigations. The relationship between skin, systemic inflammation, and associated conditions is not fully understood, and no definitive mechanisms unraveling this association have been identified. Nevertheless, there is some early evidence that systemic psoriasis therapies lead to the reduction of inflammatory cytokines and cardiovascular-related protein found in the circulation of patients with psoriasis. 39 Multiple prospective, phase IV clinical trials are currently underway to evaluate whether the treatment of moderate to severe plaque psoriasis with common systemic psoriasis therapies can reduce vascular inflammation and biomarkers of cardiometabolic risk. Improving our ability to manage psoriatic arthritis and reduce the increasing number of psoriasis-associated comorbidities through better treatment strategies will have a tremendous impact on the lives of patients living with psoriatic disease.
Footnotes
Acknowledgements
JEH would like to acknowledge support in part by the National Psoriasis Foundation/USA Early Career Research Grant. TCC would like to thank the Eighth Core Laboratory, Department of Medical Research, National Taiwan University Hospital, for their support. JEH and JGK are supported in part by The Rockefeller University CTSA award grant UL1TR001866 and KL2TR001865 from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program. TCC and JEH provided equal contribution to the manuscript.
Authors’ note
Tom C. Chan and Jason E. Hawkes are co-first authors.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JEH serves as an investigator/consultant for Pfizer Inc., Novartis, and VisualDx. JGK has been a consultant to and has received research support from companies that have developed or are developing therapeutics for psoriasis: AbbVie, Amgen, Boehringer, Bristol-Myers Squibb, Celgene, Dermira, Idera, Janssen, Leo, Lilly, Merck, Novartis, Pfizer, Regeneron, Sanofi, Serono, Sun, Valeant, and Vitae.