
Abstract
Lymphoma occurring in the central nervous system is considered primary central nervous system lymphoma (PCNSL), usually without systematic lesions. Over the last few decades, a deep understanding of PCNSL has been lacking due to the low incidence rate, and the overall survival and progression-free survival of patients with PCNSL are lower than those with other types of non-Hodgkin lymphoma. Recently, there have been several advancements in research on PCNSL. Advances in diagnosis of the disease are primarily reflected in the promising diagnostic efficiency of novel biomarkers. Pathogenesis mainly involves abnormal activation of nuclear factor kappa-B signaling pathways, copy number variations, and DNA methylation. Novel therapies such as Bruton’s tyrosine kinase inhibitors, immunomodulatory drugs, immune checkpoint inhibitors, and phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors are being evaluated as possible treatment options for PCNSL, especially for relapsed/refractory (R/R) cases. Several clinical trials also indicated the promising feasibility and efficacy of chimeric antigen receptor T-cell therapy for selected R/R PCNSL patients. This review focuses on discussing recent updates, including the diagnosis, pathogenesis, and novel therapy of PCNSL.
Keywords
Introduction
Primary central nervous system lymphoma (PCNSL) refers to an extremely aggressive subtype of extranodal non-Hodgkin lymphoma (NHL) that is limited to the central nervous system (CNS), primarily affecting the brain tissue, meninges, eyes (vitreoretinal space), and spinal cord but in the absence of systemic involvement.1,2 PCNSL is a relatively rare oncology with an overall annual incidence of approximately 0.5/100,000 in the United States; the incidence is slightly higher in males than females, and it can occur at any age, most commonly in those over 60 years old. Immunodeficiency (use of immunosuppressive drugs, acquired immunodeficiency syndrome) is a well-known risk factor. PCNSL accounts for 4–6% of all extranodal NHLs and 4% of all newly diagnosed CNS malignancies.3,4 The clinical manifestations of PCNSL often lack specificity and are generally closely related to the size, location, number, and peripheral edema of the lymphoma lesions. Most patients present with focal neurological deficits, intracranial hypertension symptoms, and nonspecific cognitive and behavioral variations; systemic B symptoms such as fever, night sweats, and weight loss are rare. Primary vitreoretinal lymphoma (PVRL) is defined as lymphoma lesions that are localized in the vitreoretinal space and may have no obvious symptoms or only mild visual abnormalities, such as blurred vision, decreased vision, or floaters. 5 The pathological type is mostly diffuse large B-cell lymphoma (DLBCL), accounting for approximately 95%, with other types being rare. Newly diagnosed patients usually receive an induction chemotherapy strategy based on high-dose methotrexate (HD-MTX) in combination with other chemotherapy drugs that have the ability to cross the blood–brain barrier (BBB), followed by autologous hematopoietic stem cell transplantation (ASCT) or whole-brain radiotherapy (WBRT) for consolidation; however, the latter is typically restricted due to the associated delayed CNS dysfunction concern. 6 PCNSL is highly invasive and progresses rapidly, leading to poor clinical outcomes. Therefore, to further improve the understanding and optimize the diagnosis and treatment of this disease, this article focuses on discussing the diagnosis, pathogenesis, and therapeutic advances of PCNSL.
Diagnosis
Traditional process
When PCNSL is highly suspected, immediate brain imaging should be performed, which can noninvasively show the location, size, and shape of intracranial lesions and aid in disease diagnosis, staging, and treatment response assessment. Magnetic resonance imaging (MRI) (enhanced + diffusion-weighted imaging) is the preferred imaging modality for identifying the characteristic radiographic features. The lesions often have clear borders and predominantly appear as isolated lesions (60–70%) and less frequently as multifocal diseases, which are usually located around the ventricles and affect deep structures such as deep cerebral white matter, corpus callosum, or basal ganglia region, with mild or moderate peritumoral edema. T1-weighted images show equal or low signal intensity; T2-weighted images show equal or high signal intensity. The lesions mostly present with homogeneous and consistent enhancement, with a few displaying ring enhancement or heterogeneous enhancement. PCNSL is a highly malignant tumor with strong proliferation ability, restricted water molecule diffusion, and a reduced apparent diffusion coefficient, distinguishing it from other primary malignant brain tumors.7,8 If the patient cannot tolerate MRI examination, computed tomography (CT) imaging is considered an alternative option. Positron emission tomography (PET)/CT imaging is usually applied for initial diagnosis and suspicion of systemic recurrence. Cases suspected of having isolated involvement in the meninges or vitreoretinal space require lumbar puncture or ophthalmologic examinations for confirmation. Lumbar puncture is performed to obtain the patient’s cerebrospinal fluid (CSF) for cell counts, glucose and protein levels, cytology, flow cytometry, and immunoglobulin heavy chain rearrangement testing. Ophthalmologic examinations, including slit lamp and ophthalmoscope examinations, are used to evaluate vitreoretinal infiltration, and vitrectomy biopsy may be necessary. 6
Imaging examinations provide the basis for disease diagnosis, but pathological diagnosis remains the gold standard, including tissue histopathology and immunohistochemical staining. Tumor tissue specimens are obtained by stereotactic biopsy or surgical resection. Microscopically, highly malignant lymphoma cells surround the blood vessels in a cuff-like arrangement, showing a diffuse infiltrative growth pattern. The pathological type is predominantly DLBCL, with overrepresentation of the activated B-cell-like (ABC) subtype. It should be noted that use of corticosteroids can lead to false-negative results by reducing the size of lesions, thereby delaying disease diagnosis and treatment; thus, it is recommended to avoid using corticosteroids before biopsy to optimize diagnosis. 1 Once a patient is diagnosed with PCNSL, PET/CT and bone marrow biopsy should be conducted, and male patients should also undergo testicular ultrasound examination. The baseline evaluations for newly diagnosed PCNSL patients based on the International PCNSL Collaborative Group (IPCG) guidelines are summarized in Table 1.
Baseline evaluations of newly diagnosed PCNSL patients.

CSF, cerebrospinal fluid; ECOG, Eastern Cooperative Oncology Group; HIV, human immunodeficiency virus; IPGG, International Primary Central Nervous System Lymphoma Collaborative Group; KPS, Karnofsky Performance Scale; LDH, lactic dehydrogenase; MRI, magnetic resonance imaging; PET/CT, positron emission tomography/computer tomography; PCNSL, primary central nervous system lymphoma.
Novel diagnostic markers
In addition to classical imaging and pathological examinations, there are a series of novel diagnostic markers for PCNSL that can avoid invasive examination, and are particularly important for patients who have difficulty in undergoing biopsy or diagnosis using classical methods. The major ones include cell-free DNA (cfDNA)/circulating-tumor DNA (ctDNA), microRNA (miRNA), neopterin, interleukin 10 (IL-10), and C-X-C motif chemokine ligand 13 (CXCL13) (Table 2). Myeloid differentiation factor 88 (MYD88) L256P, as well as transmembrane activator and CAML interactor (TACI) in combination with B-cell activating factor of the TNF family (BAFF), have excellent diagnostic efficacy, with sensitivity and specificity being 100%, which can serve as adjunctive methods for diagnosing.
Several novel CSF diagnostic markers of PCNSL.

APRIL, A PRoliferation-Inducing Ligand; BAFF, B-cell activating factor of the TNF family; CSF, cerebrospinal fluid; CXCL13, C-X-C motif chemokine ligand 13; IL-10/6, interleukin 10/6; β2-MG, β2-microglobulins; MYD88, myeloid differentiation factor 88; PCNSL, primary central nervous system lymphoma; sIL-2R, soluble interleukin-2 receptor; TACI, transmembrane activator and CAML interactor.
Cell-free DNA/circulating-tumor DNA
Liquid biopsy refers to analysis of DNA fragments produced by tumor cell apoptosis, necrosis, or secretion. Currently, MYD88 and CD79b mutations in the CSF samples of PCNSL patients have been confirmed to have diagnostic efficacy. 20 A retrospective study showed that the detection rate of the MYD88 hotspot mutation (L265P) in the CSF of 14 PCNSL patients was 86%; three of these patients did not have malignant lymphoma cells detected by cytology and flow cytometry, but the MYD88 mutation was found in CSF ctDNA through droplet digital polymerase chain reaction (ddPCR) technology. 21 Another prospective clinical study found that MYD88 mutation was detected in 15 of 17 PCNSL biopsy specimens, with a matching rate of 82% in paired tissue-CSF samples. 11 Yamagishi et al. 9 assessed the MYD88 mutation status in the CSF of PCNSL patients using ddPCR technology and developed mutation standards (cutoff: target value/total value = 0.25%), and the sensitivity and specificity for diagnosing PCNSL were both 100%. This study indicated that MYD88 mutation in CSF cfDNA can be used for diagnosis of PCNSL with high accuracy and clinical applicability. In a phase Ib clinical trial of ibrutinib for relapsed/refractory (R/R) CNSL, high-frequency mutations in the B-cell receptor (BCR) signaling pathway genes, including MYD88 L265P and CD79b Y196, were detected in patient CSF ctDNA, and the trend of change was consistent with treatment response. 22 Therefore, MYD88 and CD79b mutations have a high detection rate in the CSF cfDNA and ctDNA of PCNSL patients and can serve as noninvasive diagnostic biomarkers for PCNSL. However, a recent study showed that the detection rate of MYD88 and CD79b mutations in the blood ctDNA of PCNSL patients was significantly lower than that of stereotactic biopsy, suggesting that blood ctDNA may lack relatively reliable diagnostic value. 23
Micro RNA
miRNA refers to noncoding RNA with a length of 18–24 nucleotides that directly binds to specific sequences in the 3′ untranslated region to inhibit gene expression and participate in a series of biological processes, such as cell communication, development, and differentiation. Certain miRNAs in body fluids can serve as biomarkers to distinguish PCNSL from other CNS diseases. 24 Compared with other CNS diseases, it has been reported that miR-19b, miR-21, and miR-92a are significantly elevated in the CSF of PCNSL patients; the combination of these three miRNAs has a sensitivity of 95.7% and a specificity of 96.7% for diagnosis of PCNSL. 13 Another study confirmed that levels of miR-19b, miR-21, and miR-92a in the CSF of CNS DLBCL patients are significantly higher than those in other benign CNS diseases. 25 The combination of four miRNAs (miR-16-5p, miR-21-5p, miR-92a-3p, and miR-423-5p) in the CSF is helpful for diagnosing CNS DLBCL, with a sensitivity of 67.0% and a specificity of 100%. 14 In addition to CSF miRNAs, peripheral blood miRNAs also have potential diagnostic value for PCNSL. Previous studies have indicated that plasma miR-21 levels can help to differentiate PCNSL from glioma, with the expression levels of miR-21 in plasma and CSF correlating positively. 26 Si et al. 27 screened differential miRNAs between PCNSL and glioma patients through the GEO (GSE139031) dataset and ultimately identified 10 serum miRNAs (miR-6820-3p, miR6803-3p, miR-30a-3p, miR-4751, miR-3918, miR-146a-3p, miR-548am-3p, miR-371a-3p, miR-487a-3p, and miR-4756-5p) as potential biomarkers for distinguishing PCNSL from glioma. Therefore, CSF or peripheral blood miRNAs are novel biological markers for diagnosing PCNSL, and the combination of multiple miRNAs can further improve diagnostic efficacy.
Cytokines
IL-10 is the most extensively studied and diagnostically valuable cytokine reported to date. The concentration of IL-10 and the ratio of IL-10/IL-6 in the CSF of PCNSL patients are significantly higher than those in patients with other brain tumors (58.2 pg/mL versus 1.5 pg/mL, p = 0.001; 24.3 versus 0.6, p = 0.001), and a critical value of 8.3 pg/mL for IL-10 demonstrates a sensitivity and specificity of 59.0% and 98.0%, respectively. 10 Another study showed that the CSF IL-10 concentration (cutoff value: 2 pg/mL) has a high sensitivity of 88% and specificity of 99% for distinguishing new cases of PCNSL from other CNS diseases, with an area under the ROC curve of 0.94. Furthermore, the combination of the CSF MYD88 L265P mutation and IL-10 can significantly improve sensitivity (94%) and specificity (98%) for differential diagnosis. 11 Maeyama et al. diagnosed PCNSL based on a combination of multiple biomarkers in CSF (IL-10, CXCL13, soluble IL-2 receptor, β2-microglobulin) and evaluated diagnostic performance using a prospective clinical study cohort (n = 104). The results indicated that the sensitivity, specificity, positive predictive value, and negative predictive value were 97%, 97%, 94%, and 99%, respectively. 12 Therefore, CSF IL-10 alone or in combination with other novel markers has promising diagnostic performance in PCNSL.
Chemokines
CXCL13 is a chemokine ligand that participates in the homing process of B lymphocytes. Rubenstein et al. 15 detected concentrations of CXCL13 and IL-10 in CSF samples of 83 CNSL patients and 137 control patients (neuroinflammation, other primary, or metastatic brain tumors) and found that when the concentration of CXCL13 was >90 pg/mL, the sensitivity and specificity for diagnosing CNSL were 69.9% and 92.7%, respectively, and when the concentration of IL-10 was >16.15 pg/mL, the sensitivity and specificity were 65.4% and 92.6%, respectively. 15 Another single-center prospective clinical study analyzed the potential efficacy of CXCL13 and CXCL9 as diagnostic, therapeutic, and prognostic markers for CNSL. Concentrations of CXCL13 and CXCL9 in the CSF of CNSL patients were significantly elevated compared to those of other CNS diseases. The sensitivity and specificity of CXCL13 (cutoff value: 80 pg/mL) for diagnosing CNSL were 90.7% and 90.1%, respectively. In addition, concentrations of CXCL13 and CXCL9 decreased after treatment and increased again upon relapse, indicating that dynamic monitoring is helpful for disease status assessment. 16 Although CXCL13 performs well in diagnosing CNSL, it cannot differentiate between PCNSL and secondary CNSL (SCNSL). Moreover, setting an appropriate cutoff value for better clinical application remains an important challenge.
Pathogenesis
Gene mutations
Nuclear factor kappa-B signaling pathway
Nuclear factor kappa-B (NF-κB) is a downstream effector molecule of many common signaling pathways, including the BCR, toll-like receptor (TLR), Notch, and JAK-STAT pathways; it is involved in regulating cell proliferation, apoptosis, and immune-inflammatory responses and plays a crucial role in the occurrence and development of many malignant tumors. Current studies have demonstrated that approximately 90% of PCNSL patients have gene mutations related to the NF-κB pathway, such as in MYD88, CD79b, CARD11, TNFAIP3, and TBL1XR1. These gene mutations lead to abnormal activation of the NF-κB pathway and play a central role in the pathogenesis of PCNSL.28 –30 The MYD88 protein is an adaptor protein that regulates TLR and interleukin 1 receptor (IL-1R) signaling, activating IRAK, TRAF6, and the IKK complex and leading to inhibitor of NF-κB (IκB) phosphorylation and degradation and mediating activation of the effector molecule NF-κB. 31 CD79b belongs to the immunoglobulin superfamily and, together with CD79a, participates in BCR complex signaling, activating Bruton’s Tyrosine Kinase (BTK) and the downstream NF-κB pathway. 32 In PCNSL patients, MYD88 and CD79b mutations occur frequently, with varying frequencies in different cohorts, fluctuating between 55% and 88% and 42% and 69%, respectively.28 –30,33 Other primary brain malignancies, such as gliomas, almost never carry MYD88 or CD79b mutations, making them useful for distinguishing PCNSL. CARD11 is downstream of BTK and belongs to the Caspase Recruitment Domain family, triggering IκB phosphorylation and degradation by recruiting Bcl-10 and MALT1 to form the CBM complex, and mutation occurs in 16–30% of PCNSL patients. 32 TNFAIP3 and TBL1XR1 are negative regulatory genes of the NF-κB pathway. TNFAIP3 mutations, heterozygous deletions, and chromosomal loss occur in 6–56% of PCNSL patients and TBL1XR1 mutation in 14–36%. Mutations or deletions of negative regulatory genes enhance the activation degree of the NF-κB pathway. 34 Thus, abnormal activation of the BCR/TLR-NF-κB pathway is a key driving mechanism for the occurrence of PCNSL, providing a strong theoretical basis for targeting this pathway. Many small molecule targeted drugs have been applied in clinical practice.
Other genes
The oncogene PIM1 regulates the cell cycle and metabolism and protein transcription and translation by encoding serine/threonine kinases, thereby promoting apoptosis and mediating drug resistance; PIM1 mutations are closely related to the occurrence and development of tumors and are common in PCNSL patients, with mutation frequencies even exceeding those of MYD88 and CD79b.30,35 However, unlike MYD88, PIM1 mutations have great heterogeneity and no clear mutation hotspot. Compared to low PIM1 expression, high PIM1 expression indicates poor overall survival (OS) in PCNSL patients, suggesting that high PIM1 expression is an adverse prognostic factor for PCNSL. 28 PCNSL patients were divided into two groups based on whether they carried CD79b and/or PIM1 mutations: the CDP group (including CD79b and/or PIM1 mutations) and the non-CDP group (without CD79b and PIM1 mutations). The CDP group, although mostly composed of elderly patients, had significantly longer OS than the non-CDP group after receiving HD-MTX treatment. In addition, the tumor mutation burden of the CDP group was higher than that of the non-CDP group, and those with a higher tumor mutation burden responded better to immunotherapy. Therefore, PCNSL patients with PIM1 mutations may benefit from immune checkpoint therapy. 36 PIM1 mutations can also enhance NF-κB signaling and participate in mediating lymphoma resistance to BTK inhibitors. Currently, many protein inhibitors targeting the PIM1 family are being developed, which may provide new options for treatment of PCNSL. CDKN2A is a tumor-suppressor gene that plays a crucial role in maintaining genomic stability, and high-frequency biallelic inactivation in tumor patients is often seen. Homozygous deletion is the main mutation mode of CDKN2A and occurs in 33–83% of PCNSL patients. Abnormal CDKN2A loses its inhibitory effect on tumors and promotes the occurrence and development of PCNSL.29,37,38 In addition, CDKN2A deletion is considered an early clone event, indicating that CDKN2A mutations may be an early driving factor in PCNSL. 39 However, there is less in-depth research on CDKN2A mutations in PCNSL; with the continuous development of gene sequencing technology, the clinical significance of CDKN2A mutations is expected to be further clarified. PRDM1 is a tumor suppressor that acts in terminal differentiation of B-cells. Mutations, homozygous deletions, and chromosomal loss of PRDM1 are found in 7–20% of PCNSL patients. A small cohort study (n = 10) indicated that PRDM1 mutations are significantly associated with PCNSL prognosis. 40 Moreover, the mutation frequency of PRDM1 in SCNSL is up to 50%, which is significantly higher than that in PCNSL, suggesting that the PRDM1 gene may help to distinguish between PCNSL and SCNSL. 41
Copy number variations
Copy number variations (CNVs) are chromosomal genomic structural variations and are driving factors of many malignant lymphomas. With continuous popularization of whole-genome/exome sequencing (WGS/WES) technology, the role of CNVs in the pathogenesis of PCNSL has been gradually recognized. Zhu et al. conducted WGS on 24 cases of primary CNS DLBCL and found that amplification of 1q31.3, 2q32.2, and 2q36.3, as well as deletion of 6p21.33, 8p23.1, 22q11.1, and 22q11.21, were common. Survival analysis suggested that amplification of 1q31.3 (involving complement factor H: CFH) was associated with poor prognosis. CFH and its related family members encode soluble sugar proteins, which play a critical role in the immune response. 30 In addition, multiple omics tests on specimens from 18 PCNSL patients revealed up to 16 types of chromosomal CNVs with a frequency of over 25%, of which the most common was deletion of 9p21.3, accounting for 83%; there were various abnormalities on chromosome 6, and the incidence of deletions at 6q14.1, 6p21, 6q21, and 6q23 was 44%, 50%, 55%, and 55%, respectively. 40 Another study showed that amplification of 18q21.33 (42%) and 19p13.13 (34%) and the deletion frequency of 6p21 (39%), 6q21 (65%), 6q27 (49%), and 9p21.3 (28%) were significantly more frequent in PCNSL than in systemic DLBCL. 42 Although different cohort studies have observed different types and frequencies of CNVs, it cannot be denied that CNVs are an important pathogenic mechanism of PCNSL, with studies on deletions of 6p21, 8q12, 9p21, and 9p24.1 being relatively clear. Deletion of 6p21 leads to loss of expression of human leukocyte antigen (HLA) I/II molecules, thereby evading the T-cell-mediated immune response.29,43 Compared with other extranodal lymphomas, HLA I/II expression deficiency is more common in PCNSL and primary testicular lymphoma, which is conducive to tumor cell survival and closely related to poor prognosis. Deletion of 9p21 leads to loss of the tumor-suppressor genes CDKN2A/B, resulting in high cellular proliferative activity.29,37,38 Deletion of 8q12 leads to loss of the tumor-suppressor gene TOX. TOX is necessary for the development of various T-cell subpopulations. Its loss leads to T-cell functional defects, and downregulation of TOX is associated with poor prognosis in different cancers.37,44 Immune checkpoints are represented by programmed cell death 1 (PD-1) and its ligands PD-L1/PD-L2. Chapuy et al. found that PCNSL patients with copy number abnormalities or chromosomal translocations involving the 9p24.1 locus exhibit elevated expression levels of PD-L1/PD-L2, which binds to PD-1 on T-cell surfaces to transmit inhibitory signals and suppress their immune response. This suggests that tumor cells can escape immune surveillance and defense via upregulation of expression of immune checkpoint molecules, thus providing a theoretical basis for targeted PD-1/PD-L1 axis therapy for PCNSL. 45 In summary, immune escape caused by CNVs is an important mechanism of PCNSL occurrence.
DNA methylation
Recently, DNA methylation has been widely used for disease diagnosis, treatment monitoring, and prognosis evaluation. Whole-genome DNA methylation detection has revealed 194 genes with methylation differences between PCNSL and normal controls, significantly enriched in genes targeted by polycomb repressive complexes and genes with high CpG content in promoters. 46 In a recent study, multiomics analysis of PCNSL patients identified four robust, nonoverlapping and predictive classes (CS1–4), among which CS1 is mainly characterized by high CpG island methylation, and CS2 exhibits global hypermethylation, including the promoter and chromosome ends. Enrichment analysis found strong enrichment of CS1 in histone/chromatin protein H3K27me3 and EZH2 binding sites, showing high transcriptional activity of polycomb repressive complex 2. On the other hand, CS2 is enriched in B-cell differentiation programs, BCL11A, NF-κB, interferon regulatory factor 4 (IRF4), and BCL6, which affect B-cell differentiation programs and NF-κB activity in terms of epigenetics and transcription regulation. 42 Another study showed that high methylation of H3K27me2 and H3K27me3 is an independent risk factor for OS and progression-free survival (PFS) in PCNSL patients. 47 Thus, DNA methylation is also one of the underlying pathogenic mechanisms of PCNSL.
Standard treatment
Induction therapy
HD-MTX is the key component of induction therapy regimens for newly diagnosed PCNSL patients. The combination of HD-MTX and other drugs with the ability to cross the BBB can improve disease response rates, such as cytarabine (Ara-C), rituximab, and temozolomide (TMZ). A randomized, phase II trial comparing HD-MTX alone or in combination with Ara-C for the treatment of PCNSL patients demonstrated that higher response and survival rates were observed in the combination therapy arm. 48 Presently, HD-MTX + Rituximab + TMZ (MTR), HD-MTX + Ara-C, HD-MTX + Ara-C + Rituximab, HD-MTX + Ara-C + Thiotepa + Rituximab (MATRix), HD-MTX + Procarbazine + Vincristine + Rituximab (R-MPV), and HD-MTX + Carmustine + Teniposide + Prednisolone (MBVP) are several reported and preferred induction regimens (Table 3). 49 However, head-to-head comparative trials are relatively lacking among the above regimens except for the IELSG32 trial. This experiment indicated that MATRix significantly improved outcomes compared with HD-MTX + Ara-C and HD-MTX + Ara-C + Rituximab, with 7-year OS rates of 56%, 37%, and 21%, respectively. Moreover, the addition of rituximab to HD-MTX + Ara-C was associated with superior long-term results. 50 As such, there is no standard choice for induction therapy, and the strategy is often based on institutional and professional experience.
The treatment options of PCNSL patients.
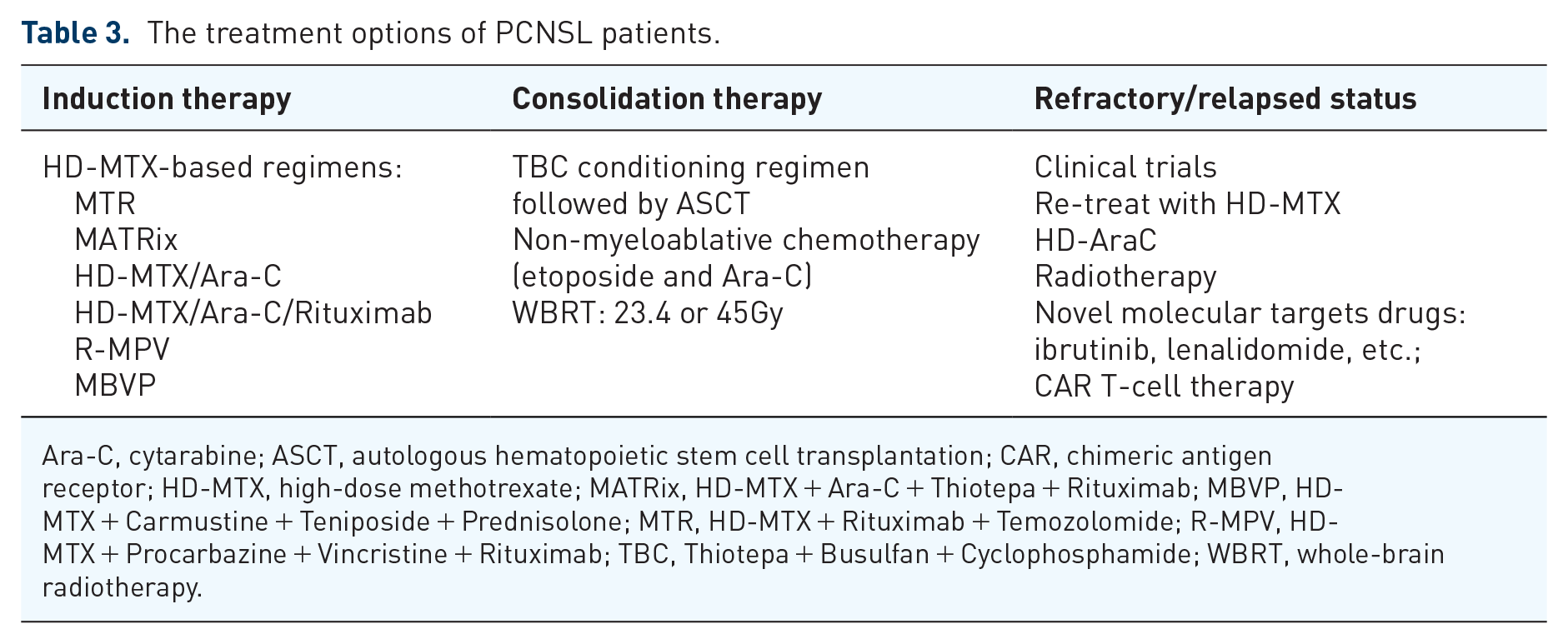
Ara-C, cytarabine; ASCT, autologous hematopoietic stem cell transplantation; CAR, chimeric antigen receptor; HD-MTX, high-dose methotrexate; MATRix, HD-MTX + Ara-C + Thiotepa + Rituximab; MBVP, HD-MTX + Carmustine + Teniposide + Prednisolone; MTR, HD-MTX + Rituximab + Temozolomide; R-MPV, HD-MTX + Procarbazine + Vincristine + Rituximab; TBC, Thiotepa + Busulfan + Cyclophosphamide; WBRT, whole-brain radiotherapy.
Consolidation therapy
ASCT and WBRT are standard consolidation therapies for suitable patients. The IELSG32 trial 51 found that ASCT and WBRT were tolerated by patients aged 70 years or younger with PCNSL, with 2-year PFS rates of 75% and 76% (p = 0.62), respectively, but a significant impairment in attention and executive functions was recorded in patients who received WBRT. Similarly, another randomized phase II study in PCNSL patients (18–60 years) treated with HD-MTX-based induction chemotherapy followed by WBRT or TBC conditioning regimen and ASCT indicated that ASCT arm achieved superior 8-year event-free survival (67% versus 39%, p = 0.03), and WBRT arm observed deteriorated balance ability (52% versus 10%, p < 0.001) and neurocognition (64% versus 13%, p < 0.001) during long-term follow-up. 52 Low-dose WBRT (23.4Gy) consolidation has been indicated to improve the PFS, but neurotoxicity still requires longer follow-up. 53 Thus, ASCT is preferred as consolidation therapy in suitable patients after HD-MTX-based chemotherapy, considering the higher risk of delayed neurotoxicity and cognitive impairment after WBRT. WBRT serves as an alternative approach for consolidation in young patients without sufficient hematopoietic stem cells (HSCs) or preference to ASCT, but it’s unacceptable in elderly patients. In addition, non-myeloablative chemotherapy may be an effective alternative option to ASCT or WBRT. The Alliance trial showed that PCNSL patients treated with EA (etoposide and Ara-C) consolidation experienced similar toxicity profiles compared with patients treated with ASCT consolidation, but with an inferior median PFS (2.4 years versus 6 years, p = 0.02), 54 suggesting no benefit of the non-myeloablative consolidative approach in comparison with ASCT.
Salvage therapy
PCNSL patients usually have an overall poorer prognosis compared with systemic lymphoma. In general, approximately 10–15% of newly diagnosed PCNSL patients are resistant to initial induction therapy, with up to half of responsive patients experiencing relapse after achieving remission. 55 Currently, standard treatment and care of R/R PCNSL have not been established, and clinical trials should be considered as the preferred options for these patients. Traditional salvage chemotherapy strategies include re-treatment with HD-MTX for late relapse individuals, HD-AraC, and high-dose systemic therapy with HSCs reinfusion in eligible patients, etc. Radiotherapy can be considered as a therapeutic decision with careful assessment because of non-lasting response and neurotoxicity. Multiple novel molecular targeted drugs and chimeric antigen receptor (CAR) T-cell immunotherapy are under investigation for R/R PCNSL patients, as discussed below. In clinical practice, salvage treatment should be determined based on published articles, the patient’s basic condition, and previous therapy experience.
Molecular targeted drugs
With continuous advancement of molecular biology research, a new era of targeted therapy for R/R PCNSL has emerged (Figure 1). BTK inhibitors and immunomodulatory drugs (IMiDs) have been reported in large clinical trials and considered as recommended salvage treatment options. However, reports on immune checkpoint inhibitors and phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitors are limited, requiring further clinical trials to determine efficacy and safety, but the current data can serve as references and offer possibilities for some R/R CNSL patients.
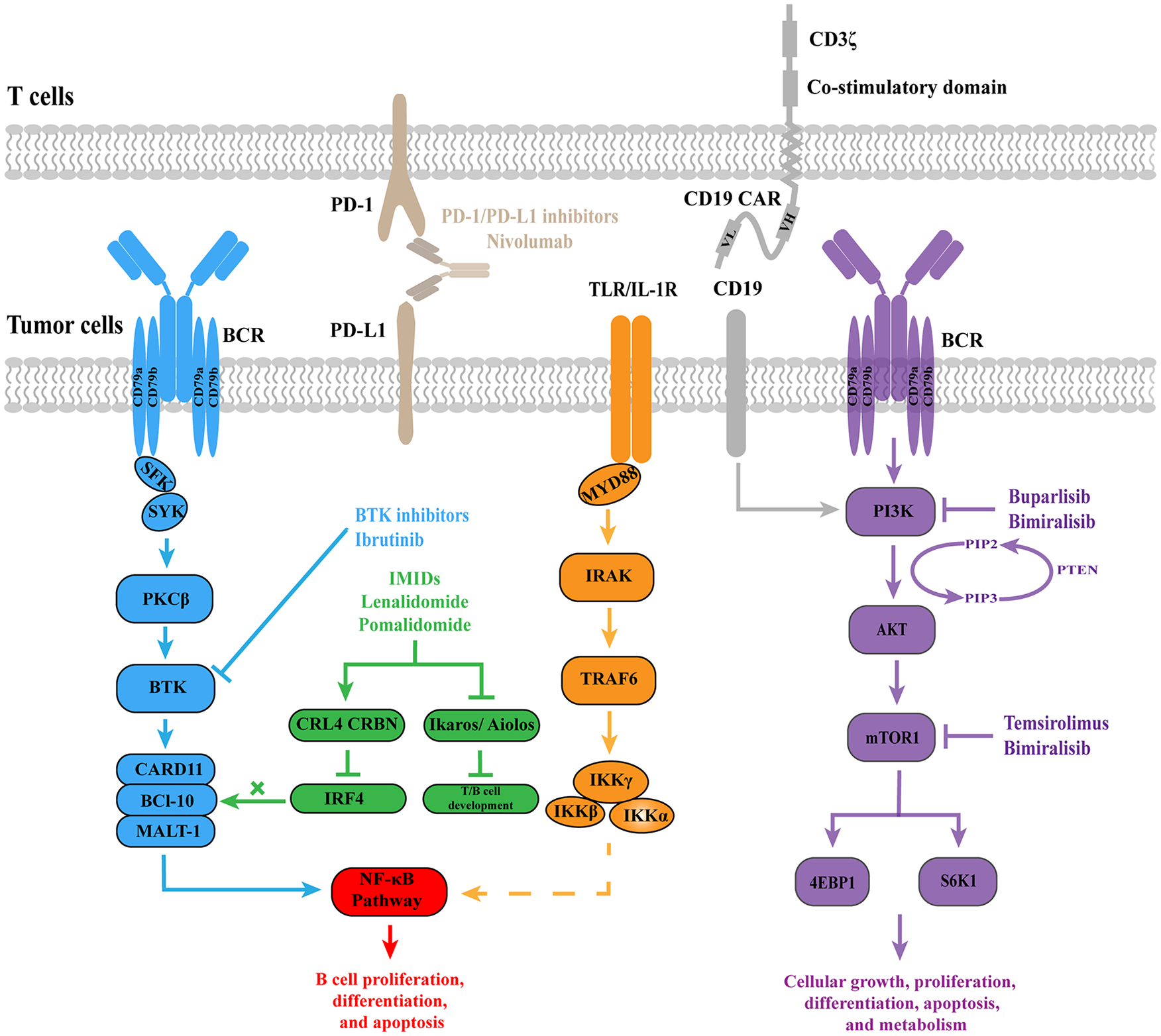
Novel therapies for PCNSL patients. These strategies include BTK inhibitors, IMiDs, immune checkpoint inhibitors, PI3K/mTOR inhibitors, and CAR T-cell immunotherapy.
BTK inhibitors
BTK belongs to the nonreceptor protein tyrosine kinase TEC family and is expressed in lymphocytes and myeloid cells. It is one of the key kinases in the BCR and TLR signaling pathways, contributing to regulation of B-cell proliferation, differentiation, and apoptosis. 56 WGS has revealed that PCNSL typically harbors CD79b and MYD88 mutations, leading to abnormal activation of the BCR and TLR signaling pathways and their downstream effector molecule NF-κB. Therefore, BTK is an ideal target for treating PCNSL.29,57
Ibrutinib is a small molecule inhibitor that can penetrate the BBB and irreversibly covalently bind to the BTK Cys-481 site, reducing its enzymatic activity and exerting antitumor effects.58–60 The clinical efficacy of ibrutinib in treating R/R CNSL has been reported in several studies. A phase II clinical trial included 52 R/R PCNSL/PVRL patients who received ibrutinib monotherapy. Complete remission (CR) was achieved in 10 of 44 evaluable patients (23%) and partial remission (PR) in 16 patients (36%), and the median OS and PFS were 19.2 and 4.8 months, respectively. 60 Thus, R/R PCNSL patients can benefit from single-agent ibrutinib therapy, but clinical remission is often neither complete nor durable, leading to the inevitable trend of combining other treatment modalities. A phase Ib clinical trial of ibrutinib in combination with HD-MTX ± Rituximab for treatment of 15 R/R CNSL patients showed clinical remission in 12 patients, including eight (53.3%) achieving CR and four (26.7%) achieving PR, with a median PFS of 9.2 months. The combination regimen was relatively well tolerated, with one patient experiencing grade 4 lymphocytopenia, neutropenia, and pulmonary infection, but no grade 5 adverse events occurred. 22 A meta-analysis showed that the overall response rate (ORR) of ibrutinib monotherapy for PCNSL was 55%; the ORR of ibrutinib in combination with chemotherapy or radiotherapy was 88% and 85%, respectively, further affirming the necessity of combination therapy. 59 In addition, ibrutinib can be used as an induction chemotherapy regimen for newly diagnosed patients. Chen et al. 61 found that the combination of ibrutinib and HD-MTX induces clinical remission in 82% of newly diagnosed PCNSL patients, with a median PFS of 7.4 months. In summary, ibrutinib significantly improves the clinical prognosis of CNSL, especially for R/R patients. Common adverse events of ibrutinib include rash, diarrhea, bleeding, atrial fibrillation, and infection, leading to discontinuation of the drug in 4–26% of patients; severe events can be life-threatening, requiring careful monitoring during use. The occurrence of adverse events is mainly related to off-target inhibition; hence, exploration of highly selective BTK inhibitors is urgently needed.62–64
Second-generation irreversible covalent BTK inhibitors with lower off-target toxicity include zanubrutinib, orelabrutinib, and acalabrutinib. One PCNSL patient with MYD88 gene mutation experienced disease progression after two cycles of HD-MTX induction chemotherapy but achieved CR after the addition of zanubrutinib for three courses of treatment. 65 In addition, three PCNSL patients experienced vitreoretinal relapse after achieving CR, and their intraocular lesions disappeared after 1 month of zanubrutinib monotherapy, with one patient experiencing manageable hypertension. 66 A retrospective study analyzed the clinical outcomes of 23 CNSL patients treated with orelabrutinib, showing an ORR of 100% for the four newly diagnosed patients and 60% for the 15 R/R patients. The long-term PFS and OS rates for the four patients receiving maintenance therapy were both 75%, and grade 3–4 hematologic toxicities were the main adverse drug reaction. 67 Clinical trials of acalabrutinib alone or in combination with other drugs for CNSL are currently in the recruiting phase (NCT04548648; NCT04906902; NCT04462328). However, despite the clinical efficacy of the first two generations of BTK inhibitors in treating CNSL, some patients still develop drug resistance and disease relapse. Mutation of BTK Cys-481 to serine (C481S) is the main cause of acquired resistance to irreversible covalent BTK inhibitors. Currently, BTK-Proteolysis Targeting Chimeras targeting this mutation site and new generations of reversible, noncovalent BTK inhibitors are being explored and developed to overcome this issue. 68
Immunomodulatory drugs
Cullin-RING ligase 4 (CRL4) is an important member of the E3 ubiquitin ligase family that regulates cellular activities by selectively ubiquitinating target proteins. 69 The IMiDs thalidomide, as well as its derivatives lenalidomide and pomalidomide, changes the substrate specificity of CRL4CRBN by binding to the CRL4 Cereblon (CRBN) subunit, leading to ubiquitination and degradation of the transcriptional regulators of T/B-cell development-zinc finger proteins Ikaros/Aiolos. In addition, they inhibit the IRF4, NF-κB, and PI3K/mTOR signaling pathways, exerting antitumor and antiproliferative effects. Furthermore, IMiDs have the ability to improve the tumor immune microenvironment, including promoting proinflammatory repolarization of tumor-associated macrophages (TAMs) and activation of T/NK cells.70,71
Lenalidomide and pomalidomide have demonstrated promising antitumor activity in patients with PCNSL. It has been reported that two of six R/R PCNSL patients achieved CR when treated with lenalidomide as a single agent, with one patient maintaining CR for over 24 months. 72 In addition to monotherapy, the French Oculo-Cerebral Lymphoma (LOC) Network and the Lymphoma Study Association reported the results of a study using rituximab in combination with lenalidomide to treat R/R PCNSL/PVRL. Among the 45 evaluable patients (34 PCNSL and 11 PVRL), 13 achieved CR/unconfirmed CR (29%) and 3 PR (7%), resulting in an ORR of 35.6%, with a median treatment duration of seven cycles. The median follow-up was 19.2 months, with a median PFS of 3.9 months for PCNSL patients and 9.2 months for PVRL patients. Grade 3–4 neutropenia occurred in 44% of patients. 73 A phase Ib clinical study of pomalidomide in combination with dexamethasone for R/R PCNSL/PVRL confirmed that the maximum tolerated dose of pomalidomide per day was 5 mg. The most common grade 3–4 hematologic toxicity was neutropenia, and the nonhematologic toxicity involved pulmonary infection. Among 25 evaluable patients, 12 achieved disease remission, resulting in an ORR of 48% and a median PFS of 5.3 months. 74 In addition, by inhibiting IRF4 to interfere with the NF-κB signaling pathway, IMiDs may have synergistic antitumor effects with BTK inhibitors. In salvage treatment of 14 R/R PCNSL patients with ibrutinib, lenalidomide, and rituximab regimen, eight patients achieved disease remission, including four CR and four PR cases, three responsive patients received consolidation therapy (WBRT: n = 2, ASCT: n = 1), and three patients discontinued treatment due to drug toxicity, with no treatment-related death. 75 These findings suggest that lenalidomide and pomalidomide are promising for treating PCNSL, and future studies will further explore the clinical efficacy and safety of their combination with other small molecule targeted drugs, particularly BTK inhibitors.
Immune checkpoint inhibitors
Immune escape is one of the major challenges in current cancer treatment, whereby tumor cells escape immune surveillance and defense by downregulating HLA expression and/or activating immune checkpoints, with PD-1 and its ligands PD-L1/PD-L2 as representatives. 76 In classic Hodgkin lymphoma (cHL), 9p24.1 locus copy number aberrations of tumor cells lead to overexpression of PD-L1/PD-L2; therefore, cHL shows good clinical response to PD-1/PD-L1 inhibitors. 77 Several studies have reported expression of PD-1 and its ligands in PCNSL tumor tissues. Chapuy et al. found that PCNSL patients also have copy number abnormalities or chromosomal translocations involving the 9p24.1/PD-L1/PD-L2 locus, allowing overexpression of PD-L1/PD-L2 to deliver inhibitory signals by binding to PD-1 on the surface of T-cells.45,78,79 However, another study indicated that PCNSL rarely shows copy number abnormalities at the 9p24.1 locus, with a low rate of PD-L1 positivity. Immune evasion is mainly achieved through HLA loss. 80 Although the specific role of the PD-1/PD-L1/PD-L2 signaling axis in PCNSL needs further clarification, immune checkpoint inhibitors have already shown preliminary efficacy in treatment of PCNSL.
Nivolumab is a humanized monoclonal antibody targeting the PD-1/PD-L1 axis that has been approved for cHL patients who relapsed or progressed after ASCT and posttransplant brentuximab vedotin. It has been reported that five patients (PCNSL: four cases, SCNSL: one case) who received salvage therapy with nivolumab achieved clinical remission, with four achieving CR and one achieving PR. Two patients experienced adverse drug reactions, including grade 2 pruritus and fatigue. Three patients maintained ongoing remission during long-term follow-up. 81 Similarly, two out of five R/R CNSL patients completely responded to pembrolizumab in combination with rituximab, and maintained response for 7 months after the initiation of pembrolizumab therapy. 82 The existing data implies that B-cell lymphomas involving the CNS are responsive to PD-1/PD-L1 inhibitors, advocating for multiple clinical studies on nivolumab and pembrolizumab alone or in combination with other drugs.
PI3K/mTOR pathway inhibitors
The PI3K/AKT/mTOR signaling pathway is widespread in the body and controls cellular growth, proliferation, differentiation, apoptosis, and metabolism. 83 Positive expression rates of phosphorylated protein kinase B (AKT), mTOR, p70 ribosomal protein S6 kinase (S6K), and eukaryotic initiation factor 4E binding protein 1 (4E-BP1) in PCNSL tumor samples were significantly higher than those in reactive lymph nodes, and expression of phosphorylated mTOR correlated positively with that of phosphorylated AKT, S6K, and 4E-BP1. 84 PCNSL can exhibit loss or mutations of the tumor-suppressor gene PTEN, which converts biologically active phosphatidylinositol 3,4,5-trisphosphate (PIP3) to inactive phosphatidylinositol 4,5-bisphosphate (PIP2), thereby antagonizing the phosphorylation of downstream AKT induced by PI3K and acting as a negative regulatory factor.84,85 The PI3K/AKT/mTOR signaling axis also participates in mediating BCR signaling and is highly activated in patients with PCNSL carrying CD79b mutations, promoting tumor cell survival. 86 Furthermore, copy number detection of PCNSL patients has shown that 37% (10/27) of patients have amplification of the PIK3CA gene, thus increasing the activity of PI3K. 87 It is evident that the PI3K/AKT/mTOR signaling pathway is aberrantly activated in PCNSL patients, and inhibitors targeting this pathway can be considered therapeutic options for PCNSL.
Temsirolimus is an intravenous small molecule mTOR inhibitor that exerts antitumor effects by inhibiting the activity of mTORC1 through binding to FK506 binding protein 12. A phase II clinical study including 37 R/R PCNSL patients with a median age of 70 years showed that treatment with temsirolimus (75 mg, qw) monotherapy resulted in an ORR of 54% and a complete response rate (CRR) of 21.5%, but long-term disease control was poor, with a median PFS of only 2.1 months and OS of 3.7 months. Common grade ⩾3 drug toxicities included hyperglycemia, thrombocytopenia, infection, anemia, and rash, with a treatment-related mortality rate of up to 13.5%. 88 Thus, the anti-PCNSL activity of temsirolimus is not sustainable, and further exploration and research are needed to prolong PFS and reduce adverse drug reactions. Buparlisib is a pan-PI3K inhibitor that can inhibit all four isoforms of class I PI3K. Clinical studies have shown that the ORR of buparlisib monotherapy for R/R CNSL is only 25%, possibly due to limited BBB permeability. 89 Bimiralisib is a dual PI3K and mTOR inhibitor with good penetration across the BBB. In vitro studies have demonstrated significant anti-lymphoma activity of bimiralisib as a single agent or in combination therapy. 90 A phase I/II clinical study evaluated the clinical efficacy and safety of bimiralisib in treatment of R/R lymphoma, showing an ORR of 14%; common grade ⩾3 drug toxicities included hyperglycemia, neutropenia, thrombocytopenia, and diarrhea, with 28% of patients discontinuing treatment due to drug toxicities. 91 The data from a phase II clinical study (NCT02669511) of bimiralisib as a salvage therapy for 21 R/R PCNSL patients have not yet been published.
Other targeted drugs
The nuclear exportin 1 (XPO1) protein assists in transport of various proteins from the nucleus to the cytoplasm; it plays a prominent role in maintaining intracellular homeostasis and is involved in development of various malignancies. XPO1 dysregulation has been linked to resistance to various standard treatments. 92 Currently, the XPO1 inhibitor selinexor has been approved for treatment of R/R multiple myeloma and DLBCL.92,93 Animal experiments have shown that selinexor monotherapy can effectively block the growth of murine brain parenchymal lymphoma, prolong survival time, exert synergistic antitumor effects in combination with ibrutinib, and reverse polarization of TAMs in the microenvironment by reducing expression of PD-L1 and signal regulatory protein alpha on M2-type TAMs, thereby improving the immunosuppressive microenvironment. 94 However, there is currently no clinical report on XPO1 inhibitor therapy for PCNSL, though a clinical trial is ongoing to evaluate the combination of XPO1 and BTK inhibitors with HD-MTX for treatment of R/R PCSNL (ChiCTR2200062154). The BCL-2 protein is expressed in the majority of PCNSL cases. 95 Phosphorylation and proteasomal degradation of IκB are prerequisites for activation of NF-κB. 96 Histone deacetylase inhibitors can silence the MYD88 L265P mutation and synergize with ibrutinib to exert antitumor effects. 97 Therefore, BCL-2 inhibitors, proteasome inhibitors, and histone deacetylase inhibitors have a theoretical basis for treatment of PCNSL, but their clinical applications require further exploration.
CAR T-cell immunotherapy
CAR T-cell immunotherapy acts as a novel technology that involves genetically modifying a patient’s own T lymphocytes to express receptors that can specifically target tumor-associated antigens (TAAs). These engineered T-cells are then expanded in vitro and infused back into the patient to exert antitumor effects. Recently, CAR T-cell immunotherapy has been rapidly developed in the field of hematological malignancies and has shown significant clinical efficacy in representative diseases, including DLBCL, acute B-cell lymphoblastic leukemia, and multiple myeloma.98–100 Six CAR T-cell products have been approved by the US Food and Drug Administration, including four targeting CD19 and two targeting B-cell maturation antigen. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) are two common complications associated with CAR T-cell immunotherapy. The reported incidence and severity of CRS and ICANS vary widely among different studies, ranging from 15% to 100% (severe cases: 0–47%) and 15% to 70% (severe cases: 0–35%), respectively. 101 Patients with neurological symptoms have a higher risk of developing ICANS after CAR T-cell treatment. 102 To avoid serious and potentially fatal CNS toxicities, early clinical trials of CAR T-cell therapy excluded hematological tumor patients with CNS involvement. In 2017, The New England Journal of Medicine published the first report on the clinical process of CD19 CAR T-cell therapy in a patient with R/R CNS DLBCL. Following the infusion, the CAR T-cells expanded well in vivo, and CSF testing showed positive results. At the 1-month disease evaluation, the patient achieved CR without the occurrence of CRS or ICANS. 103 Since then, major medical centers have started exploring use of CAR T-cell therapy for R/R CNSL (Table 4).
Clinical outcomes and complications of CAR T-cell therapy for R/R CNSL.

CR, complete remission; CRS, cytokine release syndrome; ICANS, immune effector cell-associated neurotoxicity syndrome; NA, not available; OS, overall survival; PCNSL/SCNSL, primary/secondary central nervous system lymphoma; PD, progression disease; PFS, progression-free survival; PR, partial remission; SD, stable disease.
Recently, the LOC network database indicated that 16 of 25 (64%) PCNSL patients achieved disease remission with CAR T-cell therapy. The median PFS and OS from leukapheresis were 8.4 and 21.2 months, respectively. In addition, 2 (8%) and 5 (20%) patients experienced grade ⩾3 CRS and ICANS, respectively. 117 Similarly, a multicenter cohort study reported the outcomes of CAR T-cells salvage therapy in 61 patients with SCNSL. The study reported an ORR of 68% and a CRR of 57%; the median PFS and OS were 3.3 and 7.6 months, respectively, with any grade CRS and ICANS incidence rates at 70% and 57%, respectively. 118 Meta-analysis found an ORR of 69% and a CRR of 51% for CAR T-cell therapy in CNSL by analyzing a total of 63 CNSL patients from eight publications, and the incidence rates of grade ⩾3 CRS and ICANS were 11.1% and 12.0%, respectively. 120 In the ZUMA-1 and JULIET trials, the incidence rates of grade ⩾3 CRS were 10% and 22%, respectively, and the incidence rates of grade ⩾3 ICANS were 32% and 11%, respectively.121–123 Therefore, compared with non-CNS NHL, CNSL does not pose an increased risk of CRS and ICANS following CAR T-cell therapy. Moreover, a real-world multicenter cohort study conducted by Bennani et al. 114 showed comparable rates of grade ⩾3 CRS (13% versus 6%) and ICANS (31% versus 33%) between lymphoma patients with and without CNS involvement after receiving axi-cel, and use of tocilizumab and corticosteroids was also comparable. Shumilov et al. 119 also confirmed that regardless of whether it accompanies with CNS involvement, CAR T-cell therapy exhibits similar clinical efficacy and toxicity in DLBCL patients. 119 In brief, CAR T-cell therapy is an effective and relatively safe treatment option for R/R CNSL patients, and some patients can achieve disease remission with controllable side effects. Several clinical trials are ongoing and recruiting patients (Table 5).
Several clinical trials of CAR T-cell immunotherapy for CNSL.

CAR, chimeric antigen receptor; CNS, central nervous system; NCT: national clinical trial.
Although CAR T-cell therapy has shown good clinical efficacy, challenges of treatment resistance and relapse remain unavoidable. Ghafouri et al. found that the median PFS for R/R CNSL patients treated with CAR T-cells alone was only 4.5 months. A meta-analysis revealed a relapse rate of up to 38% after achieving remission. 120 Therefore, new treatment strategies need to be developed to overcome CAR T-cell resistance and relapse. Target antigen loss has been observed in some patients who experience relapse after remission. ‘Dual targeting’ or ‘cocktail’ therapy that can recognize multiple TAAs may help to prevent relapse due to immune escape.105,108 One case of a heavily relapsed PCNSL patient receiving fourth-generation CD19/CD70 CAR T-cells co-infusion showed CR at the 1-month evaluation, with no CRS or ICANS reported, and the patient was disease-free for over 17 months. 124 However, the ‘Dual targeting’ or ‘cocktail’ therapy has not totally overcome the problem of treatment resistance. In a clinical study involving five refractory CNSL patients who received CD19/CD22 CAR T-cells ‘cocktail’ infusions, clinical responses were achieved, but the duration of response was short, and most patients experienced antigen-positive relapse. 108 Another strategy attempted by experts is to combine CAR T-cells with other treatment methods. A single arm study evaluated the clinical efficacy of ASCT combined with CD19/CD22 CAR T-cells infusion in 13 CNSL patients, including four PCNSL and nine SCNSL cases. The ORR and CRR were 81.8% and 54.6%, respectively; the median OS and PFS were not reached, and only one patient experienced severe ICANS. 125 Similarly, Xue et al. 126 compared the clinical data of CAR T-cell therapy alone or in combination with ASCT and found that the combination therapy significantly improved disease outcomes. Furthermore, PD-1 inhibitors maintenance following CD19/CD22 CAR T-cell therapy obtained superior ORR (82.9% versus 60.0%) and 2-year PFS (59.8% versus 21.3%) in r/r B-NHL. 127 The use of lenalidomide as maintenance therapy after CAR T-cell therapy has led to significant improvement in clinical outcomes for DLBCL patients. 128 Animal experiments have shown that BTK inhibitors reprogram macrophages into M1 subtypes, laying the foundation for further clinical practice of BTK inhibitors combined with CAR T-cells. 129 Overall, combination treatment strategies based on CAR T-cells, including ASCT and novel small molecule targeted drugs, can improve the clinical remission and long-term prognosis of R/R CNSL patients, and are worth further practice and observation in future clinical work.
Conclusion
PCNSL is a clinically rare and aggressive disease, and its pathogenesis mainly involves genetic mutations, CNVs, and epigenetics, particularly excessive activation of the NF-κB signaling pathway. Diagnosis of the disease relies on MRI examination and pathology, and various novel diagnostic markers are also available to aid in diagnosis. HD-MTX acts as the foundation of induction therapy for newly diagnosed patients, but treatment failure and relapse remain significant challenges. Recently, with continuous advancements in molecular biology research on PCNSL, various novel therapeutic approaches have gradually been applied in salvage treatment for R/R PCNSL. These approaches include novel small molecule targeted drugs as well as CAR T-cell immunotherapy. However, monotherapy shows a limited duration of response; combination therapy may be a key strategy to overcome drug resistance. Clinical studies of CAR T-cell therapy and combining small molecule targeted drugs with standard treatments are currently underway. Further exploration of drug resistance mechanisms is equally crucial for improving clinical treatment efficacy.