
Abstract
Acute myeloid leukemia (AML) is a heterogenous and complex disease characterized by rapid cellular proliferation, an aggressive clinical course, and generally high mortality. While progress has been made in the understanding of the genetic and molecular biology of the disease, the standard of care for patients had only changed minimally over the past 40 years. Recently, rapid movement of potentially useful agents from bench to bedside has translated into new therapies either recently approved or in clinical trials. These therapies include improved chemotherapies, mutationally targeted inhibitors, pro-apoptotic agents, microenvironment targeting molecules, cell cycle checkpoint inhibitors, and epigenetic regulators. Furthermore, advances in immunotherapy employ monoclonal and bispecific antibodies, chimeric antigen receptor (CAR) T cells, checkpoint inhibitors, and vaccines provide an alternative pathway for AML treatment. In this review, we discuss the recent results of completed or ongoing clinical trials with these novel therapeutic agents in AML.
Acute myeloid leukemia (AML) is an aggressive, heterogeneous, myeloid malignancy; in 2018 an estimated 19,520 new cases and 10,670 deaths occurred in the US. 1 The disease is particularly difficult to treat in older adults who account for the majority of patients; thus, the 5-year overall survival is only approximately 27%. 2 Since the 1970s, initial standard therapy, for those fit enough to receive it, consisted of the ‘7 + 3’ regimen, which includes 7 days of continuous infusion cytarabine and 3 days of an anthracycline. 3 Over the next 35 years, a plethora of clinical trials attempting to augment AML treatment have been performed with little change in the standard of care. However, recent data detailing the molecular ontogeny of AML have elucidated causal pathways which have led to targeted drug development;4,5 new guidelines emphasize molecular studies in both diagnostic and relapsed settings. 6 Owing to these discoveries, eight new drugs in a variety of classes were approved between 28 April 2017 and 28 November 2018 (Table 1). 7 This novel arsenal of diverse therapies, sometimes in combination with previously used chemotherapy, have drastically altered the landscape of AML treatment and has promulgated new treatment paradigms. This paper reviews the novel therapies approved for AML as well as the pipeline of compounds likely to affect treatment in the near future.
Recent approved drugs for AML with indications. 7
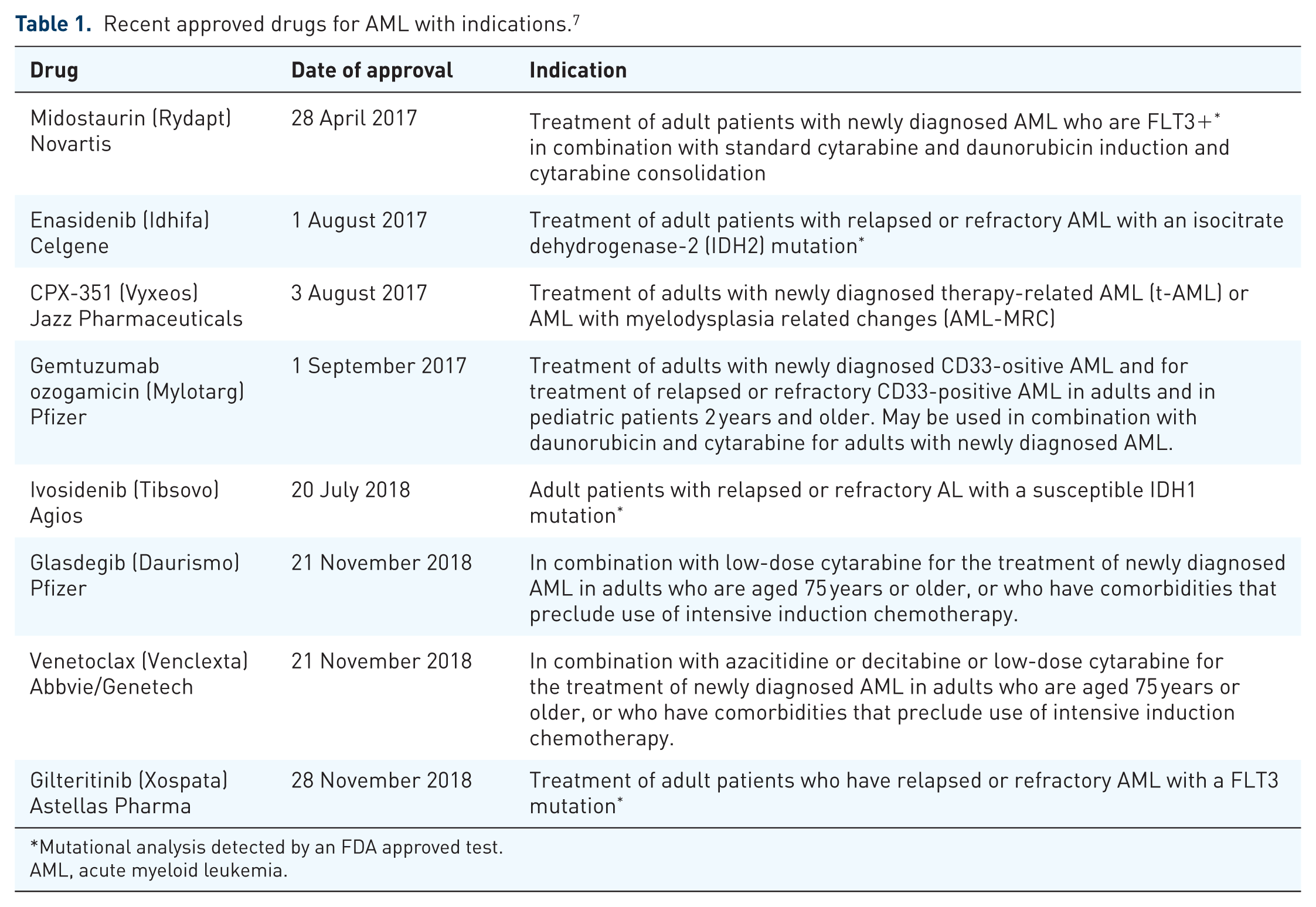
Mutational analysis detected by an FDA approved test.
AML, acute myeloid leukemia.
Conventional chemotherapy
While an anthracycline and cytarabine have been the mainstay of treatment, novel packaging of these agents has led to an important therapeutic advance. CPX-351, a liposomal formulation of daunorubin and cytarabine which releases the drugs in fixed 5:1 molar ratio, was evaluated in a randomized phase II trial followed by a randomized phase III trial compared with standard induction.8,9 In a population 60–75 years of age with newly diagnosed secondary AML, CPX-351 was associated with a superior overall remission rate (47.7% versus 33.3%, two-sided p = 0.016) and overall survival (OS; 9.56 months versus 5.95 months, HR 0.69; 95% CI 0.52–0.90; one-sided p = 0.003) with an improved safety profile except for more prolonged myelosuppression. These results led to FDA approval of this drug in adult patients of any age with secondary AML [after myelodysplastic syndrome (MDS) or prior anticancer therapy] plus those with MDS-related chromosomal abnormities and/or background marrow dysplasia.
Smoothened inhibition
The Hedgehog (Hh) signaling pathway is vital for embryogenesis and fetal development. 10 Aberrant signaling in this pathway affects the proliferation of leukemia stem cells, and upregulation results in chemoresistance in AML cell lines. 11 Furthermore, Hh signaling is vital in the maintenance of murine leukemic stem cells in chronic myeloid leukemia (CML) models.12,13 FLT3 signaling also interacts with the Hh pathway in myeloid leukemias. 14 This pathway is tightly regulated by two transmembrane proteins, patched (PTCH), which is a negative regulator, and smoothened (SMO), a positive regulator. 15
Glasdegib is an oral agent that inhibits the Hh pathway by interacting with smoothened. 16 In vitro and in vivo studies with this agent demonstrated growth inhibition of AML cell lines and human leukemia stem cells. 17 In two phase I trials in adult patients with myeloid malignancies, glasdegib was well-tolerated and was associated with a response rate as high as 49%.18,19 A multicohort phase Ib study evaluated glasdegib in combination with low-dose cytarabine, decitabine, or standard induction therapy. These combinations were deemed to be well tolerated with a recommended phase II dose of 100 mg daily. A total of 31% of patients across all three arms of the study achieved complete response (CR) or complete response with incomplete blood count recovery (CRi), with responses of 8.7%, 28.5%, and 54.5% in the low-dose cytarabine, decitabine, and standard induction arms, respectively. 20 A separate phase II study designed to evaluate the combination of glasdegib given at 100 mg daily for 28 days with standard induction cytarabine and daunorubicin in patients over 55 years of age demonstrated a CR rate of 46.4%. 21 The median survival in this older population was 14.9 months. In an important randomized phase II study of low-dose cytarabine 20 mg subcutaneously twice daily for 10 days with or without glasdegib 100 mg daily, the experimental arm demonstrated a statistically significant improved CR rates (15% versus 2.3%) and OS (8.3 months versus 4.3 months). 22 Owing to the results of this phase II study, glasdegib was approved by the FDA in combination with low-dose cytarabine for unfit AML patients on 21 November 2018. Glasdegib is currently being studied in combination with standard induction therapy in fit patients, or with azacitidine in unfit patients (ClinicalTrials.gov identifier: NCT03416179). Given the high response rates seen with use of a hypomethylating agent plus venetoclax as described in the following, the common use of the glasdegib/low-dose cytarabine combination remains unclear.
Mutationally targeted inhibitors
FLT3
FLT3 is a transmembrane tyrosine kinase that has two mutational subtypes, an internal tandem duplication which results in the duplication of 3 to over 100 amino acids in the juxtamembrane region of the protein (ITD) or point mutations in the tyrosine kinase domain, that are present in approximately 30% of AML cases. 23 At least six different FLT3 inhibitors are currently in clinical development, including sorafenib, midostaurin, lestaurtinib, quizartinib, crenolanib, and gilteritinib. Midostaurin is the only FLT3 inhibitor that, to date, has demonstrated an overall survival benefit in combination with induction chemotherapy. In the RATIFY trial, 717 patients age 18–59 with FLT3 ITD or TKD mutations underwent randomization to receive standard induction chemotherapy or induction with midostaurin 50 mg orally bid given on days 8–22. 24 Patients in remission then received high-dose cytarabine consolidation ± midostaurin according to the original phase; the trial also included a maintenance phase where placebo or midostaurin was given for 12 28-day cycles. Whether or not to perform an allogeneic transplant was left to investigator discretion, but post-transplant midostaurin was not given. Though there was no statistically significant improvement in the CR rate as defined in the protocol, the primary endpoint of OS was met, with a median OS in the midostaurin group reaching 74.7 months versus 25.6 months in the placebo group (HR for event or death 0.78, one-sided p = 0.009 favoring the midostaurin arm). This pivotal data has led to its approval in the United States with induction and consolidation chemotherapy, and for induction, consolidation, and maintenance in Europe. A similarly designed trial of chemo ± the more specific FLT3 inhibitor quizartinib (does not inhibit the TKD mutation) has recently met accrual, but results are not available (ClinicalTrials.gov identifier: NCT02668653).
A phase III trial of induction chemotherapy ± sorafenib in older adults demonstrated that sorafenib was ineffective and was associated with increased toxicity in older adults; 25 however, a similarly designed trial in younger adults showed an improved event-free survival (EFS) but not OS with use of sorafenib; there was also substantial increased toxicity in the sorafenib arm. 26 A major concern is whether a FLT3 inhibitor more specific than midostaurin would be a better drug to combine with chemotherapy in the upfront setting. Ongoing trials of chemo+midostaruin versus gilteritinib and crenolanib (ClinicalTrials.gov identifier: NCT03258931) are either planned or underway.
Responses to the nonspecific FLT3 inhibitors midostaurin 27 and lestaurtinib in relapsed refractory patients have been modest. 28 However, two second-generation FLT3 inhibitors have demonstrated a survival benefit compared with chemotherapy in prospective randomized trials in relapsed refractory patients. Quizartinib was associated with an overall survival advantage compared with physician’s choice of 27 weeks (95% CI 23.1–31.3) versus 20 weeks (95% CI 17.3–23.7), and OS HR of quizartinib to physician’s choice was 0.76 (95% CI 0.58–0.98; stratified log-rank test, one-sided p = 0.0177). 29 In a similarly designed trial that also allowed patients with TKD mutations to enroll, gilteritinib demonstrated a 29% CR or CRi rate (ClinicalTrials.gov identifier: NCT02421939), leading to its approval in the relapsed setting by the FDA in late 2018, although full results of this trial are highly anticipated.
Sorafenib, approved in hepatocellar carcinoma and renal cancer owing to its vascular endothelial growth factor receptor (VEGFR) inhibitory activity, in combination with azacitidine in an older unfit or relapsed predominantly FLT-ITD population yielded an overall response rate of approximately 46%. 30 A recently presented randomized trial of sorafenib versus placebo in the post-allotransplant maintenance setting showed a marked improvement in 2-year relapse-free survival (85% versus 53.3%). 31 A phase III study evaluating gilteritinib in the post-transplant setting is ongoing (ClinicalTrials.gov identifier: NCT02997202).
One possible reason for resistance to a FLT3 inhibitor in combination with chemotherapy is the increase in FLT3 ligand, which competes with the small-molecule inhibitor after exposure to cytotoxic stress.32,33 Based on in vitro studies, both the AKT/FLT3-ITD dual inhibitor, A674563 and MDM2 inhibitors may overcome FL-mediated resistance.34,35 Another potential mechanism for resistance is upregulation of fibroblast growth factor 2 (FGF2), which activates the mitogen-activated protein kinase (MAPK) pathway. 36
IDH2/IDH1
The isocitrate dehydrogenase (IDH) family of enzymes are involved in cellular energy generation in the oxidative decarboxylation cycle by catalyzing the conversion of isocitrate to α-ketoglutarate. While IDH1 is localized in peroxisomes in the cytosol and IDH2 resides in mitochondria.37,38 Mutant IDH has become a viable target in AML treatment. Such mutations occur in blasts from approximately 20% of AML patients [IDH1 (8%) and IDH2 (12%)]. Somatic mutations in catalytically active arginine residues in these enzymes lead to the formation of alternative or ‘neomorphic’ product, 2-hydroxyglutate (instead of alpha-ketoglutarate the reaction product generated by the wild-type enzyme), which results in pro-leukemic epigenetic changes similar to those observed in cells with TET2 mutations. The oral agents ivosidenib and enasidenib, inhibitors of mutant IDH1 and IDH2, respectively, decrease cellular 2-hydroxyglutarate by more than 90%, thus reducing histone hypermethylation and reestablishing myeloid differentiation. 39 In a phase I/II study, IDH2 mutated relapse/refractory AML patients were treated with enasidenib at 100 mg daily resulting in an overall response rate of 40.3% with a median OS of 9.3 months; the 19.3% who achieved a CR had an OS of 19.7 months. 40 Similarly, the phase I/II study of ivosidenib in IDH1 relapsed/refractory mutant AML demonstrated a CR/CRi rate of 30.4% and overall response rate of 41.6%. 41 These studies led to the approval of enasidenib and ivosidenib for relapsed AML. Further studies combining enasidenib with azacitidine in relapse (ClinicalTrials.gov identifier: NCT03683433) or ivosidenib with azacitidine in upfront AML patients (ClinicalTrials.gov identifier: NCT03173248) are ongoing. Preliminary results of a phase II study in which these inhibitors were tested in combination with standard induction chemotherapy in upfront patients with the appropriate mutation demonstrated a response rate of 93% in the ivosidenib arm and 73% in the enasidenib arm, with mutational clearance of 41% and 30% respectively. 31 A randomized phase III trial comparing induction chemotherapy to chemotherapy plus the relevant inhibitor is currently being planned.
Pro-apoptotic agents
BCL-2 inhibition
B-cell leukemia/lymphoma-2 (BCL2) is an anti-apoptotic protein that promotes leukemic blast survival through regulation of the mitochondrial apoptotic pathway. Sensitizer BCL-2 homology 3 (BH3) proteins are antagonists of these antiapoptotic proteins and therefore promote apoptosis via mitochondrial outer membrane permeabilization. 42 Venetoclax, an oral small-molecule BCL-2 inhibitor demonstrated on-target BCL-2 inhibition by BH3 profiling and an overall response rate of 19% in a single-agent trial in very advanced AML; patients with IDH mutations appeared to be more responsive (33%) to venetoclax. 43 In an effort to take advantage of the drug’s ability to help cells undergo apoptosis in the presence of cytotoxic stress, venetoclax was paired with either azacitidine or decitabine (institutional preference), in an elderly population of patients that were unfit for standard induction chemotherapy. The combinations were well tolerated, yielding a 61% CRi rate at the recommended phase II dose of 400 mg daily. 44 The phase II data revealed a CR+CRi rate of 73% and a median OS for the entire cohort of 17.5 months. 45 This combination of venetoclax with hypomethylating agents (HMA) may be becoming the new standard of care for patients who are elderly or unfit for standard induction chemotherapy. A second study evaluated venetoclax with low-dose cytarabine in a phase I/II trial in combination with low-dose cytarabine yielding CR/CRi rates of 35% for secondary AML and 71% for de novo AML. 46 Given these encouraging data, a plethora of studies have opened or proposed using venetoclax in combination with other agents, including combination with standard ‘3 + 7’ induction (ClinicalTrials.gov identifier: NCT03709758), although other ‘novel–novel’ combinations including but not limited to the multi-CDK inhibitor dinaciclib (ClinicalTrials.gov identifier: NCT03484520), gilteritinib (ClinicalTrials.gov identifier: NCT03625505), 10-day decitabine (ClinicalTrials.gov identifier: NCT03404193), and the mcl-1 inhibitor S64315 (ClinicalTrials.gov identifier: NCT03672695). A full list of the novel venetoclax trials can be found in Table 2.
AML-specific actively recruiting Venetoclax trials.

AML, acute myeloid leukemia; FLAG-IDA, fludarabine, cytarabine, GCSF, idarubicin; MRD, minimal residual disease; r/r, relapsed/refractory; TN, treatment naïve.
Myeloid cell leukemia-1 (MCL-1) inhibitors
MCL-1, like BCL-2, is capable of blocking pro-apoptotic proteins such as BAX and BAK, which do not interact with BCL-2, and upregulation may provide a resistance mechanism to venetoclax. MCL-1 inhibitors have been tested minimally in the clinic. MCL-1 inhibitory agents under investigation include AMG176 with venetoclax (in planning) or as a single agent (ClinicalTrials.gov identifier: NCT02675452), and AMG397 (ClinicalTrials.gov identifier: NCT03465540), S64315 with venetoclax (ClinicalTrials.gov identifier: NCT03672695) or single agent (ClinicalTrials.gov identifier: NCT02979366), and AZD5991 (ClinicalTrials.gov identifier: NCT03218683). 47
P53 reactivation
More than 50% of human cancers possess a p53 mutation, which is often a missense mutation that leads to protein unfolding and prevents this transcription factor from responding to cellular stress to elicit cell cycle arrest and apoptosis. p53 mutations are found in 18% of AML overall, in 70% of patients with complex karyotype, and is associated with very short median survival (5.4 months).4,48,49 APR-246 is one of the first agents to specifically target mutated p53. The drug is given IV and is converted into a Michael acceptor, methylene quinuclidinone, which covalently binds to the p53 core domain promoting refolding, and the reinstitution of wild-type p53 function and, thus, cell cycle arrest and apoptosis may occur. 50 In an ongoing phase I/II clinical trial of MDS and AML with <30% blasts, APR-246 in combination with azacitidine induced an overall response of 100% (11 of 11 patients) with a CR rate of 82%. While the duration of remission is not yet known a phase III trial of aza±APR 246 in MDS is planned as is a similar trial in older adults with AML. A further study in an AML population is being planned.
Mouse double minute 2 inhibitors
Mouse double minute 2 (MDM2) is the primary regulator of p53 stability, activity in part by controlling p53 degradation. RG7112 was the first MDM2 inhibitor evaluated in a phase I trial for all leukemias. As expected p53 mutated patients failed to have durable responses. In the AML cohort, the overall response rate was 13%. 51 A second-generation oral MDM2 inhibitor (RG7388, Idasanutlin) was evaluated in a phase I/Ib trial which demonstrated a 22% overall response rate, and in combination with cytarabine the CR+CRi rate was 24%.52,53 These data have led to a phase III study of idasanutlin plus cytarabine versus cytarabine alone (ClinicalTrials.gov identifier: NCT02545283). Given preclinical data demonstrating potential synergy between idasanutlin and bcl-2 inhibition, 54 another ongoing trial combines venetoclax with idasanutlin for relapsed or refractory AML patients (ClinicalTrials.gov identifier: NCT02670044). Preliminary data presented in 2018 demonstrated an antileukemic response rate of 37% with a median time to response and duration of response of 1.8 and 8.1 months, respectively. 55
Two other inhibitors of this pathway have been evaluated. MK-8242, a human double minute 2 (HDM2) inhibitor was evaluated in a phase I trial for refractory/recurrent AML, but only 1 of 24 patients responded. 56 A second compound, HDM201 (Novartis) which selectively inhibits the HDM2-p53 interaction has been under evaluation in p53-wt AML at multiple dosing schedules and is ongoing. The preliminary results demonstrate as CR+CRi+PR rate of 20.6%. 57 A study evaluating HDM201 and chemotherapy in both a front-line and relapsed/refractory population is planned (ClinicalTrials.gov identifier: NCT03760445).
Microenvironment targeting
Uproleselan (GMI-1271)
E-selectin is a cell adhesion molecule involved in the migration of leukocytes along vascular endothelial cells and is highly expressed on leukemic blasts, especially those with advanced disease. 58 Uproleselan (GMI-1271) is an antagonist of E-selectin which enhances chemotherapy response while ameliorating chemotherapy toxicity in animal models. In the phase I/II trial combining Uproleselan with MEC chemotherapy in relapsed/refractory patients, the CR/CRi rate was 41%, with a 30- and 90-day mortality of 2% and 9% respectively, and a grade 3/4 mucositis rate of <4%.59,60 Similarly, an elderly high-risk treatment-naïve AML population was treated with Uproleselan combined with standard induction; 68% (73% de novo, 64% sAML) achieved CR/CRi. The 30- and 90-day mortality was 8% and 12%, respectively, with no reports of grade 3/4 mucositis.60,61 A phase III randomized trial is ongoing for r/r patients comparing Uproleselan with MEC versus MEC alone (ClinicalTrials.gov identifier: NCT03616470), and a separate phase II–III trial evaluating Uproleselan with induction versus induction alone was recently activated (ClinicalTrials.gov identifier: NCT03701308).
Cell cycle checkpoint inhibitors
Whereas AML is caused in part by dysregulation of cell proliferation, a series of targets involve the cell cycle regulation and DNA repair. One mechanism of chemotherapy resistance involves cell cycle regulators’ recognition of DNA damage which delays mitosis to allow repair to be initiated. 62 Thus, inhibiting this pathway could have a synergistic effect with chemotherapy.
Aurora kinase inhibitors
Aurora kinases play an essential role in mitosis regulation, centrosome function, chromatid segregation, and bipolar spindle assembly. 63 Alisertib (MLN8273) is an oral Aurora A kinase inhibitor that increases efficacy of cytarabine. 64 A single-agent phase II trial dosing at 50 mg BID for 7 days every 21 days was associated a 17% overall response rate in unselected patients with advanced AML. 65 Given single-agent safety a phase I trial combined alisertib with 7 + 3 yielding a maximum-tolerated dose (MTD) of 30 mg bid. 66 In the phase II trial evaluating that alisertib dose plus ‘3 + 7’ in high-risk AML results included aCR+CRi rate of 64% with a 30-day and 60-day mortality rates of 8% and 13%, respectively, and a median OS of 12.2 months. 67 A phase III trial is under discussion.
Barasertib, an Aurora B kinase inhibitor delivered by IV continuous infusion, was also found to be active as a single agent and in combination with other chemotherapeutic agents. 68 A five patient single-agent phase I trial demonstrated safety with one patient achieving a CR, and a second phase I study combining barasertib with low-dose cytarabine (LODAC) demonstrated an overall response rate of 45% but at a cost of a 73% rate of significant infections.69,70 The SPARK-AML1 trial was a 2:1 randomized phase II trial comparing barasertib with LODAC in elderly unfit patients, with the barasertib arm demonstrating a CR/CRi rate of 35.4% and an OS of 8.2 month. This compound can be given in a nanoparticle delivery system (AZD2811), which may achieve improved bioavailability and is currently under investigation as a phase I clinical trial (ClinicalTrials.gov identifier: NCT03217838).
Polo-like kinase-1 inhibitors
Polo-like kinase-1 (PLK1) plays an integral role in mitosis, DNA replication, and DNA repair. If PLK1 function is impaired, polo-arrest occurs, leading to cell cycle arrest in pro-metaphase. 71 The initial PLK-1 inhibitor volasertib was studied in a randomized phase II trial low-dose cytarabine +/– the drug in untreated high-risk, older AML patients. CR+CRi rate for LODAC + volasertib was 31%. 72 A second PLK-inhibitor, rigosertib, which also inhibits PI3-kinase and the RAS/MEK/ERK pathway, has been compared in a negative large phase III trial (ONTIME) to standard care in HMA-failed MDS. Rigosertib achieved a similar OS (8.2 months, 95% CI 6.1–10.1) compared with best supportive care (5.9 months, 95% CI 4.1–9.3) that did not reach statistical significance (HR 0.87, 95% CI 0.67–1.14; p = 0.33). 73 A phase I/II single-agent trial in patients with MDS or MDS progressed to AML showed only one responder of 13 AML patients. 74
Other cell cycle inhibitors
Cyclin-dependent kinases (CDK) are a group of proteins that promote transition through the cell cycle. Perturbation in the regulation of cell cycle flow between S phase and G2/M is thought to be essential for tumorigenicity in at least some settings. 75 Palbociclib is a CDK4/6 inhibitor approved in breast cancer and also is active against FLT3-ITD AML cells. A phase I trial of single-agent palbociclib in advanced AML was disappointing; however, this trial now includes cohorts of patient treated with palbociclib plus either dexamethasone, decitabine, or sorafenib (ClinicalTrials.gov identifier: NCT03132454).76,77 A second single-agent trial limited to mixed lineage leukemia (MLL)-rearranged acute leukemias (ClinicalTrials.gov identifier: NCT02310243) is ongoing. Dinaciclib is a CDK9 inhibitor that also promotes apoptosis in MLL-rearranged AML, promoting studies in combination with venetoclax (ClinicalTrials.gov identifier: NCT03484520) and pembrolizumab (ClinicalTrials.gov identifier: NCT02684617) in this setting. 78
CHK1 is a multifunctional protein kinase and regulator of the DNA damage response. 79 In response to DNA damage, CHK1 mediates cell-cycle arrest to allow time for DNA repair, or if the damage is extensive, to trigger apoptosis. CHK1 is also essential for homologous recombination-mediated repair of double-strand DNA breaks, initiation of DNA replication origin firing, stabilization of replication forks, resolution of replication stress, and coordination of mitosis, even in the absence of exogenous DNA damage. 80 Analysis of AML patient samples has demonstrated that the CHEK1 gene is upregulated in AML with high expression associated with shorter responses and overall survival. Moreover, when these cells were treated with a CHK1 inhibitor in combination with cytarabine, DNA replication was reduced. 81 Prexasertib (LY2606368) is an ATP-competitive inhibitor of CHK1 that causes double-stranded DNA breaks in S-phase cells leading to replication catastrophe. 82 Two ongoing studies are evaluating prexasertib in the relapsed/refractory AML setting with either FLAG (ClinicalTrials.gov identifier: NCT02649764) or MEC (ClinicalTrials.gov identifier: NCT03735446).
Epigenetic regulators
Epigenetic dysregulation associated with mutations in genes such DNMT31, TET2, and ASXL1 which lead to aberrant methylation, demethylation, and acetylation contribute to myeloid leukemogenesis. Restoring the function of such mutations either directly or indirectly has remained an elusive but potentially interesting therapeutic strategy.
Disrupter of telomeric silencing 1-like
Rearrangements of the MLL gene (newly termed KMT2A) on chromosome 11q23 convey a poor prognosis in AML. 83 Mutant MLL proteins lead to high levels of histone 3 at lysine 79 (H3K79) and thereby directing disrupter of telomeric silencing 1-like (DOT1L) to aberrant targets. Inhibition of DOT1L disrupts leukemogenesis in mouse models, led to a single-agent phase I study with pinometostat (EPZ-5676), a small-molecule inhibitor of DOT1L, by continuous IV infusion. 84 Of the 51 patients with hematologic malignancies (84% AML, 82% of these with an MLL mutation) who were treated, pinometostat appeared safe; two patients, each of whom with t (11; 19) leukemia, achieved a CR. Evidence for target inhibition, based on reduction of H3K79 methylation in blasts, was also noted in all dose levels. 85 Further studies evaluating pinometostat with azacitidine (ClinicalTrials.gov identifier: NCT03701295) or with induction chemotherapy (ClinicalTrials.gov identifier: NCT03724084) in MLL mutated patients are planned.
Bromodomain and extraterminal protein family inhibitors
The bromodomain (BRD) and extraterminal (BET) protein family is integral for transcription through its association with enhancers and positive transcription factor b (P-TEFb), which promote the transition from RNA polymerase from the paused to the elongating state.86,87 BET inhibitors have preclinical activity against MLL mutant cell lines; most MLL fusion partners are members of the super elongation complex (SEC), required for downstream transcription of the proleukemic genes such BCL2, C-MYC, and CDK6, which is blocked by displacement of BRD3/4 and the SEC components from chromatin. 88 Further preclinical studies also demonstrate promising activity against NPM1 and FLT3-ITD mutant AML.89,90
Clinical studies with BET inhibitors have shown modest single-agent efficacy. In a phase I trial using the oral BET inhibitor OTX015, there were three patients with a CR among 36 AMLs treated across 6 dose levels. Dose-limiting toxicities (DLTs) included fatigue, rash, and gastrointestinal (GI) effects. 91 Other BET inhibitors undergoing evaluation include TEN-010 (ClinicalTrials.gov identifier: NCT02308761) and GSK535762 (ClinicalTrials.gov identifier: NCT01943851). Like many of the other examples noted above, combinations of BET inhibitors in combination with other agents such as venetoclax or MCL inhibitors may be optimal based on preclinical data. 92
Immunotherapy
Allogenic stem-cell and donor lymphocyte infusions represent two forms of immunotherapy that have been effective in the therapy of AML.93,94 Building on the benefit of novel agents such as monoclonal antibodies and chimeric antigen receptor (CAR) T cells seen in other diseases, multiple agents have been evaluated to take advantage of this native immune response.
Antibody-based therapy
Gemtuzumab ozogamicin
Gemtuzumab ozogamicin (GO) is a humanized anti-CD33 monoclonal antibody conjugated to the antibiotic calicheamicin, which is toxic to leukemic cells. The drug was approved in 2001 for relapsed non-chemo-eligible relapsed AML based on trials using a dose of 9 mg/m2 every 2 weeks yielding a 26% CR rate. 95 However, the increased hematologic and hepatic toxicity including high rates of veno-occlusive disease especially after allo SCT lead Pfizer to withdraw the drug from the US market in 2010. Further studies continued in Europe with the most prominent being the phase III ALFA-0701 study employing standard induction chemotherapy with or without a lower dose of GO. This study demonstrated significant improved 2-year event-free survival (40.8% versus 17.1%), relapse-free survival (50.3% versus 22.7%) and OS (53.2% versus 41.9%); the addition of gemtuzumab to chemotherapy appears especially beneficial in cytogenetically favorable risk AML. 96 Similarly, in a relapsed population, fractionated dosing of GO mitigated hematologic and hepatic toxicities, and demonstrated a CR rate of 26% and CRp of 6%. 97 Further evaluations are ongoing including use in the minimal residual disease (MRD) setting (ClinicalTrials.gov identifier: NCT03737955), and in combination with azacitidine, 98 cytarabine (ClinicalTrials.gov identifier: NCT02473146), decitabine (ClinicalTrials.gov identifier: NCT00882102), MRD (ClinicalTrials.gov identifier: NCT03737955) and multiple induction regimens.
Vadastuximab talirine (SGN-33A)
Vadastuximab talirine (VT) also targets CD33 but the cysteine residues of the antibody are conjugated to a DNA cross-linking agent pyrolobenzodiazepine (PBD) dimers via a cleavable maleimidocaproyl–valinealanine dipeptide linker. In the phase I study in advanced AML, a dose of 40 µg/kg was selected; the CR+CRi rate was 28%. 99 A second phase I trial combined VT with hypomethylating agents in an elderly population and demonstrated a CR+CRi rate of 70%, but significant myelosuppression was noted. Similar toxicity was noted in a study combining VT with 7 + 3 chemotherapy with dosing of 10–20 µg/kg on days 1 and 4. 100 This led to the phase III CASCADE trial, which was discontinued due to higher mortality in the VT arm.
Anti-CD123
CD123 is the interleukin 3 receptor alpha chain (IL-3α) and found to be overexpressed on myeloid blasts and leukemic stem cells. There have been multiple agents targeting this receptor. While the initial phase I study of the naked antibody CSL360 demonstrated minimal efficacy, 101 anti-CD123 molecules are continually being explored. SGN-CD123A, an antibody using the same linker and warhead as Vadastuximab, demonstrated preclinical promise and is currently under investigation in AML (ClinicalTrials.gov identifier: NCT02848248). 102 Similarly, the antibody–drug conjugate IMG632, a humanized anti-CD123 antibody conjugated to a mono-alkylating payload of the indolinobenzodiazepine pseudodimer (IGN) class, and is in phase I (ClinicalTrials.gov identifier: NCT03386513). 103
SL-401
SL-401 (Tagraxofusp) is a recombinant fusion protein combining human IL-3, the CD123 ligand, with a truncated diphtheria toxin that has demonstrated activity against CD34+CD123+ AML and MDS cells. 104 It is highly effective in blastic plasmacytoid dendritic cell neoplasm (BPDCN), 104 and has modest single-agent activity in AML. 105 Current studies are ongoing with combination therapy with azacitidine (ClinicalTrials.gov identifier: NCT03113643) and also as a single-agent consolidation for eradication of minimal residual disease (ClinicalTrials.gov identifier: NCT02270463). 106
Bispecific antibodies
Bispecific antibodies utilize two different variable regions, one binding to the T-cell subunit CD3 and the other to the tumor surface antigen to provide contact between the cells and create a T-cell activation against tumor cells. The success of blinatumomab, a bispecific antibody to CD19 and CD3, in acute lymphoblastic leukemia 107 has led to attempts to use this technology in AML. AMG330 is a bispecific antibody against CD3 and CD33 that demonstrates AML cell cytotoxicity, particularly in favorable-risk disease, and also has synergy with blockade of the PD-1/PD-L1 axis, thereby preventing immune exhaustion.108,109 Owing to the intriguing preclinical data, phase I clinical trials are underway (ClinicalTrials.gov identifier: NCT02520427). Similarly, flotetuzumab (MGD006 or S80880) is a dual-affinity retargeting antibody (DART), which employs two independent polypeptides fusing the heavy-chain variable domain of one antibody to the light-chain variable domain of the other to connect CD3 and CD123. Preclinical studies demonstrated a dose-dependent killing of AML cells. 110 Preliminary data of a phase I/II trial of flotetuzumab in advanced AML/MDS patients, with an acceptable safety profile, and demonstrated a CR/CRi rate of 31% (4/13 patients) with primary refractory disease, but no responses in relapsed patients (0/11). Another phase I trial in patients with a variety of hematologic malignancies is ongoing (ClinicalTrials.gov identifier: NCT03739606). 111
CAR-T-cell therapy
CAR-T-cell therapy hast had a dramatic effect on the management of lymphoid malignancies and thus while there is great interest in adapting this technology for AML, the lack of target that is ubiquitously expressed in AML yet dispensable has created challenges.112–114 In a phase I study using CAR-T cells targeting the Lewis-Y antigen, three of four patients had evidence of an ephemeral biological response. 115 A report described a CD33 directed CAR-T cell in one patient with relapsed/refractory AML; this patient had a dramatic cytokine release syndrome and experienced a decrease in blasts 2 weeks after treatment but then progression at 9 weeks. 116 Budde et al. presented data on six patients who received anti-CD123 CAR-T cells, which demonstrated safety and activity with one morphologic leukemia-free state, two CR, and two reduction in blast counts. 117 This trial is ongoing (ClinicalTrials.gov identifier: NCT02159495). NKG2DL has also been evaluated as a target for CAR-T cells. In a phase I trial in myeloma and AML, one patient had a transient response but otherwise no tumor responses were observed. 118 A second phase I trial used CYAD-01, a similar product to that used in the aforementioned study but at a much higher dose of cells evaluated 8 r/r AML patients. The drug displayed promising efficacy as well as cytokine release syndrome that was successfully managed. An overall response rate of 42% was noted (ClinicalTrials.gov identifier: NCT03018405). 119 There are currently other ongoing studies with variations of CAR-T cells from AML in both the United States and China, including UCART123 (ClinicalTrials.gov identifier: NCT03190278) which is an allogeneic or ‘off-the-shelf’ product and a novel CD33 CAR (ClinicalTrials.gov identifier: NCT03126864).
Checkpoint inhibitors
Anti-CTLA-4
Immune checkpoint inhibitors have been widely studied on solid tumors, and their migration to the hematologic malignancies has become more prominent since the success in Hodgkin’s Lymphoma, 120 and in the relapsed hematologic malignancy post-transplant setting. 121 In the latter study using ipilimumab, an anti-CTLA-4 antibody, 5 of the 12 AML patients had responses, including four patients with extramedullary disease. While a phase Ib single agent ipilimumab trial demonstrated minimal effect as monotherapy after hypomethylating agent failure, 122 further studies combining ipilimumab with decitabine (ClinicalTrials.gov identifier: NCT02890329) in the de novo and post-transplant setting are currently enrolling.
PD-1 blockade agents
Preclinically, PD-1 blockade demonstrated PD-L1 upregulation in murine AML cells in vivo with response to PD-L1 blocking antibodies. 123 Further studies have demonstrated the potential to enhance cytotoxicity in adoptive T-cell therapy by combining PD-1 with cytotoxic T-cell infusions. 124 A phase IB/II study combined nivolumab with azacitidine in 51 relapsed/refractory AML patients. Fourteen patients had grade 2 or higher immune-mediated toxicities with 12 responding to steroids; 6 of 25 evaluable patients achieved a CR, Cri, or hematologic improvement. 125 Another study in high-risk AML patients ineligible for stem cell transplant who were in CR received nivolumab every 2 weeks for 6 months, then every 4 weeks for 6 months, and every 3 months thereafter. In the 14 evaluable patients, the 12 and 18 month OS estimates were 86% and 67%, respectively (ClinicalTrials.gov identifier: NCT02532231). 126 Lastly, a phase II nonrandomized study was performed evaluating safety, efficacy, and biomarkers using azacitidine with either nivolumab or nivolumab + ipilimumab in relapsed/refractory AML. In the azacitidine/nivolumab arm, the CR/CRi rate was 22%, while the azacitidine/nivolumab + ipilimumab arm revealed a CR/CRp/CRi rate of 43%. Responders had a higher frequency of CD3+ and CD8+ cells in bone marrow by flow cytometry, making these potential predictive biomarkers for this regimen (ClinicalTrials.gov identifier: NCT02397720). 127
Nivolumab (every 2 weeks) has also been evaluated in an induction regimen containing idarubicin and cytarabine in AML or high-risk MDS (⩾10% blasts). Response rates were 77%, with responders again having higher frequency of CD3+ cells (ClinicalTrials.gov identifier: NCT02464657). 128 Further studies in relapsed/refractory AML utilizing nivolumab involve combinations with oral cyclophosphamide (ClinicalTrials.gov identifier: NCT03417154), and a phase I vaccine trial combining a novel vaccine with nivolumab and decitabine (ClinicalTrials.gov identifier: NCT03358719).
Pembrolizumab is another anti-PD-1 antibody researched for the treatment of AML. In a relapsed/refractory AML population, the group at University of North Carolina evaluated age-adjusted high-dose cytarabine followed by pembrolizumab, an anti-PD-1 antibody, on day 14, with responders receiving maintenance pembrolizumab every 3 weeks for up to 2 years (ClinicalTrials.gov identifier: NCT02768792). Preliminary data showed a CR/CRi rate of 40%; two patients went to maintenance with a median duration of CR 3.9 months. 129 Pembrolizumab was also combined with decitabine in a relapsed/refractory AML population, with 1 of 10 reported patients responding with a MRD-negative CR after 8 cycles. 130 A relapsed/refractory AML study using azacitidine and pembrolizumab in elderly patients is ongoing (ClinicalTrials.gov identifier: NCT02845297).
Vaccine therapy
Concepts being explored include vaccines to trigger an immune response to tumor-associated antigens by peptide vaccinations, or through fusion with dendritic cells that present antigen in the proper context may elicit a primary immune response.131,132 A systematic review of the 9 Wilms’ Tumor-1 (WT-1) antigen trials demonstrated correlations between induction of T cells specific to WT-1 and response, and a moderate response rate (22 responses of 67 patients). 133 The most advanced of these vaccines is the multivalent WT1 peptide galinpepimut-S. In a phase II trial, the vaccine was administered 6 times over 10 weeks followed by 6 monthly doses in patients who achieved CR1. The vaccine was deemed well tolerated, with a median disease-free survival of 16.9 months and a relapse rate of 68%. 134 Another vaccine utilized OCV-501, an HLA class II restricted helper peptide derived from the WT1 protein. The phase II trial of this vaccine conducted in AML patients over 60 in CR1. A total of 133 patients were randomized to vaccine or placebo, and although well tolerated, there was no significant difference in disease-free survival. 135
Multiple dendritic cell-based vaccines have been evaluated for AML. DCP-001 is a vaccine developed from an AML cell line having epitopes for the disease but not patient specific. In a phase I trial in patients in CR or with smoldering disease, the treatment was well tolerated, and patient with no circulating blasts had an OS of 36 months. 136 An international phase II trial utilized dendritic cells electroporated with WT1 messenger RNA, and found an antileukemic response in 43% of patients. 137 A third trial developed a personalized vaccine fusing patient-derived AML cells with autologous dendritic cells. In 17 patients who achieved a remission to chemotherapy, 12 remained alive with a median follow up of 57 months at the time of publication (ClinicalTrials.gov identifier: NCT03679650). 138
Conclusion
In summary, the elucidation of pathways that promote proliferation, inhibiting apoptosis, targeting cause mitogenesis due to mutations in specific genes, and limit the native immune system have each provided a much-needed burst of novel agents for the treatment of AML over the past 2 years. New pharmacologic breakthroughs, such as venetoclax, or targeted therapies such as midostaurin or enasidenib have transformed the paradigm of AML treatment. The intense collaboration between benchwork and bedside has prompted a more rational approach compared with the sledgehammer of conventional chemotherapy; the ongoing BEAT AML trial and planned US cooperative group studies have acknowledged the heterogeneity of AML by assigning patients to therapies bases on their molecular subtype. The plethora of clinical trials using novel small-molecule inhibitors is intriguing: while history has taught that only a small fraction of these trials will be paradigm changing, the further development of some of these novel agents will surely build on these recent successes.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
ESW has served on advisory boards for Shionogi, Inc. and Pfizer and received research funding from Lilly. RMS has served on an advisory board/consulted for AbbVie, Actinium, Agios, Amgen, Arog, Astellas, AstraZeneca, Cellator/Jazz, Celgene, Cornerstone, Fujifilm, Juno, Macrogenics, Novartis, Ono/Theradex, Otzuka/Astex, Pfizer, Roche, and Sumitomo; served on a data safety monitoring board for Argenx, Celgene, and Takeda; and received research funding from AbbVie, Agios, Arog, and Novartis.