
Abstract
Background:
DDX41 serves as a DNA sensor in innate immunity and mutated DDX41 is pathogenic, mainly for myeloid neoplasms.
Methods:
In this study, “DDX41” was searched in PubMed and Web of Science between 1 January 2015 and 29 April 2021 with individual-patient data seeking. A meta-analysis was not valid here due to the absence of a large dataset. Thirty articles were finally included in the qualitative analysis and 277 patients from 20 studies without overlap were involved in the quantitative summary.
Results:
Pooled incidence was 3.3% (95% confidence interval 2.4–4.2%) of unselected myeloid neoplasms. Patients with hematologic disorders harboring mutated DDX41 were featured as 80% males, median 66 (20–88) years old at diagnosis, 75% acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS), 64% with normal karyotype. Eighty-five percent of patients had germline variants which were nationally diverse and more of frameshift type, whereas 64% of patients had somatic DDX41 variants where p.R525H and missense dominated. ASXL1 and TP53 were the top frequent concomitant somatic mutations. Therapeutically, 70% overall response rate was obtained of hypomethylating agents in MDS, 96% complete remission of chemotherapy in AML, and 8% of relapse in hematopoietic stem cell transplant. Neither overall survival nor progression-free survival could be summed.
Conclusions:
Several significant clinical differences were observed in different diagnosis groups, familial and sporadic cases, and p.R525H compared with other somatic variants. In conclusion, myeloid neoplasms carrying DDX41 mutations were mainly older, male, MDS, and AML patients who had promising responses to treatment. Both germline and somatic DDX41 variants possessed unique characteristics and groups of interest presented certain differences worth further research. (CRD42021228886)
Keywords
Introduction
Although most myeloid neoplasms are sporadic cases affecting the elderly, those malignancies with germline predisposition are increasingly recognized to comprise a distinct subtype. 1 The World Health Organization has included myeloid neoplasms with germline predisposition into its classification since 2016. 2 Especially, DEAD-box helicase 41 (DDX41) mutation is clinically unique due to its late onset compared with other germline lesions, such as CEBPA and GATA2. 3 As increasingly recognized as clinically significant, DDX41 was more frequently added to tumor sequencing panels for identifying variants responsible for myeloid malignancies.4,5 DDX41 is a versatile protein not only functioning as a DNA/RNA sensor in innate immunity but also involved in transcription by altering the splicing process. 6 Recently, DDX41 in zebrafish was reported to be a crucial gatekeeper of over-producing hematopoietic stem and progenitor cells, by inhibiting excessive R-loops and thus preventing an inflammatory cascade. 7 This study has shed light on how DDX41 mutants lead to a clinically significant subtype of myeloid neoplasms. Moreover, deficient DDX41 in erythroid progenitors would diminish their proliferation and impair differentiation, thus manifesting anemia clinically. 8 Although previous reviews summarized some molecular and clinical features of DDX41-mutated myeloid neoplasms,9,10 a systematic review is still essential due to emerging publications on DDX41-related diseases and low prevalence in the population, to provide a comprehensive summary of their clinical characteristics by incorporating both large cohorts and individual case reports. Thus, our study aimed to report the clinical features of DDX41-mutated diseases, mainly myeloid neoplasms but also including other hematologic disorders, and describe phenotypes of different groups utilizing individual-level data. The molecular pathway of DDX41 mutation is beyond the scope of this systematic review.
Methods
A meta-analysis was not available because of a lack of large datasets, but a comprehensive systematic review is still feasible. Moreover, individual patient data (IPD) is available, thus the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement 11 was followed for this study. Two authors (Z.W. and B.H.) independently performed database search, data extraction, and quality assessment. Any disagreement would be resolved by discussion until consensus was reached. This study was registered at PROSPERO (CRD42021228886).
Database search
The term “DDX41” was searched in PubMed and Web of Science with no language restriction between 1 January 2015 and 29 April 2021 since the first article of DDX41-mutated myeloid neoplasms published in 2015. 12 We refined the document types of items from Web of Science to automatically exclude corrections, patent, books, data sets, meetings, editorials.
Eligibility criteria
Studies were included if (1) involving patients with DDX41 mutation; (2) their patients were diagnosed, including but not limited to myeloid neoplasms, aplastic anemia, lymphoma, solid cancers; (3) of case series, such as cohort studies, and case reports; (4) the site of DDX41variants, either somatic or germline, was reported.
We excluded inappropriate publication types, including reviews, comments, errata, books, patents. Studies with irrelevant subjects were ruled out, those developing vaccines utilizing the anti-viral response of DDX41 and those exploring the molecular mechanism of DDX41. Any relevant review was identified to find any additional related study, but not included in the result.
Studies that screened DDX41 mutation within unselected patients with myeloid neoplasms were selected to report the pooled DDX41 mutation incidence.
Data collection
The following individual information was collected carefully from the original articles and their supplementary files: (1) study of origin; (2) patients’ original number; (3) age at diagnosis, gender, nationality, diagnosis, family history of cancer; (4) type of DDX41 mutation, somatic and germline with sample type for germline verification; (5) DDX41 variant site (p.) along with its variant allele frequency (VAF; %) and transcript number for reporting it; (6) concomitant somatic mutation of other genes, also with specific sites and VAF; (7) karyotype; (8) treatment therapy; (9) response or clinical course; (10) overall survival (OS), months.
Information of studies selected for pooling DDX41 incidence on hematologic malignancies was also incorporated: (1) characteristics of publication (first author, publication year); (2) the number and type of screened population; (3) the number of DDX41-mutated patients. The missing data was requested by contacting the corresponding authors, otherwise recorded as not available and not included in the analysis.
Quality assessment
As there was not a specific recommended tool to assess the quality of observational studies, 13 we independently utilized two aspects specific to this study: completeness of patients’ clinical records (age at diagnosis, gender, nationality, diagnosis, family history investigation, clinical course with therapy and outcome), full information about DDX41 mutation (mutated site, VAF, origin, germline verification method).
Statistical analysis
For IPD, descriptive variables were presented with numbers and percentages. Continuous variables were provided as median with range if not specially mentioned. Familial cases were those with clear family histories of hematologic diseases and sporadic cases were those with no related family history after investigation. Groups of interest were compared, three different diagnoses (acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and others), familial and sporadic cases, p.R525H and other somatic variants, using either Fisher’s exact test or Kruskal–Wallis test. p < 0.05 was considered statistically significant.
DDX41 mutation incidence was pooled by a random-effects model commonly used when conducting a meta-analysis. The number of studies included was too small for a regular small study effect, that is, publication bias. All statistical analysis was performed using R version x64 3.6.3.
Results
Study selection
The numbers of publications identified in PubMed and Web of Science were 100 and 95, respectively. After deleting duplicates and going through title and abstract review 107 remained. We excluded 77 publications, including 41 animal studies, 20 about irrelevant subjects, 13 reviews, and three in languages other than English and Chinese. The remaining 30 were next given full-text reviews and all were considered eligible for this study, including nine case series,12,14–21 11 case reports,22–32 four reports about DDX41-related solid cancers,33–36 and six rare case reports37–42 (Figure 1). No extra study was identified from the references listed in the relevant reviews and the included studies. The reasons to include these 30 articles are presented in Supplemental material Table S1 online. Two hundred and seventy-seven patients with DDX41-mutated hematologic disorders, from 20 articles without overlap, were included in the individual patient-level dataset (Supplemental Table S2). Other DDX41-related diseases were described. Quality evaluation is presented in Supplemental Table S3.
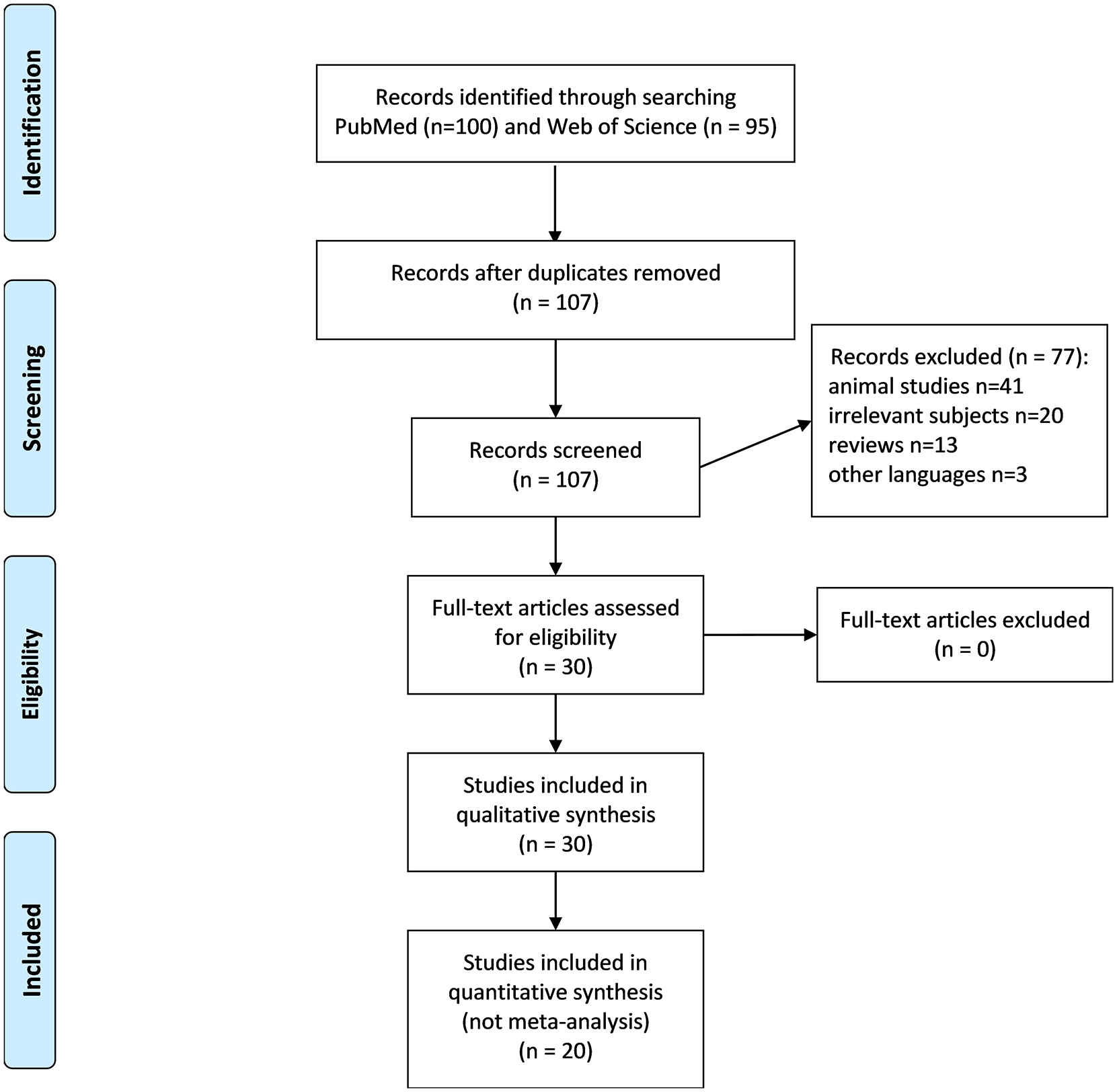
PRISMA flow diagram of selected studies.
DDX41-mutated hematologic disorders
The pooled incidence of DDX41 mutation among 10,975 screened patients with myeloid neoplasms was 3.3% [95% confidence interval (CI) 2.4–4.2%, I2 = 79.7%, p < 0.01; see Supplemental Figure S1 for forest plot]. Among 277 patients, 258 were from case series (93.1%), whereas others were from case reports (6.9%). The baseline information is collectively displayed in Table 1 and details are fully presented in Supplemental Table S2. The median age at diagnosis was 66 (20–88) years, 69.3% in the 60s and 70s [Figure 2(a)]. But DDX41 mutation was not only responsible for adult hematologic diseases, but also for pediatric leukemia, as a case report described. 39 Two hundred and twenty-two (80.1%) were male, and 52 (18.8%) were female, and gender of three cases (1.1%) was not described in the original article. Regarding nationality, most were American [115 (41.5%)], followed by 48 Chinese (17.3%), 43 French (15.5%), and 40 Korean (14.4%). Other nationality groups contained 10 cases at most [Table 1 and Figure 2(b)]. The portion of the normal karyotype was 63.9% [177/277; Figure 2(c)]. Karyotypes of 57 patients (20.6%) were described as abnormal including seven trisomy 8, six -Y, six del (5q), six del (20q), 10 complex, and 22 other less common non-complex (see Supplemental Table S2 for details).
Clinical features of 277 patients with DDX41-related hematologic diseases.

Nine cases were confirmed as non-complex but karyotypes were not clear.
AML, acute myeloid leukemia; CLL, chronic lymphoid leukemia; CML, chronic myeloid leukemia; ICUS, idiopathic cytopenia of undetermined significance; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasms; NA, not available; t-AML, therapy-related AML; t-MDS, therapy-related MDS.
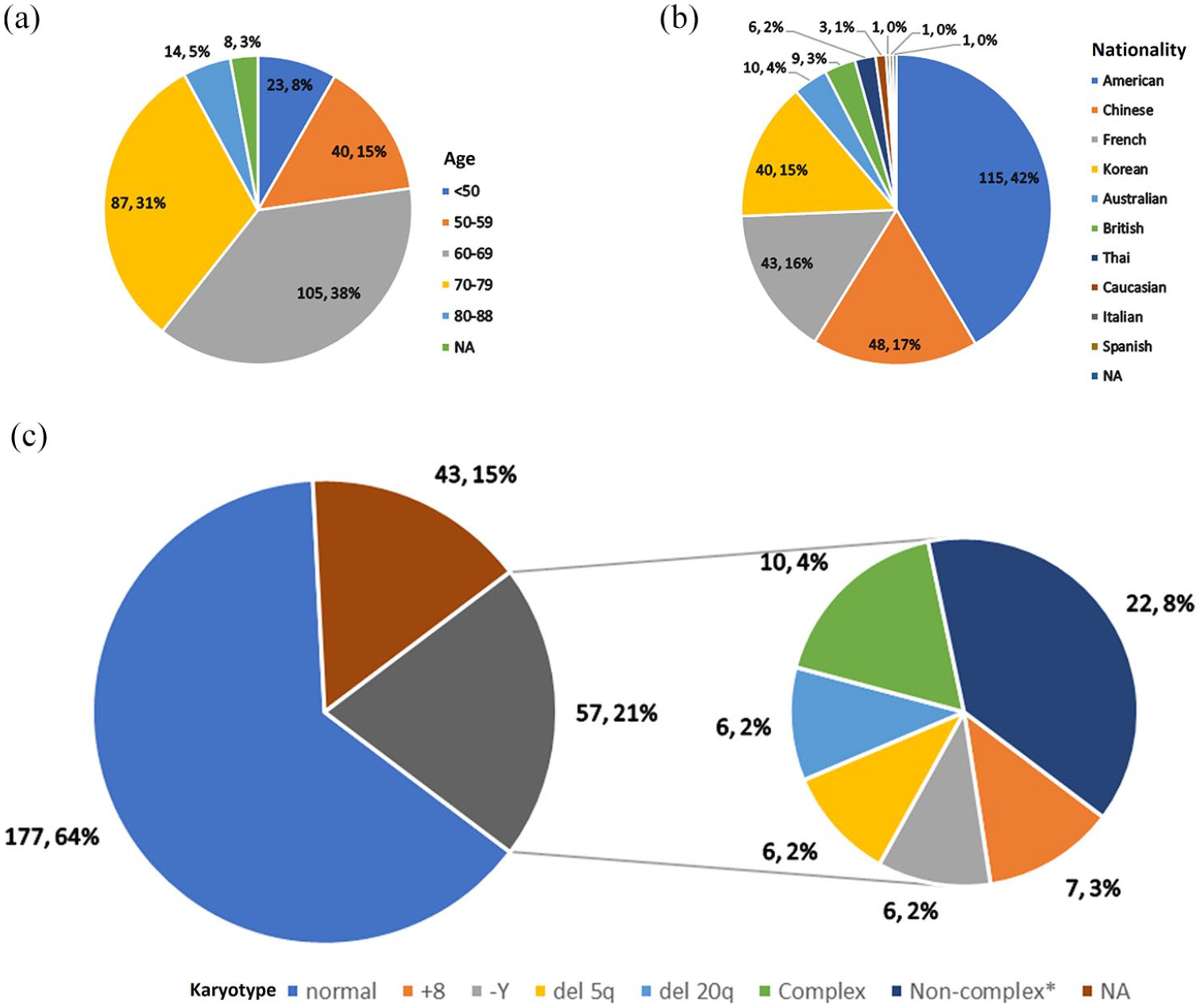
Age, nationality, and karyotype of 277 patients with DDX41-mutated hematologic diseases.
DDX41 mutations were most commonly diagnosed with myeloid neoplasms, including MDS (115, 41.5%), AML (91, 32.9%), and myeloproliferative neoplasms (n = 11, 4.0%). Though rare, other hematologic diseases were also reported, such as three follicular lymphomas, three aplastic anemia (AA). The rest relevant diseases are presented in Table 1. AML, MDS, and other diagnoses (n = 38) are compared (Supplemental Table S4). All three groups presented noticeable clinical features. AML harbored more germline p.D140fs and more cytogenetic abnormalities than MDS and other diseases (p = 0.01 and <0.001). MDS, however, was associated with more normal karyotypes. And male preference was more remarkable for AML and MDS compared with other diseases (p = 0.03). Other hematologic diseases (not AML or MDS), were diagnosed at a younger age (p = 0.06), of a higher female proportion (p = 0.03), detected fewer somatic variants (p = 0.04), and less mutated p.R525H (p < 0.001).
Ninety-four (33.9%) had clear family histories (familial), whereas 139 (50.2%) had no related family history after investigation (sporadic) and 44 were not available from original reports. A significant mean age gap was observed in that familial cases were diagnosed 4 years earlier than sporadic cases (p = 0.001; Supplemental Table S5). Some germline variants, at least p.D140fs and p.M1I, were probably more enriched in familial cases compared with sporadic cases (p = 0.07 and 0.10), whereas others were more detected in sporadic cases [odds ratio (OR) = 0.41, 95% confidence interval (CI) 0.21–0.78), p = 0.004]. This could be because p.D140fs and p.M1I are two variants described earlier, thus patients with such variants were possibly investigated more thoroughly regarding their family history. Sporadic cases were diagnosed with MDS more; however, this difference could result from a large portion (32/139, 23%) of not available diagnosis in the sporadic group. No difference was found in gender, somatic variants, or karyotype (Supplemental Table S5).
In terms of DDX41 variants, all referred to NM_016222, but in their latest version by each study (version 2–4). Suspected germline variants based on next-generation sequencing were detected in 236 patients (85.2%). Thirty-nine cases (14.1%) were demonstrated as not carrying germline DDX41 variant. Three cases harbored two germline DDX41 variants and two had no available germline information. One hundred and eighty-one probable germline variants (76.7%) were confirmed of germline origin by cultured skin fibroblasts, oral epithelial cells, clear transmitting pedigree, and others (Supplemental Table S2). The other 55 had no germline verification other than 40–60% VAF. Missense variants composed the largest part (124/239, 52%), followed by frameshift (n = 76, 32%), nonsense (n = 20, 8%), splicing (n = 14, 6%), and last in-frame deletion/duplication (n = 5, 2%). Frameshift [OR = 43 (95% CI 11–370), p < 0.001], nonsense [OR = 8.5 (95% CI 2.0–75.9), p < 0.001], splicing variants [OR = 3.8 (95% CI 1.1–21.2), p = 0.026] showed their strong associations with germline origin. The top five germline variants were p.D140fs (n = 39), p.M1I (n = 29), p.V152G (n = 12), p.Y259C (n = 10), p.A500fs (n = 7). They showed an apparent difference in different countries: p.M1I and p.D140fs in the US, p.V152G, p.Y259C and p.A500fs in Korea and China (Figure 3). DDX41 mutation was negatively detected in Hungary. 40 One hundred and thirty-five patients had both germline and somatic DDX41 mutations.

DDX41 protein map based on 20 population studies.
Somatic DDX41 variants were detected in 176 patients (63.5%) including 13 cases with two somatic variants. Sixty-six patients had no somatic DDX41 variant and 35 did not have available information about it. Missense variants still contributed the most (181/189, 96%), followed by splicing (n = 3), frameshift (n = 2), nonsense (n = 2), and last in-frame deletion (n = 1). The type of missense was significantly enriched in somatic variants [OR = 21 (95% CI 10–51), p < 0.001]. Different from diverse sites of frequent germline DDX41 mutants, the most common somatic variant was p.R525H, which accounted for 64.2% (n = 113) of the variants and was consistent in all countries (Figure 3). The other frequent somatic variants of DDX41 were p.T227M (n = 12), p.P321L (n = 9), and p.G530D (n = 6). We compared patients carrying p.R525H and other somatic variants (Supplemental Table S6). Interestingly, somatic p.R525H may link to certain germline variants; p.M1I at least [OR = 9.22 (95% CI 1.32–402), p = 0.01]. Moreover, somatic mutation sites were possibly associated with types of hematologic diseases: p.R525H could be a possible risk factor of AML [OR = 2.91 (95% CI 1.28–7.08), p = 0.007] and other somatic variants could link to less common DDX41-related hematologic diseases, such as AA [OR = 0.19 (95% CI 0.05–0.63), p = 0.003]. No other difference was found in age, gender, or karyotype.
One hundred and twenty-nine (46.6%) patients harbored at least one concomitant somatic mutation besides DDX41. Ninety-eight (35.4%) had no somatic mutation of other genes and 50 had no available report about it. DDX41 coincided with ASXL1 mutation most commonly (34/129, 26.4%), followed by TP53 (29/129, 22.5%). Other frequent concomitant lesions were TET2 (17/129, 13.2%), EZH2 (17/129, 13.2%), SRSF2 (16/129, 12.4%), DNMT3A (14/129, 10.9%), and CUX1 (11/129, 8.5%).
Clear treatment records were obtained from 155 patients in 11 studies consisting of 83 MDS, 52 AML, 20 other diagnoses. Fifty MDS patients received hypomethylating agent (HMA)-based treatment. Out of 33 with clear responding information, 23 responded (70%) and 10 were either a failure, stable disease, or not responded. Ten MDS were under observation and another eight received hematopoietic stem cell transplant (HSCT), without analyzable responding evaluation. Three MDS patients were treated with lenalidomide (LEN) and two reached complete remission (CR); the other one was without responding information, so the overall response rate (ORR) of LEN cannot be analyzed. The other 12 with various clinical courses can be further checked in Supplemental Table S2. As for AML, 24 patients received chemotherapy; 23 of them had clear responding records: 22 achieved CR (96%) and one was refractory to intensive induction chemotherapy but achieved CR after two courses of low-dose cytarabine (LDAC) and underwent LDAC chemotherapy. Seventeen AML patients were administrated HMA-based therapy whereas only six responding records were clear: five responded and one failed. Clinical courses of the other 11 AML patients were individually various and are presented in Supplemental Table S2.
In total, 39 DDX41-mutated patients received HSCT at the median age of 60 (20–70) years. Three patients, two cases of AML after 4 years and one of MDS (time not reported), got relapsed diseases (8%). For DDX41-mutated patients who are scheduled for HSCT, donor cell leukemia stemming from DDX41 mutants should be carefully ruled out, an event reported by two independent groups.37,38 Before transplant, screening DDX41 mutation would be a recommended regular preparation to prevent re-emergence of hematologic diseases.
Other DDX41-mutated diseases
From a Danish cohort, DDX41 p.R535W was identified in a 48-year-old female suspected of common variable immunodeficiency (CVID). 41 She presented with lymphocytosis and cervical dysplasia, recurred with sinusitis and pneumonia abscesses. The author proposed that this DDX41 variant might be linked to her cervical lesion. However, this case could be further investigated, for instance, on her family history or pedigree, on her karyotype, on her bone marrow aspiration and biopsy to evaluate hematopoietic stem cells and differentiate any hematologic diseases.
DDX41 has not yet been identified as a possible pathogenic variant in cases of solid cancers. Nonetheless, elevated DDX41 expression was reported to be associated with pathological grades, clinical stages, and thus worse prognosis, for example, shorter OS and less progression-free survival (PFS), at least in human colorectal cancer, 33 hepatocellular cancer, 34 breast cancer, 35 and clear cell renal cell carcinoma. 36
Discussion
DDX41 is a well-known multifunctional protein. In innate immunity, phosphorylated DDX41 senses bacterial or viral invasion and induces type I interferon, and is degraded by ubiquitination. 43 In myeloid neoplasm, DDX41 was identified as a tumor suppression gene because overexpression blocked cell proliferation whereas knockdown promoted tumor growth. 12 The molecular pathway was unveiled that insufficient DDX41 enabled R-loops to accumulate, hence triggering inflammatory signaling and finally empowering hematopoietic cells to increase. 7 Here we analyzed the clinical aspects of DDX41-mutated diseases, which is the first systematic review along with IPD about this mutation at least to date. Although unable to conduct a meta-analysis due to a large portion of missing data, we presented a larger dataset of individual patients with comprehensive searching.
This mutation was rarely detectable; in about 3% of unselected myeloid neoplasms. DDX41 mutation affected all age groups, mainly those in their 60s and 70s, and rarely children. Familial and sporadic patients present hematological malignancies at different ages. We observed that familial cases were diagnosed 4 years earlier than sporadic counterparts. Such early diagnosis could benefit patients in early intervention and presumably better prognosis. A hypothesis could not be tested yet in this study because of a lack of reports of patients’ clinical courses. Male carriers were predominant, and we noticed that such preference was more conspicuous in AML and MDS. The role that gender plays, whether just as a risk factor or linked to pathogenesis, still needs to be determined.
DDX41 mutation covered a broad spectrum of hematologic diseases, from AML and MDS, which constituted 75% of the diagnosed cases, to rare cases such as AA and lymphoma. AML, MDS, and other diseases showed some interesting differences in comparison with each other, besides different gender preference as mentioned above: more germline p.D140fs variant and more cytogenetic abnormalities in AML, more normal karyotypes in MDS, and fewer somatic variants and less mutated p.R525H in other diseases. These differences possibly implied that not only one pathway existed in developing DDX41-related diseases.
A large portion of normal karyotype was another prominent feature. According to the latest Revised International Prognostic Scoring System, normal karyotype, del (5q), del (20q), and -Y, which dominated our cytogenetic findings, are predictive of a good prognosis. 44 Hence, from the aspect of cytogenetics, mutated DDX41 is assumingly associated with longer survival.
Germline mutated sites varied among the different nationalities, as described in the results. Several other national specificities were also reported in the meeting abstract for instance, p.A500fs common in Japan, p.Q41* mainly in Germany, and p.G218D mainly in Italy. 45 Somatic mutation was largely concentrated on p.R525H, which indicated the pathogenic nature of this site to some extent, as verified by another report that p.R525H mutant may cause defects in ribosomes producing hematopoietic cells. 46 And our data showed that p.R525H was significantly associated with germline p.M1I variant and AML. Therefore, p.R525H is worth researching about its roles such as in pathogenesis and disease progression. It could potentially become a marker monitoring recurrence, as practiced in a case, 31 and a therapeutic target to prevent progression and lengthen PFS. Noticeably, frameshift, nonsense, and splicing variants preferred to be of germline origin and, on the other hand, missense variants were more likely to be somatic. This phenomenon clinically indicated that frameshift, nonsense, and splicing, these three types of mutation, deserve germline investigation such as culturing skin fibroblasts.
Nearly half of patients had at least one concomitant somatic mutation. Most of the concomitant somatic mutations followed the distribution of disease types and belonged to type II mutations which were suggested to be enriched in high-risk MDS and primary AML. 47
The efficacy of HMAs was 70% for DDX41-mutated MDS patients; much better than previously reported.48,49 Mutated AML patients also responded well to chemotherapy, with 96% CR. LEN was expected to have promising ORR from the finding that LEN can lower the expression of DDX41 mRNA in responders; 12 unfortunately, neither calculation of ORR to LEN nor comparison between LEN and HMAs was applicable owing to the rareness of cases. In terms of HSCT, late-onset for DDX41-related diseases, and thus large population of the elderly, would make worthwhile physicians’ caution about patients scheduled for HSCT. However, from data collected here, the relapse rate was lower than previous studies reported.50,51
There were some limitations to this study. First, detailed information about the clinical course was largely absent. Survival, both OS and PFS, cannot be summed because of poor resources. Second, an overall regression analysis could not be conducted to find some risk factors and dependent factors. Only comparisons between groups of interest were carried out. Finally, patients with DDX41 mutation cannot be compared with those with wild-type.
Conclusions
DDX41 is involved in multiple diseases, from hematologic diseases, mainly myeloid neoplasms, to non-hematologic CVID and solid cancers. Patients with hematologic diseases carrying DDX41 mutation were characterized as 80% of males, most diagnosed after 60, 64% of normal karyotype, and 75% of AML or MDS. Germline DDX41 variants were nationally diverse and strongly linked to a frameshift mutation, whereas somatic mutants were p.R525H-dominant and missense-preferred. Different diagnosis groups, familial and sporadic cases, and p.R525H compared with other somatic variants all presented some significant features worth research or clinical caution. Though limited in data on clinical course, this systematic review still found a 70% ORR to HMA in DDX41-related MDS, a 96% CR to chemotherapy in DDX41-related AML, and a relatively lower incidence of relapse after HSCT.
Supplemental Material
sj-pdf-1-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-pdf-1-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-2-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-2-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-3-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-3-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-4-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-4-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-5-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-5-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-6-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-6-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Supplemental Material
sj-xlsx-7-tah-10.1177_20406207211032433 – Supplemental material for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data
Supplemental material, sj-xlsx-7-tah-10.1177_20406207211032433 for Clinical features of DDX41 mutation-related diseases: a systematic review with individual patient data by Ziqi Wan and Bing Han in Therapeutic Advances in Hematology
Footnotes
Author contributions
Z.W. and B.H. performed the database search, study selection, data extraction and quality assessment. Z.W. conducted the statistical analyses and wrote the manuscript. B.H. revised the draft. All authors approved the final submitted version.
Availability of data and materials
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Beijing Natural Science Foundation (7192168), the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (2016-I2M-3-004), and the Non-profit Central Research Institute Fund of CAMS (2019XK 320047).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.