
Abstract
Hodgkin lymphoma is a special type of lymphoma in which tumor cells frequently undergo multiple genetic lesions that are associated with accompanying pathway abnormalities. These pathway abnormalities are dominated by active signaling pathways, such as the JAK-STAT (Janus kinase–signal transducer and activator of transcription) pathway and the NFκB (nuclear factor kappa-B) pathway, which usually result in hyperactive survival signaling. Targeted therapies often play an important role in hematologic malignancies, such as CAR-T therapy (chimeric antigen receptor T-cell immunotherapy) targeting CD19 and CD22 in diffuse large B-cell lymphoma, while in Hodgkin lymphoma, the main targets of targeted therapies are CD30 molecules and PD1 molecules. Drugs targeting other molecules are also under investigation. This review summarizes the actionable genetic lesions, current treatment options, clinical trials for Hodgkin lymphoma and the potential value of those genetic lesions in clinical applications.
Keywords
Introduction
Hodgkin lymphoma (HL) is a relatively rare malignant disease of the hematological system. HL can be divided into classic Hodgkin lymphoma (cHL) and nodular lymphocyte-predominant HL (NLPHL). cHL can also be further subdivided into four subtypes, namely, nodular sclerosis HL (NSHL), mixed cellularity HL (MCHL), lymphocyte-rich HL (LRHL), and lymphocyte-depleted HL (LDHL). 1 Because NLPHL accounts for only about 10% of HL, this review will focus mainly on cHL.
The main tumor cells of cHL are HRS cells (Hodgkin Reed-Sternberg), which are currently thought to originate from the germinal center, possess gene expression similar to that of CD30+ extrafollicular B-cells, and lack markers, including CD19 and CD79α. Although such cells will be quickly cleared under immune system surveillance, some of them not only survive the immune system but also transform into HRS cells through mechanisms such as constitutive activation of NFκB, JAK-STAT, and other signaling pathways. 2
Research has identified genetic lesions and related pathways for HL (Table 1), as technologies such as next-generation sequencing (NGS) have matured and become widespread, an increasing number of disease-related mutant loci are being identified. More and more patients are choosing to include genetic sequencing as part of their general screening program. Therefore, here, we summarize the genetic lesions, related pathways, and targeted therapy for HL, and by reviewing previous studies on the functions of these genes, we generated hypotheses regarding the mechanisms by which these genes increase the risk of HL and provide directions for future therapy. The genetic lesions related to HL include somatic mutations, germline mutations, and structural variations. The included literature was obtained from PubMed.
Frequencies of gene mutations and related SVs involved in different pathways in HL.

DNA, deoxyribonucleic acid; JAK-STAT, janus kinase–signal transducer and activator of transcription; NFκB, nuclear factor kappa-B.
In HL cell lines.
8/32 (gains), 4/107 (translocations).
61/108 (copy gain), 39/108 (amplification).
In a family with multiple cases of cHL.
12/15 (9q21 loss); 9/15 (16q23 loss).
Case report.
Genetic lesions and related pathways
There are many pathways related to somatic mutations and structural variations, including the JAK-STAT pathway, NFκB pathway, immune evasion, cell cycle, and DNA repair. However, germline genetic abnormalities also play an important role in HL. In a study of 153,115 patients with primary malignant hematologic disease in the Swedish Family-Cancer Database, researchers measured the familial relative risks (FRRs) of malignant hematologic disease by calculating the standardized incident ratios (SIRs) of a total of 391,131 first-degree relatives of these patients, ultimately finding that familial risk exists. 68 Similar conclusions have been drawn in several other analogous studies.69–71 In a cohort of 13,922 HL patients with 57,475 first-degree relatives in five European countries, the cumulative risk (CR) of HL in first-degree relatives of a patient with HL was 0.6%, 72 showing a three-fold increased risk over the general population [standardized incidence ratio: 3.3; 95% confidence interval (CI): 2.8–3.9]. Besides, the familial risks were higher in siblings (6.0-fold) than those in parents and/or children (2.1-fold). Some subtypes showed significantly high familial risks, especially in lymphocyte-rich (81-fold; 95% CI, 30- to 177-fold) and nodular sclerosis patients (4.6-fold; 95% CI: 2.9- to 7.0-fold). Even so, further research is still needed to better understand the potential mechanism of the familial aggregation of HL as no major high-penetrant gene has yet been identified till now. These studies are sufficient to prove that there are indeed familial predisposition and genetic risk factors for HL, representing the impact of germline genetic lesions.
Primary immunodeficiency-related genetic lesions
Epstein–Barr virus (EBV) mainly infects human B cells. It is considered the pathogen of infectious mononucleosis (IM) and is closely associated with oral hairy leukoplakia, Burkitt’s lymphoma, and so on. EBV invades the body and further infects primitive B-cells in the tonsils, wherein EBV can activate these cells to differentiate into quiescent memory B-cells (latent state). Under certain circumstances, these cells will transform into an activated state to differentiate and proliferate massively, releasing EBV to infect new B-cells, which may be associated with HL. 73 In this process, the main immune response of the organism to EBV + B-cells is performed by cytotoxic T-lymphocytes (CTLs),73,74 whereas the spread of EBV is mainly prevented by cells such as natural killer NK, γδ T, and CD8 T-cells. Thus, the function of immune cells is particularly important for the development of EBV-associated HL.
Previous studies have shown that elevated levels of anti-EBV antigen antibodies can be detected in patients with HL and that such elevated antibody levels occur before the development of HL. 75 EBV is associated with approximately 40% of cases in developed countries, and previous studies showed MCHL is more likely to be EBV-associated. 76 Furthermore, adolescent and older adult patients are particularly likely to be EBV+. 76 However, young adults with HL in developed countries are usually EBV negative. Taken together, the association of EBV and HL varies with age, subtype, and region of the world. 77
A large number of studies have reported genetic susceptibility genes associated with EBV + HL, and their pathogenesis can be broadly categorized into three stages: (1) genetic defects lead to immunodeficiency, (2) immunodeficiency causes EBV susceptibility, and (3) EBV infection triggers HL. These genes and their possible mechanisms for triggering EBV + HL will be described next, with a focus on the stage in which these genetic susceptibility genes lead to immunodeficiency (Figures 1–3).

Relationship between EBV, genetic lesions, and HL.

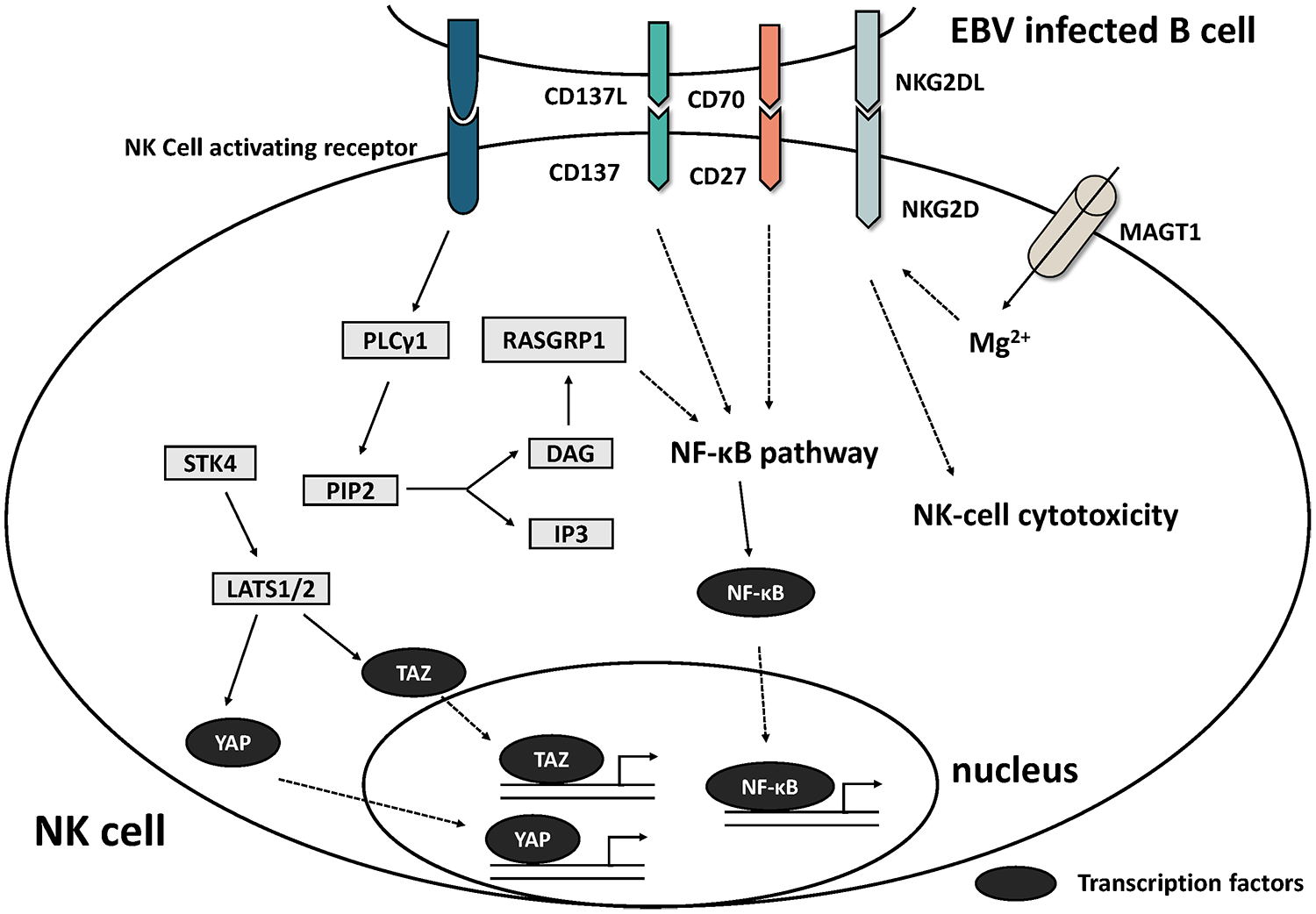
The activated pathway in T-cells after the recognition of EBV-infected B-cells.
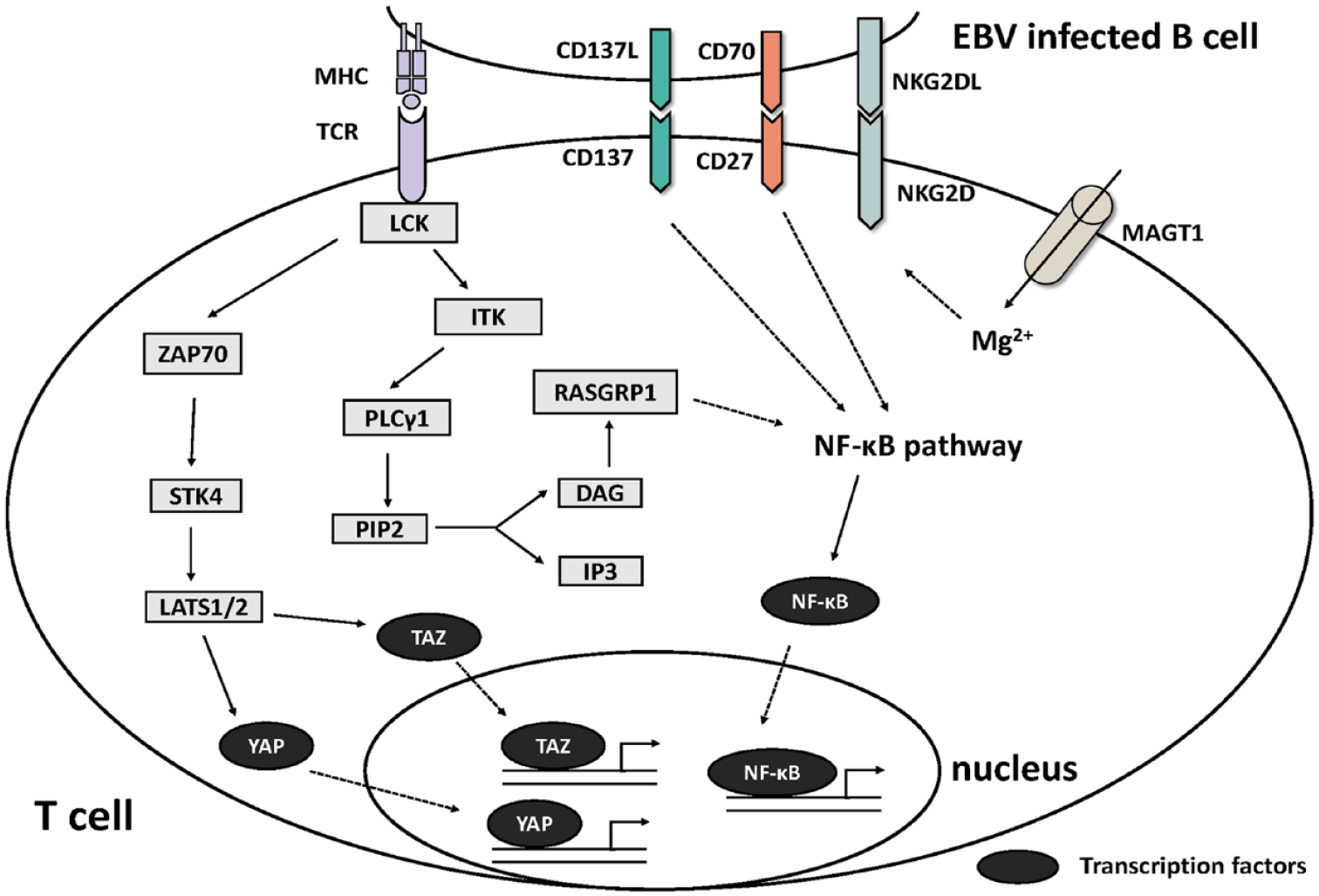
The activated pathway in NK cells after the recognition of EBV-infected B-cells.
The process of killing EBV + B-cells by CTLs is the main pathway by which immune system responds to EBV infection, a process that involves the activation, expansion, and killing of CD8 + T-cells. The proliferation of CTLs can be mediated by molecules such as TCR and CD27-CD70, in which pathways such as the MAPK pathway are involved and crossover occurs, ultimately initiating downstream signals and activating the target gene. It has been suggested that defective T-cell expansion is probably the primary explanation for a predisposition to severe/chronic EBV infection. 74
ITK is also associated with EBV + HL,3,4 and its encoded product is an interleukin (IL)-2-induced tyrosine kinase belonging to the TEC/BTK family, which is expressed exclusively in T-lymphocytes and NK cells. ITK can participate in T-cell receptor (TCR) signaling by phosphorylating and activating PLC-γ1, and the activated PLC cleaves PIP2 to generate two second messengers, namely, IP3 and DG. These two second messengers lead to processes such as the opening of calcium channels, activation of ERK (part of the MAPK pathway), the release of cytokines, and the reorganization of actin. 78 In mice, it has been shown that the absence of ITK affects CTL expansion and delays the expression of cytolytic effectors during activation. 79 Thus, ITK deficiency can lead to defective expansion and maturation of EBV-specific CTLs by interfering with TCR activation signaling. 6
The MAPK pathway plays an important role in the amplification of CTLs and NK cells. RASGRP1 has been reported to be one of the susceptibility genes of HL4,6 which is highly expressed in T-cells and NK cells, and encodes a small G protein RAS acting in the downstream RAF-MEK-ERK kinase cascade (also known as the MAPK pathway). In T-lymphocytes and NK cells, RASGRP1 is a major activator of the MAPK pathway, and RASGRP1-deficient cells show defective MAPK pathway activation accompanied by downregulation of CTPS1 expression, 74 which sustains the proliferation of activated lymphocytes by the MAPK pathway. CTPS1 encodes CTP synthase 1, and CTP is a precursor required for the metabolism of DNA, RNA, and phospholipids involved in DNA synthesis in lymphocytes. 80
CD27 encodes a protein belonging to the TNF receptor superfamily (TNFSFR), also known as TNFSFR7. CD27 binds to CD70 (also called TNFSF7), a ligand of the TNF superfamily, and is a co-stimulatory molecule for T-cell activation. The expression of CD70 is upregulated on EBV-infected B-cells, and CD70 drives EBV-specific CTL proliferation via TCR-CD27-dependent co-stimulation. Defects in CD27/CD70 are associated with EBV + HL, 4 with reports showing EBV + HL in four of six CD70-deficient patients7–9 and 3 of 18 CD27-deficient patients. 6 It was found that the expansion of EBV-specific cytolytic T-cells depends on CD70 expressed on the surface of EBV-infected B-cells via the co-engagement of TCR (actually CD3) and CD27 on T-cells. 7 Although the exact mechanism by which CD27-CD70 mediates CD8 + T-cell proliferation remains unclear, it has been suggested that CD27 can function as a co-stimulatory molecule of the TCR-dependent lymphocyte activation pathway,7,81 to some extent reinforcing the importance of CD27 and CD70 for EBV-specific T-cells. When CD70 is not available on EBV-infected B-cells or CD27 is not available on T-cells, the proliferation of EBV-specific T-cells is blocked, resulting in a reduced cytotoxic response to EBV-infected B-cells. 82
MAGT1 is a kind of magnesium ion transporter protein gene, and its loss-of-function mutation can lead to XMEN (X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia). 6 The function of MAGT1 mainly involves two aspects: driving magnesium ions into the cell and N-linked glycosylation (NLG). TCR activation was found to promote magnesium entry into cells as well as calcium release from the ER, and loss of MAGT1 impaired PLCc1 activation. However, the TCR-stimulated influx of both magnesium and calcium ions were reduced in normal human T-cells after the knockdown of MAGT1. The influx of both calcium and magnesium was restored in patients. 83 Recent studies have revealed that MAGT1, a subunit of the OST (oligosaccharyl transferase) complex, 5 is primarily localized to the ER and is a facilitator of NLG. MAGT1 deficiency negatively affects processes such as N-glycosylation. The reduced level of glycosylation of the EBV killing-related activating receptor NKG2D in such patients leads to its degradation and decreased expression in NK and T-cells. Decreased level of glycosylation and expression of CD28, CD70 and other genes were reported, 5 which affected the expansion of CTL activation and the killing of EBV + B-cells by CTLs and NK cells.
In addition, TNFRSF9 deficiency (CD137/4-1BB) affects the function of CTLs and NK cells, which may be related to HL [2/8 in reported IEI (inborn errors of immunity) patients].11,12 CD137 can be induced by CD137L (mainly expressed on dendritic cells, macrophages, activated T-cells, and B-cells) after the activation of CTLs and NK cells. Previous studies have shown that the CD137L-CD137 pathway results in the release of perforin and cytokines [such as interferon (IFN)] and that CD137-deficient patients often show a deficiency in this area. 4
Defects in CTLA4 can lead to CTLA4 haploinsufficiency (HI), an immune dysregulation disorder characterized by overactive T-lymphocytes and a generalized lymphoproliferative and autoimmune disorder. 6 Patients with CTLA4 haploinsufficiency have a dysregulated T-cell response to EBV infection secondary to excessive T-cell activation and apoptosis, senescence, and shedding.
JAK-STAT pathway and related genetic lesions
The JAK-STAT pathway plays an essential role in the occurrence of HL 20 (Figure 4). The persistent activation of the JAK-STAT pathway is also a feature of HL. Activation of these receptors activates receptor-associated JAK, which acts as a kinase to phosphorylate tyrosines in specific regions of the receptor and recruit proteins, including STAT. This pathological process can lead to hyperphosphorylation of a variety of STAT proteins, which induces carcinogenesis by transcriptionally regulating the activation of downstream targets, such as the proto-oncogene MYC, and abnormalities of this channel in cancer also influence the effect on tumor cells of cytokines secreted in the microenvironment.84,85
Graphical representation of the pathways in HL where genetic abnormalities frequently occur.
In HRS cells, cytokine receptors on the cell surface undergo conformational changes and bind to JAKs when activated by specific cytokines, causing the phosphorylation of JAKs. Phosphorylated JAKs continue to activate downstream molecules, such as STAT and PI3K, and regulate the expression of various survival molecules described above through the JAK-STAT, PI3K-ATK, Ras-MAPK, and other pathways. 20
Indeed, although constitutive activation of the JAK-STAT pathway is common in HL, mutations in the JAK gene are not. A JAK1 activating mutation that promotes constitutive activation of the JAK-STAT pathway has been reported in HL cell lines, but the mutation is relatively rare in patients. In addition, researchers have identified a gain-of-function mutation, namely, JAK2 V617 F, which maintains a sustained activation state and transmits signals to downstream signaling molecules even though no cytokine binds to the corresponding receptor.23,24 In another study involving JAK2 and HL, the investigators also identified a genetic abnormality caused by a translocation: t (4;9) (q21; p24), leading to the production of the fusion protein SEC31A-JAK2. SEC31A-JAK2 is present in approximately 3% of HL patients and promotes constitutive activation of the JAK-STAT pathway. 25 Notably, both of these JAK2 gene abnormalities are sensitive to JAK2 inhibitors, suggesting that targeted therapy with JAK2 antagonists may be effective in these patients. In a study involving 23 patients with new-onset HL, HRS cells were isolated from biopsied tumor tissues, and whole-exome sequencing was performed. The results revealed the presence of 9p/9p24.1 copy gain in approximately 13% of the cases, suggesting an association of JAK2 with HL, given that JAK2 was localized to 9p24.1. 17 Mutations in STATs, a downstream molecule of JAKs, are also more common in HL patients, 21 such as missense mutations occurring in the DNA binding domain of STAT6 (11/34), the activating mutation D661Y in the SH2 domain of STAT3, 22 and the activating mutation T628S in the SH2 domain of STAT5B.
Both SOCS and PTPN are negative regulatory molecules of the JAK-STAT pathway and inhibit the JAK-STAT pathway by dephosphorylating JAKs. Studies have found a possible association between genetic abnormalities in these genes and HL.15,16,86,87 In a study that included 105 cHL patients, researchers found that 61% of patients (64/105) had mutations in the SOCS1 gene and that patients with these mutations often had a worse prognosis than HL patients without SOCS1 mutations (P = 0.03), as judged by overall survival. In another study, PTPN1 mutations were found in 20% of HL patients (6/30) versus 67% of HL cell lines (6/9), and functional studies of mutant cells revealed enhanced JAK-STAT pathway activity and increased phosphorylation of some molecules, including JAK, in these cells, suggesting a possible association between PTPN1 gene mutations and HL pathogenesis. Similarly, abnormalities in the genes corresponding to GNA13 and ITPKB, which are regulatory molecules of the JAK-PI3 K-AKT pathway, were also found to be associated with HL. 21 In addition, the expression product of the XPO1 gene can assist molecules (transcription factors, etc.) in entering the nucleus through the nuclear pore complex, and through this mechanism, XPO1 is involved in multiple pathways regulating gene expression, including JAK-STAT and NFκB. A study found that the XPO1 E571 K mutation was present in approximately 24.2% of HL patients and that patients carrying this mutation had shorter progression-free survival (p = 0.0601), suggesting its potential value in the diagnosis and prognosis of HL.26,27
NFκB pathway and related genetic lesions
Persistent activation of the NFκB transcription factor is one of the hallmarks of HL and activated NFκB can be detected in almost all malignant HL cells. Unlike the NFκB pathway of T-cells mentioned previously, this is the NFκB pathway of HRS cells, whose downstream actions include the regulation of anti-apoptotic factors, the expression of proinflammatory CK, and B-cell reprogramming 2 (Figure 4).
Generally, the NFκB pathway can be divided into two categories: the classical pathway and the nonclassical pathway. In the classical pathway, CD40 and CD30 activate the IKK complex via TRAFs, and the activated IKK complex promotes the degradation of the NFκB factor (p50/p65) inhibitors IκBα and IκBκ. The released NFκB enters the nucleus through the nuclear pore complex and regulates gene expression. Similarly, CD40 activated by CD40L in the nonclassical pathway activates the IKK complex through MAP3K14, and the activated IKK complex promotes the conversion of p100 to p52, which in turn promotes the formation of the NFκB factor (p52/RelB).88,89
In the classical NFκB pathway, the genetic abnormalities reported thus far focus on several positive or negative regulators. The gene expression products of TNFAIP3 and CYLD are negative regulatory molecules. The coding product of TNFAIP3, A20, can degrade the classical NFκB pathway through the LUBAC (linear ubiquitin chain assembly complex)-A20 axis by ubiquitination. 90 Mutations in TNFAIP3 are more common in EBV-negative HL, with several studies finding TNFAIP3 mutations in approximately 40% of cHL patients.28–30 Follow-up functional assays have revealed the diminished function of the LUBAC-A20 axis after mutation. CYLD, similar to TNFAIP3, can also act as a tumor suppressor by encoding a deubiquitinating enzyme that inhibits the NFκB pathway, but in known studies, CYLD mutations (mostly deletions) are mainly present in HRS cell lines36,37 and are less common in HL patients. NFKBI encodes NFκB inhibitors, including IkBα and IkBε. NFκB inhibitors inhibit the NFκB pathway by binding to NFκB and inhibiting its function. Common NFKBI genetic abnormalities include SNVs and deletions,31–35 leading to a decrease in its function and promoting the persistent activation of the NFκB pathway. The coding product of REL is one of the transcription factors of the classical NFκB pathway, and the most common REL abnormality in HL patients is amplification (56/138), so there is no doubt that genetic abnormalities of REL are potentially associated with the persistent activation of the NFκB pathway.39–43 In addition, there is an important gene (BCL3) in the classical NFκB pathway that frequently undergoes CNV or translocation.39,91,92 The BCL3 protein promotes NFκB activity by binding to the p50 homodimer and increasing its activity.
In the nonclassical NFκB pathway, genetic lesions of the MAP3K14 and TRAF3 genes are relatively common. As mentioned above, MAP3K14 (NFκB-inducing kinase, NIK) activates the IKK complex after receiving CD40 signals and promotes the formation of the NFκB factor (p52/RelB). In contrast, TRAF3, an inhibitory molecule of the nonclassical NFκB pathway, leads to the degradation of NIK. In the absence of upstream signals of the nonclassical NFκB pathway, NIK is degraded by TRAF3 and kept at a low level. In contrast, in the presence of upstream signals, TRAF3 is degraded, and the level of NIK increases, mediating the conduction of the nonclassical NFκB pathway. According to the study, a common abnormality of the MAP3K14 gene is copy number gain,38,41 while the TRAF3 gene often undergoes deletions, 38 with the former promoting NIK activation of the IKK complex and the latter decreasing the inhibitory effect of TRAF3 on NIK.
In addition, there are genes whose functions are not fully defined or cannot be classified solely as classical/nonclassical NFκB pathways. For example, TNFRSF14 (CD270, HVEM) is considered to be a tumor suppressor gene in other B-cell lymphomas, and its deletions are found in approximately 20% of cHL patients.41,44 The AKT1 gene often undergoes copy number gain and is thought to be associated with the NFκB pathway. 93 NOTCH1, one of the transcription factors of the NFκB pathway, also frequently undergoes copy number variation. 93
Immune evasion in HRS cells and related genetic lesions
Previous studies have shown that the human immune system is capable of killing tumor cells, but malignant tumors often possess the ability of immune escape. The mechanisms of immune evasion are diverse and include the tumor microenvironment, genetic alterations, and so on. Specifically, in cHL, previous studies have identified chromosome 9p/9p24.1 abnormalities as one of the most common genetic abnormalities in cHL, and correspondingly, increased 9p/9p24.1 copy number often leads to increased expression of PD1 ligands in HRS cells. These HRS cells bind to PD1 receptor-positive cells and undergo immune evasion through the PD1 signaling pathway. 94 In other words, the immune evasion by HL not only prevents the immune system from killing itself but also facilitates the participation of these immune cells in the formation of the tumor microenvironment, which contributes to the survival of HRS cells.
As mentioned above, the PD1-PDL1 pathway plays a significant role in immune evasion in HL patients, and abnormalities in related genes are often associated with the amplification of 9p24.1.17,48,94 In addition, there are other immune evasion-related pathways. For example, CD4 and CD8 cells kill HRS cells through TCR-MHC I/II, but MHC copy loss is often present in cHL patients.95,96 In addition, B2M, which is one of the components of the MHC I molecule, also frequently undergoes inactivating mutations. 54 The coding product of CIITA (Class II Major Histocompatibility Complex Transactivator) is located in the nucleus and functions as a positive regulatory molecule of MHC II transcription. This study identified tandem duplication and balanced translocations of the CIITA gene (HLA-DQB1).52,53 The significance of the former is unclear, but functional assays have found that the latter can lead to CIITA inactivation and downregulation of MHC II expression. FAS (TNFRSF6, APT1, CD95) molecules carry the death domain (DD) and are involved in the formation of the death-inducing signaling complex (DISK), activating caspase molecules and leading to proteolytic cell destruction. In vitro experiments have shown that HRS cells are resistant to apoptosis induced by the FAS pathway and that this resistance is associated with mutations in FAS.49–51,97 The germline mutation p.I1711M in Dicer raises the risk of HL, 65 and the coding product of Dicer is an endonuclease containing highly conserved tandem endonuclease structures, namely, RNase IIIA and RNase IIIB, which are required for microRNA (miRNA) biosynthesis and several other RNA interference phenomena. Their mutations lead to impaired expression of tumor suppressor miRNAs.65,98
The somatic hypermutation of immunoglobulin genes in B-cells results in different B-cell receptors (BCRs). In the dark zone of the germinal centers, these B cells can be divided into two categories according to the affinity of the BCR (Figure 5). B-cells with a high-affinity BCR or non-self-reactive BCR can differentiate into plasma cells, memory B cells, and so on. B-cells expressing a low-affinity BCR or self-reactive BCR will be eliminated by Fas-mediated apoptosis in the light zone of germinal centers. 99 It has been postulated that HRS cells may be derived from B cells that escape Fas-mediated apoptosis in the dark zone, although the details remain to be uncovered.99,100 Mutation of the FAS gene may participate in this process to some extent.

The selection of B-cells in germinal centers in classical Hodgkin lymphoma.
Genetic lesions in nodular lymphocyte-predominant HL
NLPHL is a kind of follicle-derived germinal center B-cell lymphoma, 1 whose tumor cells are not HRS cells but LP cells (lymphocyte-predominant). Typical immunophenotype for NLPHL is CD20+, CD45+, CD79a+, BCL6+, PAX-5+; CD3–, CD15–, CD30–. For cHL, the type is CD15+, CD30+, PAX-5+ (weak); CD3–, CD20– (majority), CD45–, CD79a–. LP cells without CD30 and CD15 expression are consistently positive for CD20. Due to the absence of CD30 and CD15, NLPHL is not associated with EBV infection. 2 The first-line treatment of stage IA NLPHL is involved in Site Radiation Therapy. And in intermediate-stage and relapsed NLPHL, the active treatment consists of conventional chemotherapy, anti-CD20 antibodies, and radiotherapy. Individuals with NLPHL usually have a good prognosis. 101 Notably, genetic susceptibility genes associated with tumor cells have also been identified in NLPHL, including NPAT and TET2T.
Due to the mutation of SOCS1, the constitutive activation of JAK/STAT and NF-κB pathway is also shown in LP cells. 87 However, many mutations frequently found in HRS cells seem to be less frequent in LP cells, and the distinct genetic lesions of NLPHL have not been observed in cHL. 102
Three highly recurrently mutated genes (DUSP2, JUNB, and SGK1) were revealed in an analysis of 62 genes in NLPHL by targeted ultradeep sequencing. 103 DUSP2 is a negative regulator of MAP, ERK, and JNK, though the role of DUSP2 in NLPHL remains to be clarified. 104 Truncating mutations of JUNB are frequently observed in LP cells, 103 and Szremska AP’s study indicates that the JUNB possesses the tumor suppressor function in B-cells. 105 The inhibition of SGK1 can induce apoptosis of NLPHL cell line DEV, which highlights the potential value of SGK1 in the treatment of NLPHL. 103 Moreover, the translocations affecting BCL6 have been identified in one-third of NLPHL cases.106,107
In an NLPHL family containing four cousins, an investigator found a truncating germline mutation of NPAT (nuclear protein, ataxia-telangiectasia locus) associated with NLPHL. 63 The encoded product of NPAT is closely related to the regulation of the cell cycle, and NPAT can be phosphorylated by the cell cycle protein E-dependent kinase 2 (CDK2) complex. Its expression level in the G1 to S phases of the cell cycle is consistent with the activity of the cell cycle protein E-CDK2 complex, which promotes the shift of cells to S phase. Mutations in NPAT can lead to cell cycle NPAT, which also contributes to the activation of the ATM promoter, and mutations in ATM can be seen in a variety of hematological malignancies. 63 TET2T is a gene that predisposes to familial DNA demethylation. Its mutations lead to an increase in the methylation levels in the binding regions of major transcription factors involved in hematopoiesis, 60 thus affecting the function of transcription factors with a binding preference for nonmethylated DNA, including RUNX1/2/3 and PU.1, leading to hematological aberrations. The association of these NLPHL-related genes with cHL has not yet been reported.
Treatment of HL
The treatment of HL has a long history, and the most commonly used chemotherapeutic regimen, namely, ABVD (adriamycin, i.e., doxorubicin, bleomycin, vinblastine, and dacarbazine), was introduced more than 40 years ago. 108 For relapsed/refractory cHL, several novel therapies have been put into clinical use, and most of these novel therapies are related to the signaling pathways mentioned above109,110 (Table 2). As seen in Table 2, targeted therapies for HL have focused on molecules such as CD30 and PD1. The former is upregulated in HRS cells and is involved in the hyperactivation of the NFκB pathway, while the latter is often abnormal at the genetic and chromosomal levels in HL patients, leading to immune evasion. In addition, the overactivated JAK-STAT pathway is one of the common targets, for example, ITF2357 was found to kill cells with mutated JAK2 (V617F), and idelalisib and everolimus can target molecules of the PI3K/AKT pathway, which can be activated by JAK.
Clinical trials related to pathways above with exact results.

AVD, doxorubicin, vinblastine, and dacarbazine; CD, cluster of differentiation; CI, confidence interval; CMR, complete metabolic response; CR, complete remission; EFS, event-free survival; HDAC, histone deacetylase; HL, Hodgkin lymphoma; ICE, ifosfamide, carboplatin, etoposide; JAK, janus kinase; mTOR, mammalian target of rapamycin; N/A, Not available; OR, overall survival; ORR, objective response rate; ORS, overall response rate; OS, overall survival; PD, progression disease; PD1, programmed cell death protein 1; PFS, progression-free survival; PR, partial remission; SD, stable disease.
Trial reference refers IDs found at ClinicalTrials.gov.
Antibody-drug conjugate
ADCs (antibody-drug conjugates) represent the coupling of an antibody to a cytotoxic drug, which are connected by a special chemical bond and contain three parts: the monoclonal antibody, connector, and cytotoxic small molecule (payload). 110 The main choice of mAb is IgG1, which has the advantage of a strong half-life and can mediate potent ADCC and CDC processes. In addition to IgG1, IgG2, and IgG4 and immunoglobulins other than IgG can be used as mAbs for ADCs. The ADC currently approved for HL is brentuximab vedotin (BV), which has the structure of anti-CD30 mAb + protease-cleavable linker + MMAE (a microtubule inhibitor). As mentioned earlier, HL patients tend to have genetic abnormalities in the NFκB pathway and enhanced signaling in this pathway, and CD30, an upstream molecule of the NFκB pathway, is upregulated in HRS cells, so BV can be used to target and kill HRS cells.
Immunotherapy
As mentioned above, immune evasion is very common in HL patients. This process encompasses multiple signaling pathways as well as multiple genetic abnormalities, such as PD1-PDL1 and FAS. Immunotherapy has been applied to relapsed refractory HL, including pembrolizumab and nivolumab, which are PD1 antagonists that act mainly by inhibiting PD1-PDL1, which is upregulated in HL patients. The PD1-related mechanisms are described above. However, reports have shown that patients with HL treated with immunotherapy are at risk of developing an autoimmune reaction. This process may be caused by overactive T-cells, and patients often present with skin rash, pneumonitis, and so on.111–113
Ongoing HL clinical trials
In addition to conventional chemotherapy, ADC, immunotherapy, and HSCT, novel therapies for relapsed refractory HL are under investigation, such as Camidanlumab tesirine and CAR-T therapy (Chimeric Antigen Receptor T-Cell Immunotherapy).114–116 The former is an ADC based on an anti-CD25 monoclonal antibody with tesirine. 117 The latter is an emerging therapy characterized by the artificial construction of CAR-T cells that recognize any cell surface structure and produce cytotoxicity. In HL, clinical studies of CAR-T focus on anti-CD30 CAR-T (Table 3). CD30 is upregulated on the surface of HRS cells and plays an important role in the sustained activation of the NF-κB pathway (Figure 4).
Ongoing HL clinical trials evaluating CD30 CAR-T.

CAR-T, chimeric antigen receptor T-cell immunotherapy; CD, cluster of differentiation; CI, confidence interval; CR, complete remission; HL, Hodgkin lymphoma; NR, no response; OS, overall survival; PFS, progression-free survival; SD, stable disease.
Trial reference refers IDs found at ClinicalTrials.gov.
Considering the frequent genetic lesions and associated pathways in HL patients as well as in HRS cells, treatment combined with genetic testing may be of some benefit. As mentioned above, the main genetic abnormalities in HL patients are most commonly found in the JAK-STAT, NFκB, and other pathways, especially in the REL and TNFAIP3 genes. Therefore, patients with these genetic abnormalities can be treated with drugs such as inhibitors of the relevant pathways. In addition to the above mentioned treatments, there are many drugs targeting HL that are still in clinical trials (Table 4), for example, various PD1 inhibitors, such as CS1001 and TQB2450; ipilimumab, which targets CTLA-4; itacitinib, which inhibits the JAK-STAT pathway; and LMP-specific CTLs for EBV-positive lymphoma.
Ongoing HL clinical trials evaluating other novel agents.

ASCT, autologous stem cell transplant; BARF1, BamH1-A Reading Frame-1; BTK, Bruton’s tyrosine kinase; CDK9, cyclin-dependent kinase 9; CI, confidence interval; CR, complete remission; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; EBNA1, Epstein–Barr nuclear antigen 1; HDAC, histone deacetylase; HIV, human immunodeficiency virus; HL, Hodgkin lymphoma; JAK, janus kinase; LMP1, latent membrane protein 1; mTOR, mammalian target of rapamycin; OS, overall survival; PD, progression disease; PD1, programmed cell death protein 1; PFS, progression-free survival; PR, partial response; RNA, ribonucleic acid.
Trial reference refers IDs found at ClinicalTrials.gov.
LMP, BARF-1, and EBNA1-specific cytotoxic T-lymphocytes.
Potential therapeutic targets
The essential role of JAK-STAT pathway and NF-κB pathway in HL has been proved in ‘Genetic lesions and related pathways’, which emerged as promising targets for the treatment of HL, and parts of the novel therapies were listed in Tables 2–4. And the inhibitors of positive regulators, transcription factors, and some kinases (Figure 2) are also promising targets for the treatment of HL. For example, lestaurtinib, a multikinase inhibitor, can inhibit the phosphorylation of JAK2, STAT5, and STAT3.118,119 PIM serine/threonine kinases are downstream molecules of JAK-STAT and NF-kB pathways, and pan-PIM inhibitor targeting PIM kinases is included in the preclinical setting. 120 As shown in Figure 2, the PI3K-AKT pathway is constitutively active in HRS cells, providing a strong rationale for targeting PI3K/AKT/mTOR in cHL patients.121,122 Genetic lesions lead to the loss of function of some molecules in HL, especially those negative regulators and tumor suppressors. Recently, Boice et al. 123 have restored the tumor-suppressive effects of TNFRSF14 in a xenograft mouse model through the HVEM ectodomain delivered by CD19 CAR-T cells.
Immune-related genetic lesions and pathways may broadly get involved in anti-EBV immunity and the immune surveillance of B-cells. The alteration of those genes such as PD1 could enable malignant B cells to escape from T-cells, and the related abnormalities molecules can be optimal targets for therapy since PD1 and CTLA4 inhibitors are under clinical trials. For EBV-associated HL with MAGT1 mutation, the role of magnesium supplementation in vivo and in vitro is being investigated. 5 Moreover, the idea of using EBV-related antigens (LMP, EBER, and EBNA1.1) as therapeutic targets was suggested 20 years ago. 124 In primary immunodeficiency-related HL with genetic lesions (such as ITK3, MAGT1, RASGRP14, CD27, CD70, TNFRSF9, and STK4), the restoration of normal immune function is helpful, which could be achieved through allogeneic BMT.125,126 Furthermore, gene therapy is a promising direction for future research.
Future perspectives
Although the effect of some genetic aberrations that involve genes like JAK has been elucidated, the exact function of many genetic lesions remains less well defined. To solve these problems, animal models and in vivo studies are necessary. Furthermore, it may be meaningful to analyze the data from different HL groups. For example, sequencing of relapsed/refractory HL cases will be important to explore the mechanisms in resistance, and the sequencing data from newly diagnosed patients would hint the causes of HL. Moreover, the appearance of ctDNA and single-cell sequencing also will be helpful in identifying treatment-dependent patterns of clonal evolution and mirroring HRS cell genetics.
The introduction of the concept of precision medicine and the development of technologies such as sequencing have gradually highlighted the association between a patient’s genetics and disease, which is reflected in various aspects, such as diagnosis and prediction of prognosis. It has been found that next-generation sequencing of patients’ circulating tumor DNA (ctDNA), for example, can assist in determining the genetics and outcome of HL patients at different clinical stages. 127 When combined with the common genetic lesions of HL mentioned above, ctDNA monitoring allows for the identification of the early-stage patients with refractory HL as well as the detection of residual disease in patients during treatment or remission. Besides, the cell genetics of HL can be mirrored by ctDNA, which could be further exploited as an easily accessible method to distinguish cHL genotypes. Fortunately, consistent with the assumption, Camus et al. 128 and Spina et al. 127 detected the variants of ctDNA to characterize the genetic features of cHL at the time of diagnosis, which greatly facilitate the assessment of early treatment response.
In addition, considering the existence of mechanisms that can trigger chromosomal instability, such as POT1 gene abnormalities, as well as the association between primary immunodeficiency, EBV, and HL mentioned in the previous article, it can be speculated that specific genetic lesions may be associated with the development of HL. Therefore, by monitoring the presence of specific genetic lesions, it might be possible to predict the risk of patients developing hematological malignant diseases such as HL. In addition, for patients who already have HL, targeted therapies that target abnormal pathways involved in these abnormalities may be a new direction for future treatment.