
Abstract
Pulmonary arterial hypertension (PAH) is a chronic disease that results in narrowing of the small pre-capillary pulmonary arteries leading to elevation of pulmonary artery pressure and pulmonary vascular resistance, subsequent right ventricular failure, and if unchecked, death. Advances in the treatment of PAH over the last two decades have markedly improved survival. These improvements reflect a combination of changes in treatments, improved patient care strategies, and varying disease phenotypes in the PAH population. Currently approved therapies for PAH are directed at the recognized abnormalities within the pulmonary vasculature and include endothelin receptor antagonists, phosphodiesterase-5 inhibitors, soluble guanylate cyclase stimulators, and prostacyclin pathway agents. Most of these drugs have been approved on the basis of short-term trials that mainly demonstrated improvements in exercise capacity. More recently, long-term, event-driven trials of novel drugs have been performed, demonstrating new efficacy parameters. There have also been exciting advances in the understanding of right heart failure pathophysiology in PAH that have the potential to inspire the development of right ventricular targeted therapy and continued discoveries in the heterogeneity of disease and response to treatment has great potential for developing more ‘personalized’ therapeutic options. In this article, we review the current available data regarding the management of PAH, with an emphasis on the pharmacologic therapies and discussion of novel therapeutic directions for the treatment of this fatal disease.
Keywords
Introduction
Pulmonary hypertension (PH) refers to a group of diseases with various etiologies. The most recent World Health Organization (WHO) clinical classification of these different etiologies was established in 2013 (Table 1). 1 PH is defined as a mean pulmonary artery pressure (mPAP) ≥25 mmHg at rest. Group 1 or pulmonary arterial hypertension (PAH) specifically refers to those patients with a mPAP ≥25 mmHg, a pulmonary artery wedge pressure ≤15 mmHg, and pulmonary vascular resistance (PVR) >3 Wood units. These hemodynamic parameters should fit within the appropriate clinical context, to ensure that other secondary causes of PH such as left sided heart disease or chronic lung disease have been ruled out.
Classification of pulmonary hypertension (Nice, 2013). 1
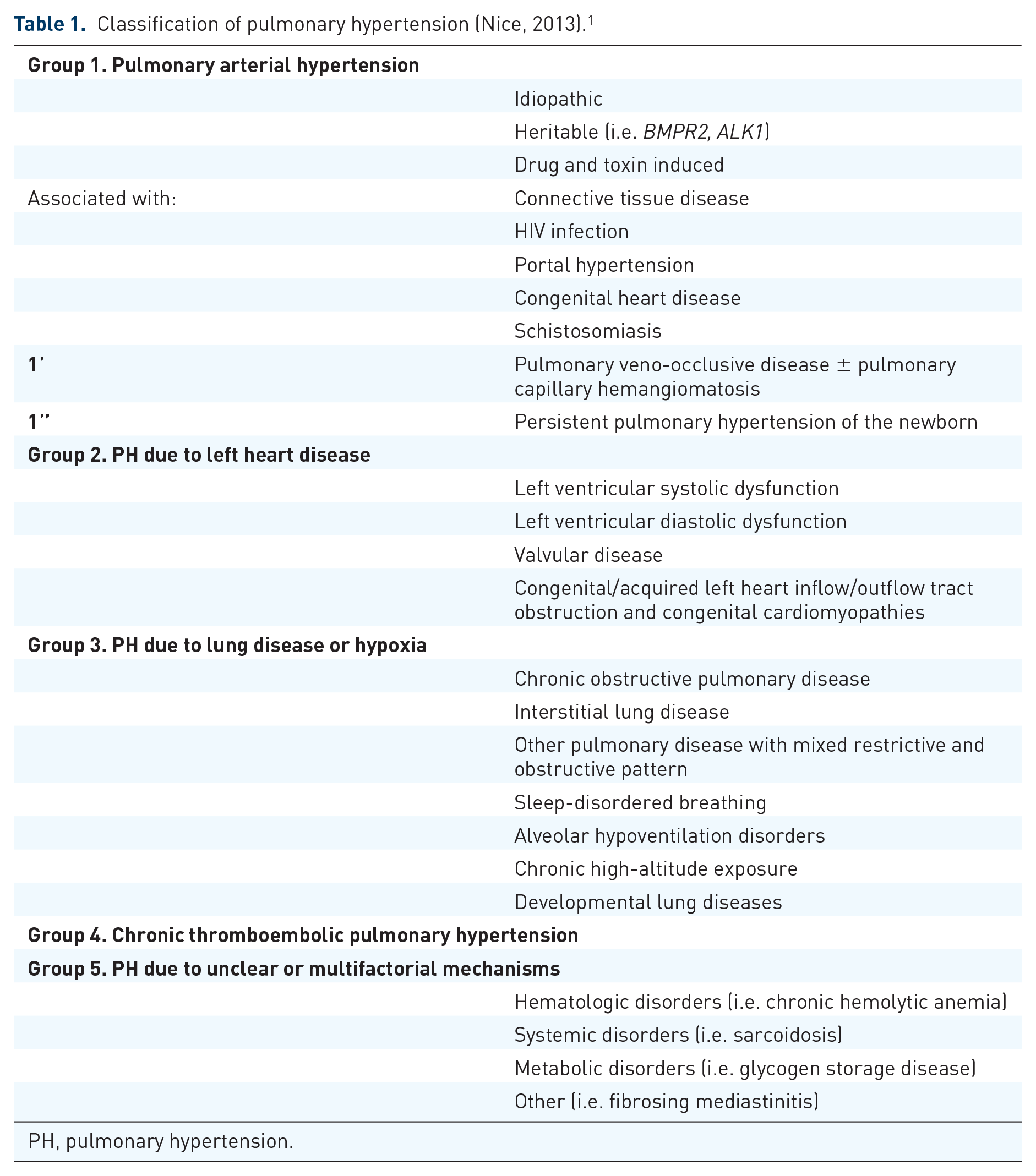
PH, pulmonary hypertension.
PAH can be caused by a diverse and disparate array of diseases and conditions, but despite the underlying cause, there appears to be a final common pathway causing the pathologic changes. Histologically there are characteristic alterations to the pulmonary vasculature including intimal fibrosis, increased medial thickness, and pulmonary arteriolar occlusion with excessive endothelial cell proliferation and formation of plexiform lesions. 2 This pulmonary vascular disturbance is further characterized by an imbalance in production of endothelial vasoactive elements such as nitric oxide (NO), prostacyclin (PGI2), and endothelin-1 (ET-1). 3 These changes result in narrowing of the small pulmonary arteries and arterioles leading to elevation of PVR, subsequent right ventricular failure, and death.
PAH is an uncommon condition, with registries from France and the United Kingdom reporting an incidence rate of 1.1–2.4 cases per million per year and a prevalence of 6.6–15.0 cases per million per year. 4 It is a female predominant disease with registries finding as high as a 4:1 prevalence in women. Although rare, PAH is a chronic and life-threatening disease. Advances in the treatment of PAH over the last two decades have markedly improved median survival from 2.8 years to approximately 9 years after diagnosis. 5 These improvements most likely reflect a combination of changes in treatments, improved patient care strategies, and varying disease phenotypes in the PAH population.
Currently approved therapies for PAH are directed at the recognized abnormalities within the pulmonary vascular pathways and include endothelin receptor antagonists (ERAs), phosphodiesterase-5 inhibitors (PDE5i), soluble guanylate cyclase stimulators, and prostacyclin pathway agents. Most of these drugs have been approved on the basis of short-term trials of only 12–16 weeks’ duration that mainly demonstrated improvements in exercise capacity. There is a notable lack of head-to-head comparisons of different therapeutic groups. More recently, seminal long-term, event-driven trials for novel drugs have been performed and may represent a more robust assessment of efficacy; though these studies have their own inherent problems.6,7There is also growing evidence supporting the efficacy of combination therapy in the treatment of PAH that has the potential to cause a paradigm shift in clinical practice. 8 Despite these rapid advances in PAH therapy, further research is required, as the disease remains deadly and disabling. Along these lines, exciting advances have been made in the understanding of right heart failure pathophysiology in PAH that have the potential to inspire the development of right ventricular (RV) targeted therapy and continued discoveries in the heterogeneity of disease and response to treatment has great potential for developing more ‘personalized’ therapeutic options.
This manuscript will review the current available data regarding the management of PAH with an emphasis on the available pharmacologic therapies. We will also briefly discuss novel therapeutic directions for the treatment of this fatal disease.
Current therapies
Supportive management
The supportive care of patients with PAH is a mainstay of treatment, although are heterogeneous or lacking data directly supporting many of these therapies. Basic and important treatment paradigms in the general management include supplemental oxygen, diuretic therapy, pregnancy avoidance, and exercise. Other pharmacologic interventions such as therapeutic anticoagulation and digoxin may also be considered.
Hypoxia is a potent pulmonary artery vasoconstrictor and oxygen therapy has been shown to counter this effect and decrease PVR. 9 Admittedly, there are no randomized control data confirming the benefits of long-term oxygen therapy in PAH. Consensus of experts however recommends oxygen supplementation to maintain oxygen saturation above 90%. 10
Optimizing intravascular fluid status to minimize RV dilatation with the use of diuretics is similarly essential in the management of PAH, though there are again no randomized controlled trials (RCTs) evaluating the benefit, type, or dose of diuretic to administer. It is important to routinely monitor renal function and chemistry panels in patients on diuretic therapy.
Similarly, there is a theoretical benefit of digoxin in the treatment of PAH but limited data to support the routine use. The pleiotropic effects of this medication on cardiac function have been largely extrapolated to PAH and right heart failure from its use in left sided systolic heart failure.11,12 There have been no studies on the long-term effects of digoxin in PAH and because of limited human data, the use of digoxin in patients with PAH remains controversial. When used, the patient must be monitored for potential toxicity.
Data on the use of oral anticoagulation in PAH are heterogeneous.13–15 The most recent 2015 European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines make a level IIb-C recommendation that oral anticoagulant treatment may be considered in patients with idiopathic (IPAH) or heritable (HPAH) disease, and PAH due to use of anorexigens. 16
Despite improved survival in PAH with newer therapies, pregnancy with this disease is associated with a clearly elevated morbidity and mortality. 17 The current recommendations advise that all patients should be counseled to avoid pregnancy, with a thorough explanation of risks to both mother and fetus, and permanent contraception should be strongly considered. Estrogen-containing contraception is not recommended because of the increased risk of venous thromboembolic disease, drug–drug interaction with PAH specific therapy, and the possible deleterious effects of estrogen on the pulmonary vasculature. 18
Patients with PAH should be encouraged to be active but avoid excessive physical activity that leads to distressing symptoms. 19 When physically deconditioned, patients may undertake supervised exercise rehabilitation at a center experienced in PAH care. 20 It is also highly encouraged that these patients are cared for at specialized centers, which has demonstrated a variety of benefits including better clinical outcomes, facilitation of participation in support groups, and inclusion in registries. 21
Calcium channel blockers
Calcium channel blockers (CCBs) inhibit the calcium influx into vascular cells leading to relaxation of smooth muscle and vasodilatation. This can be a useful therapy in few select patients with PAH. They are not specific therapies for PAH however, and in current practice, therapy with CCBs should be reserved only for those patients with a positive response to acute vasodilator testing performed in the catheterization laboratory. A positive response is defined as a decrease in mPAP by >10 mmHg, to <40 mmHg, with preserved cardiac output after administration of a vasodilator such as inhaled nitric oxide (NO), and sustained clinical response to treatment with CCBs. Notably, only 6–7% of patients with PAH have a vasodilatory response. 22 Commonly used agents include amlodipine, nifedipine, and diltiazem; choice of agent is made on the basis of the patient heart rate. The benefits of this therapy are that vasodilator responders have excellent long-term survival, it is relatively inexpensive, and taken orally. The main side effects include lower extremity edema, hypotension, and bradycardia, which may result in the need for progressive up-titration of the medication. CCBs are contraindicated in nonresponders as they may increase morbidity and mortality in this group. 23
Prostacyclin pathway agents
Prostacyclin (PGI2) is an endogenous substance produced by endothelial cells with several effects on the pulmonary vascular bed such as potent vasodilation, as well as anti-proliferative, anti-thrombotic, and anti-inflammatory activity. 24 Thromboxane A2 is a potent vasoconstrictor and promotes proliferation. In PAH, the balance between these molecules is shifted toward thromboxane A2, favoring thrombosis, proliferation, and vasoconstriction. 25 Additionally, prostacyclin synthase is decreased in the small- and medium-sized pulmonary arteries in PAH. 26 Prostacyclin synthetics and analogs have therefore been designed as therapeutic agents in PAH.
Epoprostenol, a synthetic PGI2, was the first PAH-targeted therapy approved by the United States (US) Food and Drug Administration (FDA) in 1995. It was shown that compared with conventional therapy, the continuous intravenous (IV) infusion of epoprostenol produced symptomatic and hemodynamic improvement at 12 weeks, as well as improved survival in patients with severe primary PH. 27 Additional RCTs reported improvements in exercise and symptoms in patients with scleroderma spectrum disease. 28 A meta-analysis revealed a 70% mortality risk reduction with this treatment. 29 To date, it is the only treatment that has demonstrated a reduction in mortality in PAH. Despite the clinical benefits, the short half-life (3–5 min), lack of stability after reconstitution necessitating ice packs for cooling, and continuous and uninterrupted IV administration of this medication made its use onerous. In 2010 the US FDA approved Veletri® (Actelion Pharmaceuticals Ltd.), a temperature stable formulation of epoprostenol, which obviated the need for ice packs and made the use of this drug much less burdensome. The most serious adverse events related to epoprostenol involve the indwelling tunneled catheter, such as site infection, sepsis, and catheter associated venous thrombosis.27,28,30 There is also a potential risk of fatal rebound PAH if the pump malfunctions or infusion is otherwise disrupted. Common side effects, which are similar for all of the PGI2 agents, include headache, flushing, jaw pain, and diarrhea.27,28,31 Epoprostenol is approved for use in patients with WHO functional class III–IV who do not respond adequately to other therapies and is the first-line therapy in advanced disease with high-risk features.
Treprostinil is another prostanoid analog, with some similarities to epoprostenol, but with a longer half-life [~4.5 h for subcutaneous (SC) or IV formulation]. It is clinically stable at room temperature for ~72 h. It has multiple formulations allowing multiple routes of administration including SC, IV, oral, and inhalation. SC treprostinil was approved for the treatment of PAH in 2003. An RCT tested continuous SC infusion of the medication in a 12-week double-blind placebo-controlled multicenter trial of 470 patients and demonstrated improvement in hemodynamics, symptoms, quality of life, and modest but significant improvement in 6-min walk distance (6MWD) by 16 m (p = 0.006) in the treatment group. The most common side effect was infusion site pain (85% of patients) that led to discontinuation of therapy in 8% of patients. 32 The US FDA approved the use of IV treprostinil in 2004 based on bioequivalence for those patients unable to tolerate the SC injection. 33 Several prospective studies have demonstrated in the short term that IV treprostinil is a well-tolerated and effective therapy that provides similar beneficial hemodynamic improvements to epoprostenol.34–36 An RCT of 44 patients with PAH in 2010 demonstrated significantly improved exercise capacity, dyspnea and functional class in patients treated with IV treprostinil monotherapy compared with placebo. 37 The side effects are similar to epoprostenol and the benefits include less frequent need for drug reservoir changes, improved quality of life and treatment satisfaction. 38 The TRIUMPH study evaluated the use of inhaled treprostinil in addition to background therapy (concurrently on bosentan or sildenafil) in 235 patients with WHO functional class III or IV. The addition of inhaled treprostinil, which was administered four times per day, significantly increased 6MWD and quality of life compared with placebo, but had no effect on time to clinical worsening, functional class, or dyspnea. 39 Oral treprostinil has also been evaluated as both monotherapy and add-on therapy in PAH patients in the FREEDOM studies of >300 patients with PAH. As monotherapy, there was improved exercise capacity at 12 weeks but no significant change was detected in the combination therapy studies. There was also no significant change in other efficacy parameters.40–42 Oral treprostinil therapy is only approved as monotherapy in treatment-naïve patients with PAH. Inhaled treprostinil is approved for either add-on or combination treatment.
Iloprost is the third synthetic analog of PGI2 and was US FDA-approved in 2004 for the treatment of PAH. It has improved stability at room temperature compared with epoprostenol. It is available in oral, inhaled, or IV formulations, but is only approved in the US as inhaled therapy. It has a relatively short half-life (20–25 min) and no active metabolite; therefore, it has to be administered very frequently. The inhaled form was studied in the AIR trial, which evaluated 203 patients with PAH or chronic thromboembolic PH (CTEPH), randomized to iloprost versus placebo (6 or 9 times per day). There was a beneficial effect of the drug on exercise capacity, PVR, and symptoms after 12 weeks of therapy. 43 Two RCTs evaluated the role of inhaled iloprost as combination therapy with bosentan yielded opposing results.44,45 A prospective study analyzed the long-term efficacy of inhaled iloprost on 76 patients with PAH and demonstrated that only a small percentage of patients could be stabilized. Despite iloprost being generally well tolerated, with the most common adverse events being flushing, cough, and jaw pain, chronic iloprost inhalation as monotherapy appears to have a limited role. 46 The IV formulation of iloprost has been studied but not in a randomized fashion and some analyses suggest a high rate of treatment failure and poor survival. 47 IV therapy is not currently available in the US or Europe.
Selexipag is a newly approved orally available, nonprostanoid, highly selective agonist of the prostacyclin (IP) receptor. It is chemically distinct from PGI2 and other synthetic prostanoids. The mechanism of action is believed to be similar to the synthetic prostanoid agonists as its main active metabolite is highly selective for the human IP receptor. However, it has a much longer half-life, ~7.9 h, than the other prostacyclin analogs. 48 Selexipag was studied as an oral alternative to the previously available prostacyclin analogs for the treatment of PAH. A phase II study of 43 patients with symptomatic PAH (on background therapy with an ERA ± PDE-5i) were randomized to receive either placebo or selexipag, with dosage up titrated to 800 μg twice daily, for 17 weeks. There was a statistically significant 30% reduction in PVR with selexipag compared with placebo. 49 This was followed by the GRIPHON trial that evaluated the long-term efficacy of selexipag with an event-driven study with the primary endpoint including death, hospitalization for PAH, need for supplemental oxygen, atrial septostomy, lung transplantation, IV prostacyclin, or clinical worsening. The RCT recruited 1156 patients with PAH, half with IPAH, who received placebo or selexipag in individualized doses (max dose 1600 μg twice daily) for a median duration of 70 weeks. Participants were allowed to remain on prior stable PAH therapy including ERA, PDE-5i, or both (~80% of those enrolled). Overall, there were 41.6% primary endpoint events in the placebo group and 27% in those treated with selexipag, with a hazard ratio in the treatment group of 0.60 [99% confidence interval (CI): 0.46–0.78, p < 0.001]. Most of the events (~82%) were described as disease progression and hospitalization. There was no statistically significant difference in mortality. In addition, selexipag moderately improved 6MWD. These findings were significant regardless of the presence of background therapy. The adverse events observed with selexipag were consistent with those typically observed with prostacyclin therapies including headache, diarrhea, and nausea. 7 The findings from GRIPHON demonstrate the feasibility of assessing the long-term effects of novel therapeutic agents in PAH and appear to also support the benefit of using combination therapy.
Endothelin pathway agents
Endothelin-1 is an endogenous protein that promotes vasoconstriction and proliferation by binding to two receptors, ET-A and ET-B, located in the pulmonary vasculature. Type A receptors are mainly expressed in pulmonary vascular smooth muscle cells (PVSMCs) and contribute to ET-1-induced contraction and proliferation. ET-B receptors are expressed in the endothelial cells and lead to ET-1 clearance and vasodilation. In PAH, both ET-1 and its receptors are found to be up-regulated which led to the development of ERAs as therapy in this disease. 50
Bosentan is a competitive, specific dual receptor (ET-A and -B) antagonist. It was approved in 2001 by the US FDA for the treatment of patients with PAH. It is administered orally and has a half-life of about 5 h. It is metabolized by the CYP2C9 and CYP3A4 enzymes but also induces them, leading to drug–drug interactions, importantly with other PAH therapies such as sildenafil. The efficacy and safety of bosentan has been evaluated in RCTs in multiple types of PAH. BREATHE-1 was the initial RCT of 213 patients that demonstrated treatment with bosentan for 16 weeks resulted in an improved 6MWD by a mean of 44 m (p < 0.001). Bosentan also improved the Borg dyspnea index and functional class, and increased the time to clinical worsening. 51 The EARLY study also found that long-term treatment with bosentan for up to 1 year in patients with WHO functional class II was associated with improvement in hemodynamics and delayed time to clinical worsening.52,53 The most common adverse event with bosentan therapy is reversible and usually asymptomatic elevation in transaminases (≥3 times upper limit of normal). Postmarketing surveillance showed significant elevation in around 10% of patients annually leading to 3% of patients discontinuing the medication. 54 Other potential down sides to bosentan therapy include the teratogenicity (absolute contraindication in pregnancy) and drug interactions including all usual estroprogestative contraception, as well as commonly used medications such as cyclosporine, simvastatin, warfarin, and rifampin. Importantly all of the ERAs, including bosentan, have the potential for drug interactions with many antiretroviral therapies used to treat HIV and AIDS.
Ambrisentan is an oral selective ET-A receptor antagonist with a bioavailability and a half-life (9–15 h) that allows once a day dosing. The selectivity of ambrisentan is theoretically beneficial to PAH patients by allowing the preservation of ET-B receptor function such as vasodilation. The US FDA approved this drug for use in PAH in 2007. The two large RCTs, ARIES-1 and ARIES-2, ran concurrently in ~400 patients randomized to placebo or oral ambrisentan (ARIES–1: 5–10 mg, ARIES–2: 2.5–5 mg). These studies demonstrated improvement in 6MWD, time to clinical worsening, dyspnea scores, WHO functional class, and B-type natriuretic peptide (BNP). 55 A long-term 2-year study called ARIES-E demonstrated sustained improvements in exercise capacity and a low risk of clinical worsening and death in patients with PAH. 56 Ambrisentan is generally well tolerated and has a low risk of aminotransferase abnormalities. The most common adverse events include peripheral edema, headache, and nasal congestion. Overall, ambrisentan has a lower potential for liver toxicity and drug–drug interactions than bosentan, including oral contraceptives, but is teratogenic.57,58
Macitentan is a novel, oral, tissue-targeting dual ERA, ET-A and ET-B, with sustained receptor occupancy. The molecular structure is in a new chemical class called sulfamides, compared with the other ERAs with a sulfonamide structure. It has a half-life of 17.5 h, and is therefore suitable for once-daily dosing. 59 The US FDA approved it in 2013 for the treatment of PAH after the SERAPHIN study. This trial assessed the long-term safety and efficacy of macitentan and has the distinction of being the first event-driven trial evaluating long-term morbidity and mortality as the primary endpoint in PAH. SERAPHIN included 742 patients who were randomly assigned to receive placebo or macitentan at 3 or 10 mg once-daily dosing. Patients were allowed to remain on prior stable oral or inhaled PAH therapy, with the exception of ERAs, which included 64% of those enrolled. The primary endpoint was the time from initiation of treatment to the first occurrence of a composite endpoint of death, atrial septostomy, lung transplantation, initiation of treatment with SC or IV prostanoids, or worsening of PAH. Both doses of macitentan demonstrated reduced risk of morbidity and mortality by 30% in the 3 mg dose group and 45% in the 10 mg dose group when compared with placebo. This difference appeared to be driven by the primary endpoint of clinical worsening and notably mortality alone was not statistically different. Macitentan also improved exercise capacity and dyspnea scores. These differences were dose-dependent and significant regardless of the presence of background therapy. Adverse events included headache, nasopharyngitis, and anemia. There was no difference in liver test abnormalities between drug and placebo groups and currently no guidelines recommend routine hepatic panel testing for this agent. 6 Studies have demonstrated that despite significant induction of CYP3A4, macitentan is markedly less prone to drug–drug interactions, including oral contraceptives, than bosentan. 60 Similar to the other ERAs, it is teratogenic and pregnancy is a formal contraindication.
Nitric oxide pathway agents
NO is produced in the endothelial cells from L-arginine by activity of the enzyme NO synthase. NO then activates soluble guanylate cyclase (sGC) in the smooth muscle cells to catalyze cyclic guanosine monophosphate (cGMP), which leads to vasodilation, inhibition of platelet aggregation and smooth muscle cell proliferation. In PAH, the NO bioavailability in the pulmonary vasculature is decreased. Phosphodiesterases (PDEs) are enzymes which inactivate cGMP. PDE-5 is an isozyme abundantly expressed in lungs and known to be up-regulated in PAH. PDE-5 inhibitors therefore preserve cGMP levels and promote effects of endogenous NO, including pulmonary artery smooth muscle cell relaxation and possibly growth inhibition of vascular smooth muscle cells, making this class of medications beneficial in treating PAH. Currently, two PDE-5i and one soluble guanylate cyclase stimulator are approved by the US FDA for the treatment of PAH.
Sildenafil has a half-life around 4 h and requires three times a day dosing. It was evaluated for efficacy and safety in PAH in the SUPER-1 and open label extension SUPER-2 trials. The pivotal RCT SUPER-1 included 278 patients with PAH and demonstrated significant improvement in exercise capacity, functional class, and hemodynamics when compared with placebo. 61 When treatment was extended to 3 years in SUPER-2, 60% of patients improved or maintained functional class, 46% maintained or improved 6MWD, and there was a 79% 3-year estimated survival rate. 62 Sildenafil is predominately metabolized by CYP3A4 enzyme leading to multiple important drug–drug interactions, especially with bosentan, which is a CYP3A4 inducer.52,63 Due to the vasodilator effects, there is a risk of synergistic hypotension when co-administered with other drugs such as nitrates and CCBs. The most common side effects include headache, flushing, dyspepsia, and epistaxis. Sildenafil is currently available in oral, including suspension, and IV formulations. Physicians must also be aware of the rare but serious effects such as sudden decrease or loss of vision or hearing that have been reported with sildenafil.
Tadalafil was the second PDE-5i US FDA- approved for use in PAH. It has a once-daily dosing with a half-life of 17.5 h. The efficacy was investigated in the 16-week PHIRST-1 trial and an extension phase of 1 year in PHIRST-2. PHIRST-1 included 405 patients with PAH, either treatment-naïve or on background therapy with bosentan, who were randomized to placebo or tadalafil. The treatment increased exercise capacity in a dose-dependent fashion with 40 mg reaching statistical significance. The medication also led to clinical improvement, better quality of life, and increased time to clinical worsening at this dosing. 64 The long-term tolerability and sustained efficacy over 1 year was demonstrated in the continuation study PHIRST-2. 65 The side effects of tadalafil appear to be class specific for PDE-5i and include headache, flushing, nasal congestion, dyspepsia and myalgia, which are a reflection of vasodilatory effects on the capillary smooth muscle in other parts of the body. 66
Riociguat is a guanylate cyclase stimulator that acts directly on sGC even in the absence of NO to enhance activity. Enhanced activity of sGC leads to increased cGMP, which mediates vasodilation through the activation of cGMP-dependent protein kinases. Thus, riociguat can work in synergy with NO or in an NO-independent mechanism to promote vasodilation. 67 The PATENT-1 trial studied 443 patients with symptomatic PAH who were randomized to placebo or riociguat at a dose of up to 2.5 mg 3 times a day over 12 weeks. There were favorable effects of treatment on 6MWD, PVR, WHO functional class, dyspnea, and time to clinical worsening. 68 The RCT CHEST-1 evaluated 250 patients with inoperable CTEPH (systematic assessment was undertaken by an adjudication committee composed of experienced pulmonary endarterectomy surgeons at expert centers to determine operability of disease) or continued disease despite pulmonary endarterectomy, randomized to placebo or riociguat at a dose of up to 2.5 mg 3 times a day for 16 weeks. Treatment with riociguat significantly improved exercise capacity and hemodynamics in patients with CTEPH. 69 Based on these studies, riociguat is the first US FDA-approved drug to treat CTEPH. There are open-label extension phases of both trials, PATENT-2 and CHEST-2, which are ongoing to assess the efficacy and safety of long-term use. The most serious adverse events associated with this drug include syncope and hemoptysis. Other side effects are similar to PDE-5i and include headache, flushing, orthostatic hypotension, and nasal congestion. When initiating therapy, hypotension requires gradual up-titration of the medication and frequent monitoring. Combining PDE-5i and guanylate cyclase stimulators is potentially harmful, as demonstrated in the PATENT-PLUS study showing unfavorable safety signals with sildenafil plus riociguat, high rates of discontinuation due to hypotension, three (18%) deaths, and no evidence of a positive benefit–risk ratio. 70
Combination therapy
As there are currently three imbalanced signaling pathways in the pulmonary vasculature implicated in the pathogenesis of PAH, there is obvious potential for combination therapy targeting multiple pathways simultaneously. This combined approach has successfully been employed in the treatment of other serious and chronic diseases such as congestive heart failure, HIV infection, and cancer. There have been multiple studies to date evaluating both initial (up-front) and sequential combination therapy with varying results.45,64,71– 75 In 2011, a meta-analysis of the existing six trials, in a total of 858 patients, found efficacy in the strategy of sequential combination therapy when there is a suboptimal response to the initial monotherapy. In general, combination therapy reduced the risk of clinical worsening with relative risk of 0.48 (95% CI: 0.26–0.91, p = 0.023), increased the 6MWD significantly by 22 m and reduced mPAP and PVR. The incidence of serious adverse events was similar in the two groups with relative risk 1.17 (95% CI: 0.40–3.42, p = 0.77). The reduction in all-cause mortality was not statistically significant. 76 The only up-front combination therapy study included in this analysis was BREATHE-2. 71 This was a small extension of BREATHE-1 that failed to demonstrate any significant difference between patients treated initially with the combination epoprostenol and bosentan as compared with epoprostenol alone, perhaps because it was underpowered for this endpoint. Since that time, there have been multiple additional studies looking at sequential combination therapy. The PATENT trial, as above, showed improvement in functional class, hemodynamics, and quality of life when riociguat was added to background ERA or prostanoid therapy. 68 Alternatively, the recent COMPASS-2 trial evaluating the addition of bosentan to sildenafil therapy and FREEDOM trials adding oral treprostinil to PDE-5i ± ERA failed to show any significant effect on primary endpoints.40,41,72
The AMBrisentan and Tadalafil in Patients with Pulmonary Arterial HypertensION (AMBITION) study investigated a novel up-front dual combination therapy of ambrisentan and tadalafil. It was a unique study design in treatment-naïve PAH patients comparing combination therapy and monotherapy with the two agents as a pooled group. The primary efficacy endpoint was time to clinical failure that included death by any cause, hospitalization for worsening PAH, disease progression, or unsatisfactory clinical response to therapy. A total of 500 patients were enrolled and randomized in a 2:1:1 fashion to up-front combination or monotherapy then followed for at least 24 weeks. A primary endpoint event occurred in 18% of the combination therapy group and in 31% of the pooled monotherapy group. The hazard ratio for the primary endpoint in the combination therapy group versus the pooled monotherapy group was 0.50 (95% CI: 0.35–0.72, p = 0.03). The main driver of the reduced endpoint in the combination group was a reduction in hospitalizations for PAH. At week 24, the combination therapy group had statistically significant greater reductions from baseline in BNP, higher percentage of clinical response, and greater exercise capacity (6MWD + 26 m in the combination therapy group) when compared with the pooled monotherapy group. The side effects that occurred more frequently in the combination therapy group than in either monotherapy group included peripheral edema, headache, nasal congestion, and anemia. At 3 year follow up, 68% of the combination therapy group versus 56% of the pooled monotherapy patients were free from events. 8 The 2015 ESC/ERS guidelines now recommend ambrisentan and tadalafil as initial combination therapy with grade B evidence in WHO functional class II or III. 16
The past variations in study results of combination therapy may reflect a true difference in drug action, individual patient response to therapy, or simply trial design. The AMBITION trial supports a new paradigm for treating PAH up-front with two agents simultaneously, targeting two separate pathways, with the potential for altering disease progression. There is great promise with this approach but further studies are needed to evaluate different combinations of different classes of drugs to improve the generalizability of this approach and define what the ideal up-front combination of agents may be.
Future potential therapies and approaches
Over the last 2–3 decades as described, there have been great advancements in the understanding of the mechanisms underlying the development of PAH and design of beneficial therapies targeting these known signaling abnormalities. Other potential targets of therapy are under active investigation including mitochondrial dysfunction, altered inflammation/immunity, vascular remodeling, RV adaptation, and sex hormones. There is also great interest in seeking out ways to personalize therapy for patients with PAH with the potential to predict response to therapies and individualize treatment approaches.
Mitochondrial modulators
There is evolving evidence that the pulmonary vascular cells and RV myocytes in PAH have abnormalities in energy metabolism including lower oxygen consumption and higher glycolytic rate resembling the metabolism of cancer cells.77,78 Mitochondrial dysfunction may underlie this glycolytic shift (Warburg effect) in PAH. Most experimental evidence regarding the impact of blocking the glycolytic shift has focused on the inhibition of pyruvate dehydrogenase kinase (PDK). There are animal studies of the drug dichloroacetate (DCA), which is an analog of acetic acid that inhibits mitochondrial PDK, demonstrating reversal of monocrotaline-induced PAH and RV remodeling, as well as prevention of the formation of neointimal lesions.79–81 This is a promising area to target as it may positively impact both the pulmonary vascular and RV dysfunction in PAH. A phase I open-label two-center study in patients with PAH WHO functional class III–IV on background therapy with ERA or PDE-5i who were randomized to doses of DCA up to 12.5 mg/kg twice a day for 16 weeks was recently undertaken. The primary endpoint was the safety and tolerability of DCA. No results have been published to date. 82
Piperazine derivatives promote glucose oxidation in mitochondria through the inhibition of beta oxidation. Ranolazine is in this class and has typically been used as an anti-angina therapy in coronary artery disease. A prospective, open-label, pilot study of treatment with ranolazine for 3 months in 11 patients with PAH demonstrated significant improvement in functional class, reduction in RV size, improvement in RV strain during exercise at 3 months, and a trend toward improved exercise. Ranolazine was not associated with improvement in invasive hemodynamics. 83 Another piperazine derivative, trimetazidine, is currently in a phase II trial evaluating the effects on RV function in PAH. 84 As our knowledge of the metabolic derangements in PAH expands, there will undoubtedly be additional glycolytic enzymes identified as targets for pharmacologic intervention.
Anti-inflammatory and immune modulatory therapy
A significant number of patients with IPAH have laboratory evidence of autoimmunity and inflammation. 85 It has recently been shown that PAH patients have altered perivascular inflammatory cell infiltration in pulmonary vascular lesions, including increased macrophages (CD68+), macrophages/monocytes (CD14+), mast cells (toluidine blue+), dendritic cells (CD209+), T-cells (CD3+), cytotoxic T-cells (CD8+), and helper T-cells (CD4+). Furthermore, blocking a key chemokine (stromal derived factor-1) for recruitment of these inflammatory cells was associated with improvement of hemodynamics and pulmonary vascular remodeling in a monocrotaline-induced PH model. 86 Immunosuppressive therapy is routinely administered in patients with connective tissue disease associated with PAH and has been shown, in conjunction with vasodilators, in mostly observational studies to improve functional class and hemodynamics in patients with lupus and mixed connective tissue disease.87,88 Several inflammatory and immune cell modulators including mycophenolate mofetil, leukotriene inhibitors, and anti-CD20 antibody have been investigated as therapeutics in PAH and demonstrated benefit in animal models.89–91 Further studies and experimental models are needed to determine the benefit of pre-existing or novel therapeutics that target the different cell types involved in this immune/inflammatory disturbance. Presently there is ongoing recruitment for a multicenter phase II RCT evaluating the use of monoclonal antibody to CD20 (rituximab) in the treatment of systemic sclerosis-associated PAH. 92
Anti-proliferative therapy
In PAH there is maladaptive vascular remodeling characterized by medial hypertrophy, intimal and adventitial thickening, and the formation of occlusive plexiform lesions. 2 Therapies targeting this vascular proliferation have been studied, with a focus on the anti-neoplastic receptor tyrosine kinase inhibitors (TKIs). Animal studies of platelet-derived growth factor receptor inhibitors such as imatinib, dasatinib, and nilotinib, which inhibit cellular proliferation and promote apoptosis of PVSMCs, were able to demonstrate reversal of this pathologic vascular remodeling.93,94 A multicenter phase III trial of imatinib as add-on therapy in 200 patients with PAH (IMPRES) over 24 weeks demonstrated significantly improved 6MWD and PVR when compared with placebo. However, there was no improvement in functional class, time to clinical worsening, or mortality. Severe adverse effects occurred in 44% of patients, including a significant increase in subdural hematomas. 95 Despite promising results in this study and follow up studies on right heart function, 96 the side effects and resultant high rate of discontinuation of treatment (~1/3) represent a barrier to the clinical use of this medication in PAH. Multikinase inhibitors, such as sorafenib and sunitinib, have also been evaluated in both animal models demonstrating reversal of vascular remodeling and limited human trials with improved exercise and RV function, but side effects again appear to limit the utility of these therapies.97–99 Given the malignant-like transformation in the pulmonary vascular cells in PAH, anti-neoplastic drugs, like TKIs, that target this maladaptive remodeling have great potential benefit but further studies are needed to find balance between their anti-proliferative properties and off-target effects resulting in decreased tolerability.
Right ventricle targeted therapy
Survival in patients with PAH is very closely related to RV function and there is significant individual variability in RV adaptation to elevated pulmonary pressures and increased afterload that determines clinical outcome.100–102 The remodeling of the RV in response to PAH is a complex process that depends not only on the severity of the pulmonary vascular disease but also on the interplay between neuro-hormonal activation, coronary perfusion, and myocardial metabolism.103–106 Adaptive remodeling to chronic pressure overload is characterized by concentric remodeling with increased protein synthesis and preserved function, whereas maladaptive remodeling is associated with more eccentric cardiomyocyte hypertrophy and fibrosis leading to worse function with RV dilatation and failure. 106 The presently approved therapies for PAH can lead to reverse remodeling of the right heart but this is mainly through their vasodilatory and afterload reducing effects. Drugs that are commonly used in left heart failure, such as beta-blockers, have been minimally evaluated with varying effects on right heart function.107,108 Targeted treatment strategies for PAH that promote RV adaptation could preserve cardiac function and improve outcomes.
As described above, there is metabolic remodeling in the cardiomyocytes characterized by decreased fatty acid oxidation and increased glycolysis. There is potential for therapies targeting these metabolic derangements, such as DCA, and the results of the completed phase I trial are eagerly anticipated.80,82 The development of cellular hypertrophy and myocardial fibrosis is also associated with increased oxidative stress with the excessive generation of reactive oxygen species (ROS) and lipid peroxidation. 109 Therapeutics targeting the generation of ROS have been investigated. For example, allopurinol, a xanthine oxidase inhibitor, demonstrated improved myocardial contractility and amelioration of hypoxic PAH in rats.110,111 Recent work has targeted the thromboxane-prostanoid (TP) receptor, which is found with increased density in the RV of patients with PAH and when activated contributes to hypertrophy and decreased cardiac function. They demonstrated antagonism of the TP receptor was cardioprotective against RV pressure overload in a mouse pulmonary artery banding model. 112 Other potential nonpharmacological RV-targeted therapies that are under investigation include exercise therapy, cardiac resynchronization, atrial septostomy, and mechanical RV support. 113 Given that PAH is clearly a cardiopulmonary disease, it would be beneficial for new and ongoing drug development to be experimentally tested for effects on both the pulmonary vasculature and pressure-overloaded right heart.
Hormonal manipulation
The best established clinical risk factor for most forms of PAH (including idiopathic, heritable, connective tissue disease-associated) is female sex; however, women with PAH have better outcomes than men, leading many in the field to investigate sex hormone influences on the disease.4,5,100,114,115 Manipulation of estrogen in animal models of PH has shown contradictory outcomes leading to the ‘estrogen paradox’ of pulmonary vascular disease. 116 In humans there may also be a differential effect of sex hormones on the pathogenesis of PH and the RV response to stress. Previously it was shown that higher levels of E2 were associated with better RV systolic function in women without cardiovascular disease using hormone replacement therapy. 117 In contrast, a recent study demonstrated higher levels of estradiol (E2) in men with PAH and higher levels associated with shorter 6MWD. 118 Aromatase (which converts androgens to estrogen) is produced in PVSMC of both female animal models of PH and women with PAH. Administration of the aromatase inhibitor anastrozole (AN) reduced pulmonary pressures, pulmonary vascular changes, and indices of RV hypertrophy in experimental PH.119,120 Together, these findings suggest that sex hormones appear to influence the development and progression of the disease and hormonal manipulation, such as drugs that target components of the estrogen pathway such as CYP19A1 (AN) or estrogen receptors (tamoxifen), may be a novel therapeutic strategy in PAH.
A recently completed two-center small phase II RCT was the first to evaluate the effect of hormonal manipulation with AN on PAH. A total of 18 patients (postmenopausal women and men) with PAH were randomized in a 2:1 fashion to receive either 1 mg daily of AN or placebo for 3 months. AN reduced circulating levels of E2 by 40% without changing other sex hormone levels. There was no effect on lab (NT-proBNP levels) or echocardiographic measures of RV function. AN did significantly increase 6MWD at 3 months. There was no difference in quality of life, functional class, or side effect profiles. This study demonstrates the feasibility and safety of AN administration in PAH, but larger and longer studies with clinically important endpoints are needed to further elucidate the effectiveness of this treatment for PAH. 121 As many of these drugs are widely used in the treatment of cancer, there is great translational potential for both preclinical and clinical research in the treatment of pulmonary vascular disease.
Personalized medicine
While substantial progress has been made in treating PAH by targeting key molecular pathways, heterogeneity in response to treatment is a challenge in continuing to improve outcomes in this disease. Recently many investigators have taken a pharmacogenomics approach to better understand variable response patterns to these therapies. Pharmacogenomics is the study of how genetic variations influence the response to drugs, through correlation of gene expression or single nucleotide polymorphisms with the efficacy and toxicity of the drug. 122 This concept has been applied to the field of oncology with great success: breast cancer with ErbB2/Her2 amplification and lung cancer with epidermal growth factor receptor (EGFR) mutations.123,124 Much like cancer, PAH is a heterogeneous disorder that appears morphologically similar, but likely has different and distinct genomic variations that may account for varying responsiveness to the same therapeutic agents.
Previously, gene microarray of peripheral blood mononuclear cells of 16 PAH patients and 5 controls identified a signature set of 106 genes that discriminated with high certainty (p ≤ 0.002) between patients with PAH and normal individuals. Supervised clustering analysis also generated a list of differentially expressed genes between patients with idiopathic and secondary causes of PH. 125 Further studies also identified inflammatory and immune-related genes including interleukin-7 receptor and chemokine receptor 7 as differentially expressed in patients with scleroderma-associated PH when compared with normal controls. 126 A proof of concept study evaluated two distinct animal models, Vasoactive Intestinal Peptide (VIP) gene -/- mice and monocrotaline-treated rats, and their response to VIP therapy. These two models had different gene expression alterations before and after therapy, as well as varying responses to treatment in both PAH features and gene expression. 127 In a study comparing patients with both vasodilator-responsive (VR) PAH and vasodilator-nonresponsive (VN) PAH, investigators utilized microarray of immortalized circulating lymphocytes to determine differentially regulated genes that could predict patient vasodilator response. The two principal genes that have been previously implicated by others in the pathobiology of PAH, DSG2 (involved in Wnt/β-catenin signaling and PVSMC growth) and RHOQ (encodes a cytoskeletal protein linked to insulin-mediated signaling), were found to account for the majority of variance in gene expression between the two groups. These results were validated in peripheral blood samples of a separate cohort. 128 Additionally, the relationship between genetic variation and clinical response to selective endothelin receptor (ET-A) antagonist sitaxentan has been evaluated. They identified a gene variant GNG2 that was associated with a significant increase in rate of achieving the predetermined composite endpoint of improved functional class and 6MWD, as well as an association with being homozygous for the minor allele T/T and earlier clinical benefit. 129 All of these findings are encouraging but await validation from further studies to fully realize the utility of using a pharmacogenomics approach in the treatment of PAH. Eventually, treatment selection and clinical trial inclusion may be based on individual patient gene expression patterns and associated targeted therapies.
Conclusion
This is an exciting time to be a clinician treating PAH. Over the course of the last 20 years, a multitude of new therapies have been developed to treat this deadly disease and have greatly improved survival. 130 In the last 3 years, there have been advances in the pre-existing vasodilatory therapies and design of drugs targeting novel pathways. The completion of long-term, event-driven trials provides both investigators and clinicians with robust data on the sustained benefit of these agents, that is likely more useful and applicable to the PAH patients currently being treated. The use of up-front combination therapy has the potential to alter disease progression, but further inquiry is needed in to the different combinations of drug classes prior to the widespread implementation of this treatment strategy. Importantly, IV epoprostenol remains the only PAH approved therapy to demonstrate mortality benefit to date. There is enormous potential for the development of therapeutics targeting mitochondrial dysfunction, altered inflammation/immunity, vascular remodeling, hormonal manipulation, and RV adaptation. As our knowledge and understanding of the underlying mechanisms affecting the pulmonary vasculature and RV in PAH grows, there is great potential for not only targeted but also individualized therapies for the heterogeneous group of patients with the diagnosis of PAH.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.