
Abstract
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening thrombotic microangiopathy driven by severe ADAMTS13 deficiency with consequent ultra-large von Willebrand factor-mediated microthrombosis. Over the last two decades, understanding of the ADAMTS13–vWF axis has transformed management and made TTP a model for precision medicine. This narrative review (adult focus) synthesizes contemporary evidence on diagnosis, treatment, genetics, and emerging therapies in immune-mediated TTP. We summarize a pragmatic, biomarker-guided algorithm integrating early therapeutic plasma exchange and corticosteroids, upfront rituximab to suppress anti-ADAMTS13 autoimmunity, and caplacizumab to block vWF–platelet interactions; define escalation strategies for refractory disease (e.g., proteasome inhibition); and formalize treat-to-target de-escalation anchored to ADAMTS13 recovery. We emphasize the role of rapid ADAMTS13 assays for front-door triage, routine ADAMTS13 monitoring in remission, and preemptive rituximab at biochemical relapse to avert clinical recurrence. Precision diagnostics are extended by genetic markers (e.g., HLA associations) and emerging biomarkers that may refine relapse risk. Finally, we discuss near-term opportunities—point-of-care ADAMTS13 assays, longer-acting vWF-pathway inhibitors, and data-driven risk tools—that can further individualize timing and intensity of therapy. Collectively, a biomarker-first strategy offers a clear path to fewer exacerbations, fewer relapses, and more consistent outcomes.
Plain language summary
Thrombotic thrombocytopenic purpura (TTP) is a rare medical emergency where tiny blood clots form throughout the body, causing organ damage and dangerously low platelets. In the past, many people with TTP did not survive. Today, outcomes have dramatically improved because treatment is based on understanding the exact cause of the disease—a lack of the enzyme ADAMTS13. This approach is known as precision medicine. When TTP is suspected, treatment must be started immediately. The standard first step is plasma exchange, which removes harmful antibodies and replaces the missing enzyme. Corticosteroids help calm the immune system, and rituximab, a medicine that removes antibody-producing B-cells, reduces the chance of relapse. A newer drug called caplacizumab stops blood clots from forming by blocking von Willebrand factor. When combined, these treatments help platelets recover faster, lower the risk of complications, and improve survival. Long-term care focuses on preventing relapse. Even when patients feel well, TTP can return if ADAMTS13 levels fall again. Regular blood tests allow doctors to detect relapse early and treat it before symptoms appear. Giving rituximab during remission can restore enzyme levels and prevent repeat attacks. Research is moving quickly. Future therapies like recombinant ADAMTS13 and longer-acting clot-blocking drugs may reduce the need for plasma exchange and make treatment more personalized, safer, and easier to access. Genetic and biomarker testing may also help identify which patients are most likely to relapse. Overall, TTP is now a condition where most patients survive and live well when treatment is rapid, targeted, and guided by biomarkers. Precision medicine continues to expand options and improve long-term outcomes.
Keywords
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a rare, life-threatening thrombotic microangiopathy (TMA) characterized by microangiopathic hemolytic anemia (MAHA), severe thrombocytopenia, and widespread microvascular thromboses.1,2 The underlying cause is a severe deficiency of ADAMTS13, which is a metalloprotease responsible for cleaving ultra-large von Willebrand factor (vWF) multimers. In the absence of treatment, acute TTP is highly fatal. Historically, mortality exceeded 90%, but the utilization of plasma exchange and other therapies has transformed TTP into a mostly survivable condition (now >90% 30-day survival with appropriate treatment).3,4 This dramatic improvement is largely due to precision medicine approaches, that is, leveraging the specific molecular mechanism of TTP to develop targeted therapies that target ADAMTS13 and anti-ADAMTS13 antibodies. 1 This narrative review focuses on adult TTP (primarily the immune-mediated form) and discusses how precision medicine has advanced its management through understanding the pathophysiology of TTP, current treatment strategies (plasma exchange and immunosuppression), as well as emerging targeted therapies (e.g., caplacizumab and recombinant ADAMTS13). It also reveals an algorithmic approach that can guide the decision-making process of when to initiate treatment, which agents to utilize, and how to prevent relapse.
Methods
Relevant literature was identified through searching major electronic databases, PubMed, Web of Science, and Google Scholar, focusing on studies published between January 2000 and October 2025. The search employed key terms such as “TTP,” “precision medicine,” “ADAMTS13,” “biomarkers,” and “targeted therapy,” along with related concepts to capture the breadth of recent research. Thematic grouping was used to present insights into core domains: diagnostics, therapeutic strategies, relapse prevention, and future directions in precision medicine for TTP. The review was written and structured in accordance with published guidance for narrative literature reviews, with an explicit description of search approach and thematic synthesis (Green et al. 5 ).
Pathophysiology and diagnosis of TTP
TTP results from an extreme deficiency of ADAMTS13 activity (<10% of normal), leading to accumulation of ultra-large vWF multimers that spontaneously bind platelets and form microthrombi. In immune-mediated TTP (iTTP), which accounts for most adult cases, ADAMTS13 is inhibited by autoantibodies (most commonly IgG, but IgA/IgM have also been reported) and/or immune complexes. In contrast, congenital TTP (cTTP; Upshaw–Schulman syndrome) is caused by biallelic pathogenic variants in the ADAMTS13 gene, leading to inherited severe enzyme deficiency. cTTP may present at any age, and adult presentations (including during pregnancy or other physiologic stress) can be fulminant rather than “mild.”3,4 iTTP is also known historically as Moschcowitz syndrome.
Clinically, patients present with thrombocytopenia and MAHA, often accompanied by organ ischemia (classically affecting the brain, heart, and kidneys) due to microthrombi. Because untreated TTP can rapidly be fatal, prompt diagnosis is critical. The definitive diagnostic test is an ADAMTS13 activity assay – a level <10% is both highly sensitive and specific for TTP.6–8 Obtaining ADAMTS13 results can take days in some settings, so clinicians rely initially on clinical judgment and scoring systems (such as the PLASMIC score) to identify probable TTP and start treatment empirically. A high PLASMIC score or French TMA score indicates a high likelihood of severe ADAMTS13 deficiency, warranting immediate therapy. If the pretest probability is intermediate, expert opinion favors treating for TTP rather than delaying therapy, given the life-threatening nature.9,10 In recent years, rapid ADAMTS13 assays with turnaround times under 1 h are being developed, which may soon allow more immediate confirmation of the diagnosis and help avoid unnecessary plasma exchange in mimicking conditions. Once available, these point-of-care assays will be a major advance in the precision diagnosis of TTP.11,12
From a molecular perspective, the understanding of TTP’s mechanism has revealed genetic risk factors that may inform precision medicine. Certain HLA class II variants have been associated with increased susceptibility to immune TTP and higher relapse risk; for example, the HLA-DR/DQ locus tagged by rs6903608 has been reported as a risk marker in White/European ancestry cohorts. Additional HLA (Human Leukocyte Antigen) associations have been described in Japanese patients (Sakai et al. 13 ). Population studies also show a higher incidence in females and in individuals of African ancestry, likely reflecting a combination of host genetics, immune response genes, and environmental triggers. 2 These insights reinforce that while iTTP is defined by an immune anti-ADAMTS13 response, host genetics can influence disease susceptibility and may ultimately support risk stratification. 14
Current treatment strategies in iTTP
Management of acute immune TTP revolves around three complementary strategies: (1) replenishing the missing ADAMTS13 through therapeutic plasma exchange (TPE), (2) halting ongoing microthrombosis through administration of caplacizumab, and (3) suppressing the pathogenic autoimmunity through immunosuppressive therapy such as corticosteroids or rituximab. The standard of care has evolved to incorporate all three approaches in a precise, targeted manner, drastically improving patient outcomes. For an overview of these precision Medicine Therapies for Immune Thrombotic Thrombocytopenic, see Table 1.
Precision medicine therapies for immune thrombotic thrombocytopenic purpura.

iTTP, immune thrombotic thrombocytopenic purpura; TPE, therapeutic plasma exchange; vWF, von Willebrand factor.
TPE and supportive care
TPE with plasma replacement has historically been the backbone of acute iTTP treatment and was the first intervention to significantly improve survival. 15 TPE removes circulating anti-ADAMTS13 autoantibodies and ultra-large vWF multimers while simultaneously replenishing ADAMTS13 and physiologic plasma proteins, including vWF of normal multimer composition. In most settings, TPE is initiated immediately when iTTP is strongly suspected (particularly if rapid ADAMTS13 testing is unavailable), alongside corticosteroids. However, in experienced centers with rapid ADAMTS13 testing and close monitoring, TPE-sparing strategies using caplacizumab plus immunosuppression have been reported in selected patients. 16 TPE carries procedure-related risks including allergic reactions to plasma, citrate-related hypocalcemia, and central venous catheter complications (bleeding, thrombosis, and infection) that should be discussed and mitigated.
Together, TPE plus corticosteroids can induce clinical response in most patients. In the caplacizumab era, outcome definitions have been refined to distinguish early disease control from immune remission. An international working group consensus defined key outcomes including clinical response, exacerbation (recurrence within 3 months after stopping TPE), and relapse (recurrence after 3 months). 17 Historically, 10%–20% of patients experienced early exacerbation during TPE taper or hematologic refractoriness (failure to achieve platelet recovery despite ongoing TPE). Importantly, pivotal trials of caplacizumab and recombinant ADAMTS13 enrolled patients upfront as adjuncts to standard therapy rather than restricting to only high-risk or refractory subgroups.18,19,20
Rituximab and immunosuppressive therapy
Because iTTP is driven by pathogenic B-cell production of anti-ADAMTS13 antibodies, targeting the immune root of the disease is a precision strategy to induce durable remission. Rituximab, a monoclonal antibody against CD20 on B-lymphocytes, has emerged as an effective immunomodulatory treatment in TTP. It is often added to front-line therapy or used in early relapse. Rituximab helps eliminate the autoantibody-producing B-cells, thereby addressing the cause of the ADAMTS13 deficiency. Although rituximab was initially introduced in refractory cases, multiple studies and a recent meta-analysis have demonstrated that using rituximab during the initial TTP episode can significantly reduce the rate of relapses compared to plasma exchange and steroids alone. In fact, patients who receive rituximab as part of first-line therapy have shown fewer relapses and longer remission periods.21,22 Based on this evidence, many experts now incorporate rituximab early (e.g., weekly infusions for four doses) in adult iTTP, even though it is an off-label use, because it improves outcomes by precisely targeting the antibody-mediated pathogenesis.
Beyond steroids and rituximab, additional immunomodulatory therapies have been used as salvage, particularly for refractory autoimmunity (persistent anti-ADAMTS13 inhibitor despite adequate first-line therapy) rather than hematologic refractoriness alone. Proteasome inhibition with bortezomib has supporting case series and systematic reviews in refractory iTTP.23,24 More recently, anti-CD38 therapy (daratumumab) has been reported for severe, multi-relapsing or rituximab-refractory iTTP with promising responses, consistent with a plasma-cell directed strategy. 25 Obinutuzumab (a type II anti-CD20 antibody) has also been reported as an alternative for patients with rituximab resistance or intolerance. 26 These agents are not routine first-line therapy but may be considered in selected refractory cases in expert centers.
Overall, the use of immunosuppressive therapy in TTP is a prime example of precision medicine: by identifying that anti-ADAMTS13 antibodies are the culprit, therapies like rituximab (B-cell depletion) or bortezomib (plasma-cell ablation) directly target the source of disease, thus improving remission maintenance and a reduction in TTP relapses.21–24
Caplacizumab: Targeted vWF inhibition
Perhaps the most transformative advance in iTTP therapy has been the development of caplacizumab, a nanobody targeting the vWF A1 domain. By blocking the vWF–platelet interaction, caplacizumab rapidly controls ultra-large vWF-mediated microthrombosis and reduces early ischemic complications.18,19 Caplacizumab entered clinical practice in 2018, and randomized trials (TITAN and HERCULES) demonstrated faster platelet recovery and fewer exacerbations compared with placebo when added to standard therapy. Consistent with consensus outcomes, caplacizumab primarily addresses the early goal of clinical response, while immunosuppression targets the longer-term goal of ADAMTS13 remission. 17 Administration is intravenous for the first dose, followed by daily subcutaneous dosing. Treatment duration should be individualized using serial ADAMTS13 activity (treat-to-target): caplacizumab is typically continued until ADAMTS13 recovery to reduce the risk of early exacerbation, with ADAMTS13-guided stopping/extension strategies supported by cohort data. 28 Because caplacizumab induces a functional (acquired) vWF deficiency, mucocutaneous bleeding is more common, and bleeding risk should be monitored and balanced against thrombotic risk.18,19
TPE-free acute management has evolved from isolated case reports to a more systematically described approach in selected patients. A large multicenter retrospective cohort from Germany and Austria reported outcomes with caplacizumab and immunosuppression without routine TPE in carefully selected iTTP patients, with rescue TPE used when needed. 16 In addition, the MAYARI study has been completed (results presented at ISTH 2025; publication pending), further supporting the feasibility of caplacizumab-based regimens without upfront TPE in expert settings. 29
The impact of caplacizumab is evident in both clinical trials and real-world data. For example, an international study reported that caplacizumab-treated patients required fewer plasma exchange sessions and had dramatically lower relapse rates in the short term than historical controls. In one multicenter cohort, only 9% of patients who received caplacizumab experienced a TTP recurrence after their acute episode, versus 62% of patients in a pre-caplacizumab era control group. This highlights that caplacizumab not only accelerates recovery but also protects against early relapse while the drug is being administered. Notably, the HERCULES trial design involved using caplacizumab for 30 days after the last plasma exchange; some patients in the caplacizumab arm did relapse shortly after the drug was stopped, especially if their ADAMTS13 had not yet recovered.19,30 This finding taught clinicians that while caplacizumab is highly effective at preventing microthrombi, it does not address the underlying autoimmunity; therefore, discontinuing it before autoantibodies are eliminated can lead to recurrence. The current practice to maximize precision is to continue caplacizumab until ADAMTS13 activity substantially recovers (e.g., >20% or even into normal range). This individualized duration, guided by the patient’s own biomarker recovery, has proven to minimize relapses as indicated by the low relapse rates when ADAMTS13-guided dosing was used. 30
While caplacizumab offers clear benefits, it does come with considerations. The drug’s mechanism of inhibiting platelet adhesion inherently increases the risk of bleeding. In fact, several trials noted that mucocutaneous bleeding events (e.g., epistaxis and gingival bleeding) were more frequent in caplacizumab-treated patients than in placebo-treated patients, although major bleeding was uncommon. Clinicians must be vigilant about bleeding risk, especially if patients have to undergo invasive procedures while on caplacizumab. Another important consideration is cost: caplacizumab is an expensive biologic therapy. A cost-effectiveness analysis in 2020 suggested that routinely adding caplacizumab for all iTTP patients might not be cost-effective in some healthcare systems at its current price. 19 This has led to some debate on whether caplacizumab should be given empirically to every TTP patient or reserved for those with the most severe presentations or risk factors. Nevertheless, most experts advocate its use in all confirmed iTTP cases, given the high stakes of the disease and the substantially improved outcomes. In summary, caplacizumab exemplifies precision medicine by focusing on a critical molecular interaction (vWF–platelet binding) in TTP’s pathogenesis, thus offering a targeted way to protect organs from ongoing ischemic damage while other therapies take effect.
Long-term management and relapse prevention
Surviving an acute episode of TTP is only part of the journey—patients require careful long-term follow-up to ensure durable remission. A striking feature of iTTP is its tendency to relapse: even after an initial episode is successfully treated, the autoantibody can recur months or years later, precipitating another TTP crisis.31,32 Precision medicine has a role here by identifying biomarkers that predict relapse and guiding preemptive therapy. The most important predictor of relapse is the ADAMTS13 activity during remission. Many survivors have their ADAMTS13 activity monitored regularly (e.g., monthly for the first 6 months, then every 3–6 months for a period, and eventually at least annually for life).31–34 Studies have shown that if a patient’s ADAMTS13 activity becomes severely deficient again (<10%–15%) while they are clinically well, a relapse is almost inevitable without intervention. Even a drop to <20% has been associated with a high risk of impending relapse. Therefore, routine ADAMTS13 monitoring is an essential component of personalized care in TTP—it can detect subclinical recurrence of the autoantibodies before the patient has overt TTP.31,33
If a significant decline in ADAMTS13 is detected (for instance, two readings <20% or a confirmed <10% in an asymptomatic patient), clinicians will often initiate preemptive therapy to abort a full-blown relapse. Typically, this involves giving rituximab in the remission phase. Prophylactic rituximab has been shown to raise ADAMTS13 activity back to normal in approximately 80% of such patients, thereby averting a clinical relapse in those cases. A single rituximab infusion (or a short course) can sometimes suffice to durably restore ADAMTS13 if given at the right time. Evidence for the efficacy of preemptive rituximab comes from multiple cohorts where it significantly reduced relapse rates compared to observation.21,22 Some patients who do not adequately respond to rituximab (or who have contraindications) have been maintained on low-dose cyclosporine immunosuppression to prevent relapses, or in very refractory cases, splenectomy has been performed with success as mentioned earlier. 27 The goal is always to keep the patient’s ADAMTS13 above the danger threshold so that TTP does not clinically recur.
In addition to ADAMTS13 activity, ADAMTS13 conformation has emerged as a promising precision biomarker for relapse risk. An “open” ADAMTS13 conformation reflects ongoing anti-ADAMTS13 immune activity and has been associated with subsequent clinical relapse in cohort studies, including work from the Vanhoorelbeke group.35,36 By contrast, evidence for broad multi-marker panels (e.g., complement markers) remains preliminary and should be interpreted cautiously; at present, ADAMTS13 activity monitoring remains the cornerstone of relapse surveillance.31,32
Psychologically and neurologically, many TTP survivors have persistent issues (e.g., mild cognitive impairment and depression), which also require long-term management, though those aspects are beyond the scope of this review. It’s worth noting that addressing the chronic consequences of TTP (some of which may relate to microvascular damage sustained during acute episodes) is an emerging field.32,34 As precision treatments reduce acute mortality and relapses, attention is turning to improving quality of life for survivors—an area for future multidisciplinary research.
Adult iTTP management algorithm (biomarker-guided)
Based on the aforementioned information, we have created a pragmatic, biomarker-guided algorithm for adult immune-mediated TTP that aligns bedside actions with ADAMTS13 activity, caplacizumab timing, and preemptive rituximab triggers. The flowchart shows an approach to patients with high clinical suspicion of TTP, early interventions and targeted therapy, refractory pathways, treat-to-target de-escalation as well as remission surveillance and relapse prevention. See Figure 1 for the full algorithm.
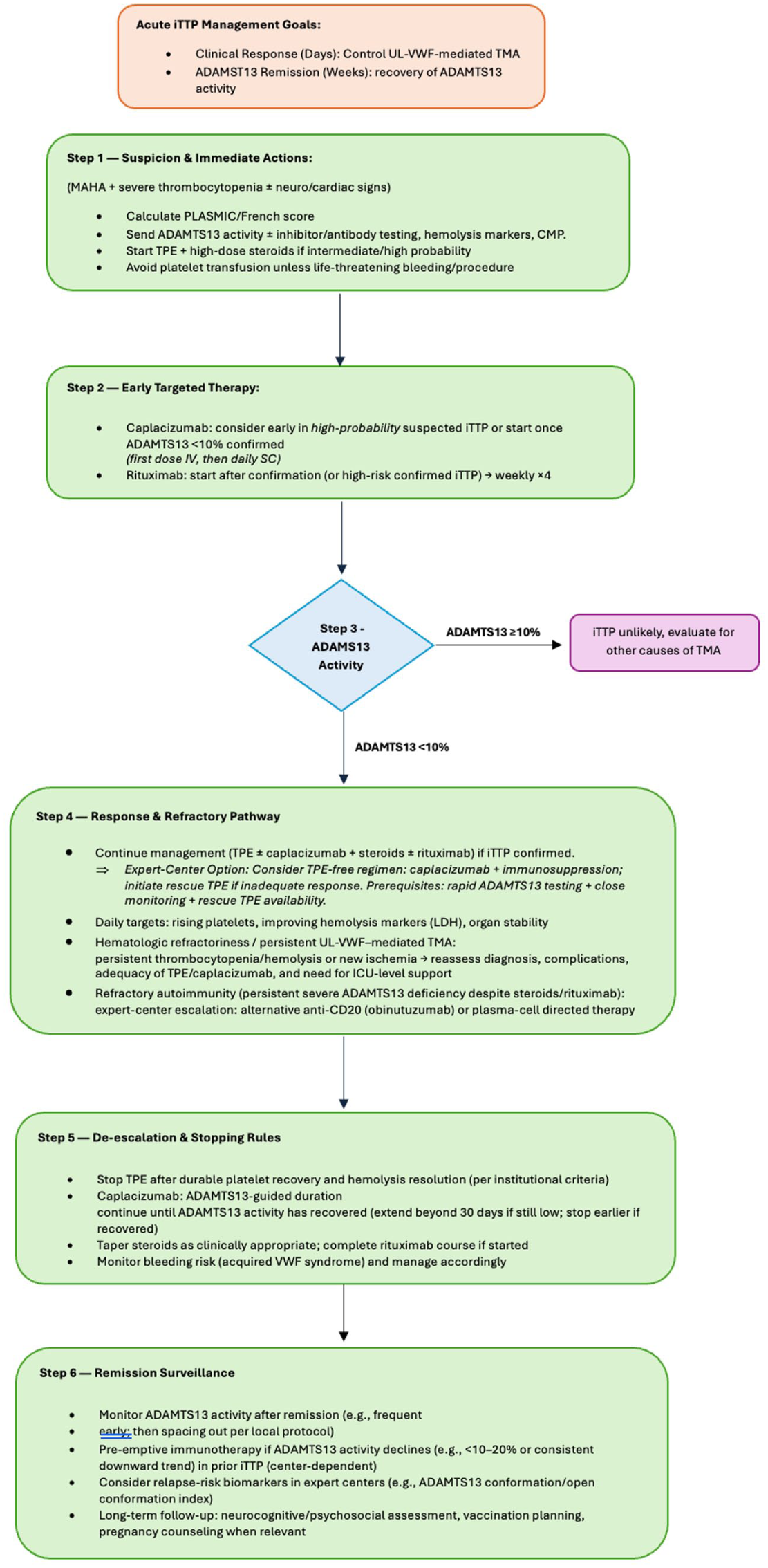
Precision-informed algorithm for iTTP. Management is guided by clinical probability and urgent ADAMTS13 activity testing. ADAMTS13 activity <10% supports iTTP and triggers iTTP-directed therapy with ADAMTS13-guided caplacizumab duration; ⩾10% prompts evaluation for alternative TMAs. The algorithm distinguishes hematologic refractoriness from refractory autoimmunity and includes an expert-center TPE-free option requiring close monitoring and rescue TPE capability.
Emerging and future therapies
The landscape of TTP treatment continues to evolve rapidly, with several promising therapies on the horizon that further exemplify precision medicine. One major area of development is novel agents to replace or augment ADAMTS13. As discussed, recombinant ADAMTS13 is in trials and could become a key therapy not just for cTTP but possibly as an adjunct in immune TTP. The idea would be to give rADAMTS13 to acutely restore enzyme activity (instead of plasma exchange) while concurrently using immunosuppression to handle the autoantibodies. Early-phase research demonstrated that recombinant ADAMTS13 can function in the presence of inhibitors if given in high enough doses. This could revolutionize acute treatment by providing a standardized, pathogen-free enzyme replacement (avoiding the logistical challenges of plasma). However, the results of ongoing trials to determine if this approach is effective and safe in practice are yet to be revealed.37,38
Another line of research is extending the concept of vWF-pathway inhibition. Caplacizumab has proven the validity of targeting the vWF–platelet interaction; now, scientists are working on longer-acting vWF inhibitors. One example is an aptamer called BT200, which binds vWF and has shown the ability to inhibit vWF in preclinical models. 30 Unlike caplacizumab (which requires daily injections), an extended half-life molecule could reduce dosing frequency. The ultimate goal would be a convenient agent that any hospital can quickly administer to TTP patients to immediately safeguard against microthrombosis.
On the immunosuppression front, there is interest in therapies that more specifically target plasma cells (which rituximab does not eliminate) or the autoantibodies themselves. Bortezomib is one such agent already used in refractory cases; future trials might formally evaluate its use earlier in the course for certain high-risk patients (for instance, those who still have high inhibitor levels after standard therapy).24–27 Anti-B-cell activating factor or other novel autoimmune drugs could potentially find a role if TTP is linked with other autoimmune features. 27 There’s also ongoing research into tolerization strategies—for example, giving small amounts of ADAMTS13 (when recombinant form is available) to retrain the immune system to accept it, akin to allergy desensitization.20,37,39 This is speculative but rooted in the precision idea of addressing the exact immune trigger.
An intriguing concept that could be explored is whether TTP could one day be managed without plasma exchange altogether. Currently, plasma exchange is mandatory in most cases, however, it can be considered that, in the availability of a rapid diagnostic test confirming TTP within minutes, an immediate injection of a vWF inhibitor (caplacizumab or successor) to stop new thrombosis; a dose of recombinant ADAMTS13 to correct the enzyme deficit; and potent immunotherapy (rituximab or even newer agents) to begin eliminating antibody production, thus rendering plasma exchange unnecessary. There have been case reports hinting at this future—for example, a patient who could not receive plasma (due to religious objections or allergy) was successfully treated with caplacizumab plus immunosuppressive therapy alone. That patient recovered from TTP without TPE by using targeted treatments to cover each aspect of the disease process.40,41 While this is not yet standard and plasma exchange remains the backbone, it suggests that as precision therapies advance, we may rely less on nonspecific interventions like plasma exchange. This would be particularly beneficial in resource-limited settings or special circumstances (since plasma exchange is intensive and not without risk).
Finally, it’s important to consider that precision medicine is not only about drugs and genes, but also about patient stratification and personalized decisions. Ongoing efforts to refine risk stratification in TTP (who will relapse, who might need prolonged caplacizumab, who might tolerate tapering therapies sooner, etc.) are a form of precision medicine. With more data, we might develop algorithms that integrate clinical features, lab results (like ADAMTS13 kinetics), and genetic markers to tailor the aggressiveness of therapy to each patient. For instance, a patient whose ADAMTS13 autoantibody is particularly inhibiting might benefit from earlier addition of plasma-cell-targeted therapy; another patient with specific HLA markers might need longer immunosuppression to maintain remission. Machine learning models are even being explored to predict TTP outcomes using multi-parameter data—a modern extension of precision medicine.32–35
Implementing a precision medicine approach in under-resourced settings requires pragmatic adaptation. Key steps include early clinical risk stratification (e.g., PLASMIC score) while arranging centralized or referral-based ADAMTS13 testing, standardized protocols for timely initiation of TPE and corticosteroids when iTTP is suspected, and prioritized use of high-cost agents (such as caplacizumab) for patients with confirmed severe ADAMTS13 deficiency or high thrombotic risk. Regional laboratory networks, point-of-care assays as they become available, and shared-care follow-up models can help extend ADAMTS13-guided monitoring and preemptive immunosuppression to settings where access is limited.
In summary, the future of TTP management is likely to become even more individualized. New targeted therapies (like rADAMTS13 and next-generation vWF inhibitors) and refined monitoring will work hand in hand. The vision is that no patient should die or suffer irreversible complications from TTP—a goal that seems achievable given the trajectory from essentially 0% survival a century ago to >90% survival today. TTP’s story exemplifies how understanding the molecular basis of a disease can directly lead to life-saving interventions.
Conclusion
Precision medicine has fundamentally reshaped the approach to TTP. By pinpointing the key molecular defect in TTP—the ADAMTS13/vWF axis—researchers and clinicians have devised therapies that specifically target the disease mechanism, dramatically improving patient outcomes. What was once a nearly fatal disorder is now effectively treatable in most cases. Innovations such as caplacizumab (targeting vWF) and rituximab (targeting B-cells) were made possible by insights into TTP’s pathophysiology, and they showcase the power of translating research “from bench to bedside.” TTP can be viewed as a model for rare disease treatment development: within a span of roughly 20 years, the discovery of ADAMTS13 and its role led to multiple tailored therapies that turned TTP into an eminently manageable condition.
Moving forward, continued research and clinical vigilance are required. The therapies discussed need to be made accessible and used optimally (e.g., using ADAMTS13 activity to guide therapy duration is a current best practice). Future trials will determine the optimal combination of these treatments and whether some patients can be managed with less invasive approaches. Importantly, the principles learned from TTP—rapid diagnosis, targeted treatment, and individualized follow-up—can be applied to other thrombotic and hematologic diseases. For clinicians treating adult patients, it is crucial to stay updated, as guidelines for TTP are evolving with each new piece of evidence. In conclusion, precision medicine in TTP has already saved many lives and holds the promise of even better outcomes ahead. With emerging therapies like recombinant ADAMTS13 on the horizon and an ever-deepening understanding of the disease, we are moving toward an era where no TTP patient should die from this condition and where treatment can be tailored to ensure long-term remission for all.
Finally, immune reprogramming strategies are being explored for iTTP. Early translational concepts include CAR-T (Chimeric Antigen Receptor-T cell) or other cellular therapies targeting autoreactive B-cell/plasma-cell compartments, although clinical experience remains limited. 13
Footnotes
Acknowledgements
The authors would like to acknowledge the Hematology Department at the National Center of Cancer and Research Hospital at Hamad Medical Corporation for scientific support, and also acknowledge the Qatar National Library for funding the article.