
Abstract
There is a worldwide epidemic of cardiovascular diseases causing not only a public health issue but also accounting for trillions of dollars of healthcare expenditure. Studies pertaining to epidemiology, pathophysiology, molecular biology, gene identification and genetic linkage maps have been able to lay a strong foundation for both the diagnosis and treatment of cardiovascular medicine. Although the concept of ‘epigenetics’ is not recent, the term in current usage is extended from the initial concept of ‘controlling developmental gene expression and signaling pathways in undifferentiated zygotes’ to include heritable changes to gene expression that are not from differences in the genetic code. The impact of epigenetics in cardiovascular disease is now emerging as an important regulatory key player at different levels from pathophysiology to therapeutics. This review focuses on the emerging role of epigenetics in major cardiovascular medicine specialties such as coronary artery disease, heart failure, cardiac hypertrophy and diabetes.
Keywords
Introduction
Cardiovascular disease (CVD) is considered to be the leading cause of death in all countries [Murray and Lopez, 1997], despite significant disparities related to socioeconomic strata and gender [The Lancet, 2013]. During the past century, there has been dramatic progress in the diagnosis, prevention and treatment in different fields of cardiovascular medicine [Nabel and Braunwald, 2012; Polonsky, 2012], which in turn reduced global and cause-specific mortality [Abi Khalil et al. 2012; Laribi et al. 2012; Nichols et al. 2013; Yeh et al. 2010]. However, current scientific knowledge does not completely explain the complex pathophysiology underlying CVD and other pathways are constantly looked for. Over the past decade, epigenetics has initiated a new era in genetic medicine capable of giving a different approach to human disease [Feinberg, 2010; Portela and Esteller, 2010]. Some international initiatives, including the Human Epigenome Project (HEP) and the International Human Epigenome Consortium (IHEC) have even been launched to catalogue the human epigenome and correlate its relation to pathophysiology [Abbott, 2010; Rakyan et al. 2004]. Epigenetics in CVD is the focus of this review.
Mechanisms of epigenetic imprinting
The term epigenetics refers to all the heritable changes in gene expression regulation other than nucleotide sequence and chromatin organization that depend on the DNA sequences itself [Egger et al. 2004; Rodenhiser and Mann, 2006]. Epigenetic inheritance is an essential mechanism that allows the stable propagation of gene activity states from one generation of cells to the next [Kelsey and Feil, 2013]. Epigenetic mechanisms have been involved in the differentiation of many cell types from progenitor or primary cells and with whom they share the same DNA sequence [Ji et al. 2010; Miao et al. 2008]. They also represent a stable cellular memory for the differentiation state of a cell population. The major epigenetic features of human cells include DNA methylation, post-translational histone modifications and RNA-based mechanisms including those controlled by small noncoding micro-RNAs [Wilson, 2008] (see Figure 1).
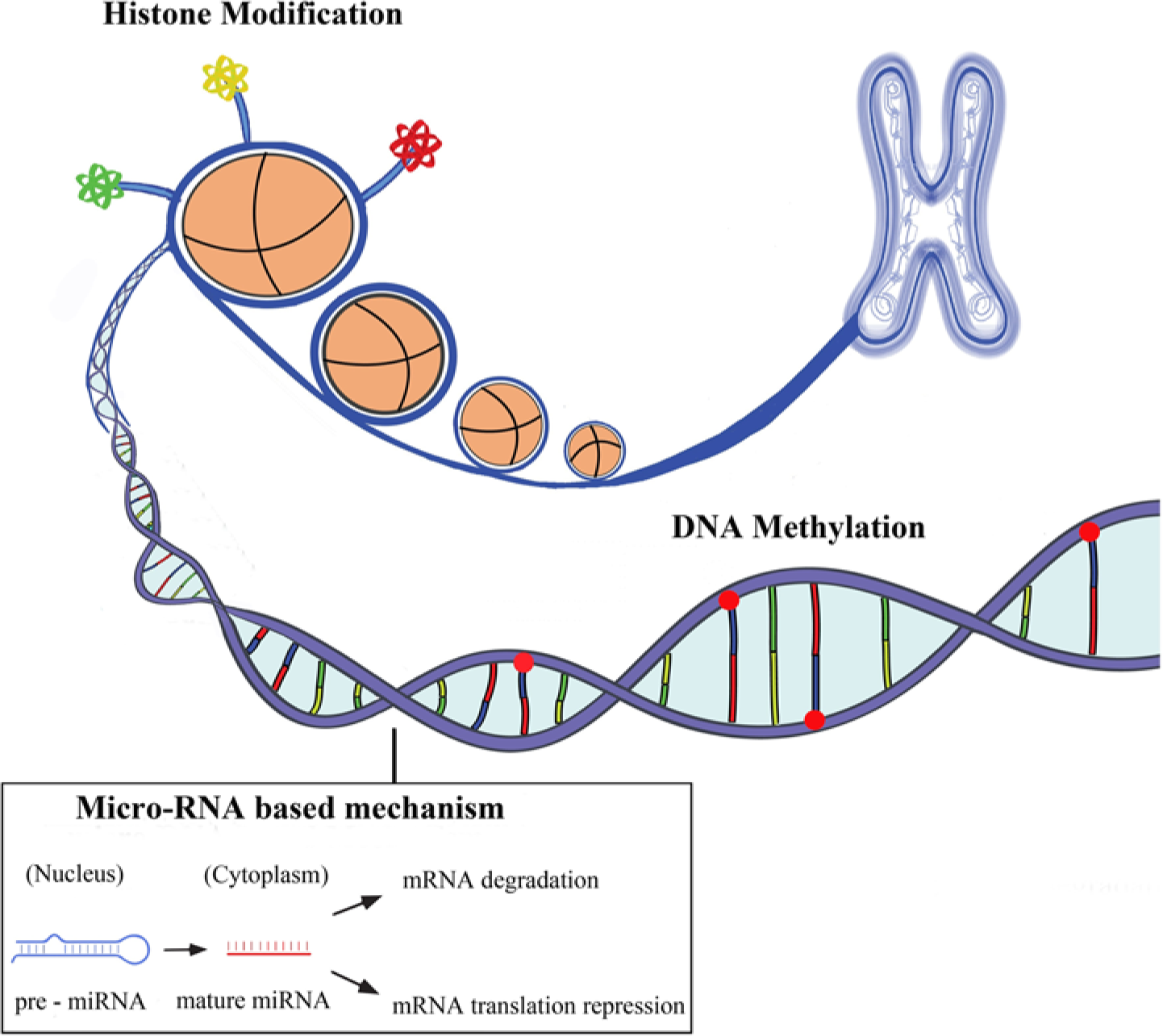
The three main areas of epigenetics are DNA methylation, histone modifications (acetylation, methylation, phospholylation, etc.) and micro-RNA based mechanisms. These three processes are distinct but are interrelated and control gene expression.
DNA methylation
This primarily happens at specific dinucleotide sites along the genome or at sites where methyl groups attach to cytosine bases followed by guanine (CpG islands). Other chemical modifications to cytosine resulting in hydroxymethylation, formylation and carboxylation also occur but their interrelation with methylation and their impact on epigenetics is not completely understood [Iurlaro et al. 2013]. In fact, 40% of genes contain CpG-rich islands and up to 70% of all CpG dinucleotides in the genome are methylated [Bird, 2002; Wilson, 2008]. Methylated CpGs act as docking sites for methyl binding proteins which have the ability to oligomerize through the DNA in order to recruit chromatin remodeling complexes that, in turn, cause chromatin condensation and gene inactivation and silencing [Ng et al. 1999; Nikitina et al. 2007; Suzuki and Bird, 2008]. Non-CpG island methylation has also been reported to influence protein–DNA interactions, gene expression and chromatin structure and stability [Fouse et al. 2010]. Non-CpG methylation was recently found to be prevalent in human embryonic stem cells [Lister et al. 2009; Ramsahoye et al. 2000] and in neurons [Guo et al. 2013].
Changes in DNA methylation patterns have been observed in association with CVD, inflammation, autoimmune diseases, infections and cancer [Bierne et al. 2012; Dang et al. 2013; Esteller, 2008].
Histone modification
Eukaryotic DNA is wrapped around an octamer of the core histones H2-A, H2-B, H3 and H4, thus building the fundamental unit of chromatin, the nucleosome [Berger, 2007]. These chromatin marks are unstable and they change rapidly in response to any external stimuli, and any permanent changes to DNA can lead to the development of defective organs or the development of a disease. Histones undergo a variety of post-translational modifications, mainly targeting amino acid residues of the N-terminal tails that protrude from the chromatin fiber [Natsume-Kitatani et al. 2011]. Among the different modifications, histone acetylation, methylation and phosphorylation are the most relevant ones to have been associated with transcription and gene expression [Li et al. 2007]. Post-translational modifications of histones also include their binding to specific proteins, called readers, which secondary interfere with chromatin function and mediate processes such as gene expression, apoptosis, and DNA damage repair [Jenuwein and Allis, 2001].
Micro-RNAs
Micro-RNAs (miRNAs) have recently emerged as regulators of gene expression that can simultaneously regulate genes in different biological networks. miRNAs have launched a new area in translational biology at a time when processing of a product was thought to be only by translation and transcription. These small (20–40 nucleotides) noncoding RNAs are highly conserved across species, highlighting the evolutionary importance of these molecules, as modulators of genes expression [Friedman et al. 2009]. More than 2500 miRNAs are currently identified in the human genome (http://www.mirbase.org/) and over 60% of all human genes are predicted to be regulated by these small RNAs (Zhang, 2008).Among miRNAs, many long-coding RNAs had already been identified years ago. Although initially neglected because of a false presumption of a lack of function, recent data show that they also enhance gene expression by modulating chromatin and organization of protein complexes across chromosomes [Geisler and Coller, 2013; Ørom and Shiekhattar, 2013].
Epigenetics in CVD
Epigenetics has been initially studied in patients with CVD for its prominent role in inflammation and vascular involvement [Castro et al. 2003; Stenvinkel et al. 2007]. Furthermore, epigenetic studies in cardiovascular medicine revealed a significant number of modifications affecting the development and progression of CVD. In addition, epigenomics are also involved in cardiovascular risk factors such as smoking [Breitling et al. 2011, 2012; Buro-Auriemma et al. 2013], diabetes (see below), hypertension [Rivière et al. 2011; Smolarek et al. 2010] and age [Fuke et al. 2004].
Epigenetics in heart disease
Cardiac hypertrophy
In experimental models of epigenetics, Cardiac Hypertrophy (CH) has been linked with histone acetylation implicating both histone acetyltransferases (HATs) and histone deacetylases (HDACs). The activity of HATs seems to have a positive role in CH as exemplified by the HAT activity of the transcriptional co-activators CREB binding protein (CBP) and p300. Overexpression of CBP or p300 in cardiomyocytes resulted in hypertrophy, whereas overexpression of mutant CBP and p300 lacking HAT activity did not [Gusterson et al. 2003]. However, the activity of HDACs has been reported in both prohypertrophic and antihypertrophic pathways, resulting in conflicting data. Class IIa HDACs have been shown to suppress CH whereas some animals and in vivo models showed that HDAC inhibitors have a protective role against hypertrophy [Antos et al. 2003; Kee et al. 2006; Kook et al. 2003]. CH has also been linked with histone methylation, in particular with H3K9 [Haddad et al. 2003; Majumdar et al. 2008; Zhang et al. 2011] and H3K4 methylations [Bingham et al. 2007; Stein et al. 2011] in animal models. In a different experimental mice model, hypermethylation associated to hydroxymethylation of epidermal growth factor receptor (EGFR) gene was associated with aortic valve calcification and subsequent ventricular hypertrophy [Barrick et al. 2009]. Parallel to histone modifications, miRNAs are also involved in CH. In neonatal cardiomyocytes, overexpression of miR-23a, miR-23b, miR-24, miR-195 or miR-214 induced CH whereas overexpression of miR-133 inhibited the phenotype [Carè et al. 2007; Latronico and Condorelli, 2009].
Heart failure
Heart failure (HF) results from a complex genetic predisposition and multiple environmental factors. In a series of patients with HF, Movassagh and colleagues reported that three angiogenesis-related genes were differentially methylated, irrespective of the etiology: angiomotin-like 2 gene (AMOTL2) was hypomethylated whereas the 5′ promoter region in the platelet/endothelial cell adhesion molecule gene (PECAM1) and the gene body of Rho GTPase-activating protein 24 (ARHGAP24) were hypermethylated [Movassagh et al. 2010]. The altered epigenomics of the three genes in endstage HF may reflect common epigenetic pathways in heart remodeling and vasculature [Movassagh et al. 2011]. Dilated cardiomyopathy represents about one in three cases of HF. Interestingly, Nguyen and colleagues reported in a mouse model that cardiac specific knockout of Dot1L, the gene encoding for the HKMT Dot1L which catalyzes H3K79 methylation in mammals, caused the appearance of a phenotype similar to dilated cardiomyopathy [Nguyen et al. 2011]. Histone modifications are also involved in HF; in a genome-wide histone methylation of heart tissues, Kaneda and colleagues reported that tri-methylated histone H3H4 and H3K9 were altered in HF [Kaneda et al. 2009]. Micro-RNAs have a particular importance in epigenetics of HF as shown in experimental models highlighting their role in driving gene expression change during the course of the disease [Ikeda et al. 2007; Latronico and Condorelli, 2009].
Arrhythmias
The contribution of epigenetics to heart rhythm disorders has recently been highlighted in common pathologies. Atrial fibrillation is the most prevalent arrhythmia in aging populations. In transgenic mice programmed to develop cardiac hypertrophy, Liu and colleagues showed that an injection of a specific HDAC inhibitor reverses atrial fibrosis and diminishes atrial fibrillation vulnerability following an electrical stimulation [Liu et al. 2008]. In other models, deletion of both HDAC-1 and HDAC-2 from the myocardium resulted in early rodent death by fetal arrhythmia [Montgomery et al. 2007]. Among the complex genetic background of the long QT syndrome, it is thought that a mutation interrupting the CpG island would prevent methylation and silence the paternal allele of the KCNQ1 gene in cardiac tissue [Bokil et al. 2010]. Micro-RNAs are also emerging as regulators of the normal electrophysiology of the heart and could also be implicated in arrhythmias [Kim, 2013]. Among the miRNAs that have been widely explored in experimental models, miR-1 was reported to be essential in normal electrophysiological conduction and its deletion was associated with high rate of sudden death [Zhao et al. 2007]. Overexpression of miR-208a is also associated with arrhythmia and heart death [Oliveira-Carvalho et al. 2013] whereas increase in miR-133a leads to prolonged QT intervals. Several other miRNAs such as the miR-212, miR-17-92, miR-155, miR-181 and miR-181a have been associated with regulation of heart rhythm through regulation of ion channels, transporters and cellular proteins [Kim, 2013].
Epigenetics in vascular diseases
Atherosclerosis and artery disease
Both DNA methylation, miRNAs and epigenetic mechanisms have been described in atherosclerosis. The era of DNA methylation in atherosclerosis was launched initially by two different groups who showed coexisting hypomethylation with atherosclerotic lesions in mice and rabbits [Hiltunen et al. 2002; Laukkanen et al. 1999]. Further studies showed that hypomethylation could even be detected at very early stages of atherosclerosis, even before the appearance of the anatomical manifestation of the disease [Lund et al. 2004]. In human atherosclerosis, hypermethylation of atheroprotective estrogen receptor α (ESR1) and estrogen receptor β (ESR2) in vascular smooth muscle cells were described by two independent groups [Kim et al. 2007; Post et al. 1999]. In cultured tissues of aorta and coronary arteries with varying degrees of atherosclerosis, Zhu and colleagues showed that methylation of the monocarboxylate transporter (MCT3) gene suppresses its transcription and hence allows the passage of smooth muscle cells and the progression of atherosclerosis [Zhu et al. 2005].
The contribution of miRNAs to atherosclerosis has been extensively investigated in the past few years. In fact, miRNAs are key players in programming and modulating gene expression of relevant cell types involved in atherosclerosis. They mediate inflammation, cholesterol influx, cellular differentiation and lipid uptake (Table 1).
Micro-RNAs involved in atherosclerosis.

miRNA, micro-RNA.
Clinical studies are aligned to experimental ones. In a follow up of elderly subjects, Baccarelli and colleagues showed that hypomethylation of long interspersed nucleotide elements was associated with higher incidence of ischemic heart disease events [Baccarelli et al. 2010]. Friso and colleagues showed that hypomethylation of the promoter of coagulation factor VII was associated with coronary artery disease in patients with angiographically proven coronary lesions [Friso et al. 2012].
Diabetic vascular disease
Chronic exposure to hyperglycemia induces specific chromatin changes and transcriptional responses, indicating epigenetic events that mediate signaling pathways relevant to diabetic CVD [El-Osta, 2012]. Genome-wide analysis of human vascular cells showed that histone modifications and DNA methylation occur simultaneously during hyperglycemic conditions and predict gene expression [Pirola et al. 2011].
Cell culture studies allow a direct comprehension of epigenetics and gene expression. The transcriptional co-activator PPARGC1A coordinates gene expression, which in its turn stimulates secretion of mitochondrial reactive oxygen species in different tissues in humans [Puigserver and Spiegelman, 2003]. In pancreatic β-cells of patients with type 2 diabetes (T2D), the PPARGC1A promoter is more methylated compared with nondiabetic individuals [Ling et al. 2008]. As a result, its expression is reduced, which confirms the theory that epigenomics have a critical part in regulating gene expression. In addition, Mutsktov and colleagues showed that insulin gene in human islet-derived precursor cells displayed high levels of histone modifications (H4 hyperacetylation and dimethylation of H3 lysine 4), which are typical of active genes [Mutskov et al. 2007].
It is already known that inflammation is part of the complex biochemical process of initiating and further developing cardiovascular complications of diabetes. Exposure to hyperglycemia could induce epigenomic changes in inflammatory pathways. The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a protein complex that controls DNA transcription and regulation of the activity of genes implicated in inflammation and atherothrombosis [Miao et al. 2004]. In individuals with T2D, chronic uncontrolled hyperglycemia enhances NF-κB activity in white blood cells, therefore affecting the activity of cytokines [Shanmugam et al. 2003]. HATs that acetylate lysine amino acids by transferring an acetyl group from acetyl CoA to form e-N-acetyl lysine on histone proteins interact with NF-κB, which causes hyperacetylation of inflammation-related genes promoters such as tumor necrosis factor-α (TNF-α) and cyclooxygenase-2 [Bird, 2007; Hofmann et al. 1998]. Other histone transferases that work in a similar way to HATs could also interfere in the regulation of pro-inflammatory genes by recruiting NF-kB to gene promoters. In animal models, such as the ones done on diabetic db/db mice, Reddy and colleagues reported that a hyperglycemic milieu enhances the expression of inflammation-related genes and precipitates pro-atherogenic responses in smooth muscle cells of the vascular system [Reddy et al. 2008]. This was also confirmed by Villeneuve and colleagues in a similar model [Villeneuve et al. 2008]. They reported that the expression of inflammatory genes in vascular cells cultured in high glucose was increased whereas that of H3K9me3, a histone H3 known to protect against the biochemical state of diabetic inflammation, were decreased at their promoters. Most importantly, El-Osta and colleagues reported that even a transient exposure to high levels of glucose induced sustained epigenomic changes in the p65 subunit promoter of the NF-κB, modifying thereafter the epigenomics of cultured aortic endothelial cells [El-Osta et al. 2008]. That might explain, at least partially, the irreversibility of cardiovascular complications of long-standing uncontrolled diabetes often encountered in clinical care. Finally, MicroRNAs are also implicated in the pathophysiology of diabetes and its complications and could even be potential pharmacological targets [Mao et al. 2013].
Conclusion
Epigenetics is likely to be involved in the biology of major disciplines of cardiovascular medicine. To date, there are no significant reports of epigenetics contribution in clinical practice or therapeutics in CVD whereas in cancer, DNA methylation inhibitors, histone methylation inhibitors and histone deacetylase inhibitors have been successful so far [Gal-Yam et al. 2008]. Understanding the epigenetic mechanisms, their interactions and alterations using translational research approaches or large human cohorts promises to make a significant contribution to our understanding of CVDs.
Footnotes
Acknowledgements
The author would like to thank Marie-Joe Dib, BS for editorial and technical support.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author declares no conflict of interest in preparing this article.