
Abstract
Acquired haemophilia A (AHA) is a rare, serious bleeding disorder most often encountered in elderly patients. The mainstay of haemostatic management is with bypassing agents (BPAs) including recombinant activated factor VII (rFVIIa) and activated prothrombin complex concentrates (aPCCs). Their major limitation is incomplete efficacy, potential risk for thrombosis and the lack of routine laboratory assays for monitoring treatment response. Plasma-derived porcine FVIII (pd-pFVIII, Hyate C®), first used in the 1950s for the management of congenital haemophilia, has sufficient sequence homology to be haemostatic in humans, but the lack of complete homology facilitates efficacy even in the presence of human allo- and autoantibodies against human FVIII (hFVIII). In a small phase II/III study, recombinant porcine FVIII (rpFVIII, Obizur®, OBI-1, susoctocog alfa) was shown to be safe and effective for the management of bleeding episodes in patients with AHA with anti-porcine FVIII (anti-pFVIII) antibody levels of 20 BU/ml or less. Treatment outcome was judged on clinical response and FVIII levels after an initial fixed dose of 200 IU/kg. The rise in FVIII levels showed considerable inter-individual variability and was significantly influenced by the presence of anti-pFVIII antibodies. Based on the baseline levels of anti-pFVIII antibodies and response to treatment, three potential patient groups were identifiable. In the first group, the absence of cross-reacting antibodies was associated with supra-therapeutic FVIII levels, fewer infusions and lower rpFVIII utilization per treatment episode. The second group had patients with low levels of cross-reacting anti-pFVIII antibodies (0.8–5 BU/ml) with near-normal response to rpFVIII. The last group had higher titres of anti-pFVIII antibody (10–30 BU/ml) associated with lower FVIII levels, more infusions and higher consumption of rpFVIII. We propose a new treatment algorithm for the haemostatic management of AHA that includes the potential first-line clinical use of rpFVIII that takes into account availability of anti-pFVIII antibody results, titre of anti-pFVIII antibodies and severity of bleeding episode.
Introduction
AHA is a rare bleeding disorder that develops due to circulating autoantibodies to endogenous FVIII. It presents more frequently in the elderly and can result in life-threatening bleeding, with a mortality rate ranging between 3.3% and 22%.1–3 It has an estimated incidence of 1.48/million/year with a median age at presentation of 75–80 years. 2 However, the age distribution is biphasic, with a small peak in patients aged 20–30 years that reflects an association with pregnancy. The majority of cases are idiopathic (50%), with the remainder seen in the context of autoimmune disorders, malignancy and post-partum.2,4 In contrast to congenital haemophilia, bleeding tends to occur in soft tissues and mucous membranes, with joint bleeds being unusual and severe and fatal bleeding events remain a significant risk while the autoantibody to FVIII persists.
The haemostatic management of this condition with BPAs can be challenging. They are not as effective as FVIII replacement in congenital haemophilia in the absence of inhibitors and, in addition, a routine laboratory assay cannot easily monitor their efficacy. They also carry a risk of thrombotic events, of concern especially in a cohort of elderly patients who often present with multiple comorbidities.
Recombinant porcine FVIII (rpFVIII) was developed as a treatment option for the management of bleeding episodes in AHA. Results from phase I, phase II and phase II/III studies and case series both in congenital haemophilia A with inhibitors and AHA have demonstrated the safety and efficacy of this product in the treatment of bleeding episodes. This now gives clinicians an alternative to BPAs in the management of this challenging condition. This article reviews haemostatic management of bleeding episodes in AHA, including the evidence base for porcine FVIII in this condition, the development of rpFVIII and the results of clinical studies and real-world case reports of rpFVIII. We also propose a potential treatment algorithm for its clinical use as a first line agent.
Evidence base for haemostatic therapy
Definitive treatment of AHA involves eradication of the autoantibody by means of immunosuppression but, as patients are at risk of severe and fatal bleeding, haemostatic therapy is required to treat bleeding episodes until eradication therapy is successful. 5 There are no randomized controlled trials of haemostatic therapy in AHA and so current clinical practice is based on retrospective, observational studies.
A 2-year national surveillance study conducted by the UK Haemophilia Centre Doctors Organisation (UKHCDO) between 2001 and 2003 collected data on 172 patients. 2 Bleeding status was known for 143 patients, of whom 13 (9%) died as a result of bleeding at a median of 19 days after diagnosis (range 1–146 days). Fifty-one patients did not receive haemostatic therapy. One-third each were treated with aPCC, rFVIIa and hFVIII (an option in the presence of low-titre inhibitors) and about 5% were treated with desmopressin or pd-pFVIII.
The largest published observational dataset to date is the EACH2 registry (European Acquired Haemophilia registry). One of the primary aims of this registry was to report on the management of the first bleeding episode. 3 It was established in 2003 and was a multi-centre web-based registry comprising data collected over 5 years from 501 patients treated at 117 centres across 13 countries. The data confirmed that the severity of bleeding is highly variable and, as seen in the UKHCDO survey, does not correlate with FVIII or inhibitor levels at presentation. Bleeding was observed in 96.2% of patients, with a bleeding mortality rate of 3.3%. Thirty per cent of patients did not require haemostatic therapy, but 61.2% experienced a bleeding event that necessitated treatment. Most patients received rFVIIa (174/307; 56.6%), with equal numbers receiving aPCC and FVIII replacement therapy.
In this registry, the efficacy of BPAs was compared to strategies to increase FVIII levels (FVIII concentrate or DDAVP). Both in unmatched and matched populations, BPAs were significantly more effective (bleeding control 91.8% versus 69.6%; p < 0.003) compared to strategies aimed at increasing FVIII levels. Further, there was no significant difference in the efficacy between rFVIIa and aPCC (93%, p = 1).
The principal adverse event associated with the use of BPAs was thrombosis (rFVIIa 2.9%, aPCC 4.8%), with a higher prevalence of arterial events compared to venous events (myocardial infarction 6, stroke 1 and venous thromboembolism 4). A previous study had reported an incidence of thrombosis of 6.9% in the context of rFVIIa used to treat bleeding events in AHA. 6 The development of thrombotic events underlines the importance of avoiding unnecessary treatment in patients with mild or superficial bleeding. 3
Limitations of bypassing therapy
The ideal haemostatic agent is one that is effective, can be monitored and has a low risk of adverse events. There are several issues with BPAs, aside from their considerable cost. 7 Although they are more effective than the alternatives, haemostasis is not achieved in the same predictable fashion that can be expected when using factor replacement in patients without inhibitors, although current efficacy rates are still reported to be around 92%. There are a proportion of patients in whom bleeding is not controlled by initial therapy and mortality continues to be observed secondary to uncontrolled bleeding. Importantly, there are no routine means of monitoring adequacy of haemostatic therapy with BPAs that are as reliable as measuring FVIII level to ensure attainment of haemostatic levels of treatment. In addition, there is an ever-present risk of thrombotic events. 8 The use of FVIII concentrate can be considered in those with a low-titre inhibitor; however, they are likely to have decreased recovery and reduced half-life due to increased clearance in the presence of an inhibitory antibody and, further, have shown to be less efficacious.2,3
Development of porcine FVIII concentrate and rationale for use in AHA
In 1937 a component of human plasma termed ‘antihaemophilic globulin’ (AHG) was described by Patek and Taylor, 9 which when injected into patients with haemophilia, shortened the clotting time. Due to the limited availability of human plasma as a source of AHG, alternatives were sought and bovine plasma was found to be effective. However, repeated treatment was often associated with severe allergic reactions and thrombocytopaenia. 10 Subsequently, other sources were pursued and in 1954 the first use of porcine-derived AHG was described in a patient who was bleeding and had become refractory to bovine AHG. Initially it was used both in patients with and without inhibitors, but with the development of cryoprecipitate and subsequently concentrates of fractionated human plasma, its use in patients with haemophilia declined.
Human and porcine FVIII share close sequence homology with an A1-A2-B-AP-A3-C1-C2 domain sequence with the porcine A2 and C2 domains being 84% and 76% homologous with the corresponding human domains respectively. FVIII inhibitors most commonly bind to the A2 and C2 domains, preventing formation of the tenase complex required for normal haemostasis. The lack of complete homology accounts for the low incidence of cross-reactivity with human allo- and autoantibodies against hFVIII. 11
Plasma-derived porcine FVIII (Hyate:C®)
Hyate:C® was first used in 1984. It was reported to be effective with minimal cross-reactivity although, as with bovine AHG, it was still associated with allergic reactions and thrombocytopaenia. The latter was found to be caused by residual traces of porcine von Willebrand factor (pVWF), 12 which binds to platelet receptor Gp1b leading to activation of the GpIIb/IIIa receptor resulting in platelet aggregation. 10 However, the concentrate was key to enabling surgery in patients with inhibitors, something previously felt to be too risky to undertake. As a natural extension of its use, it was also used as a treatment option in patients with AHA as aPCC was the only available BPA at the time.
The key studies published in the late 1980s and early 1990s summarized the experience of pd-pFVIII in both patients with congenital haemophilia and inhibitors and those with AHA. They documented successful treatment in both categories of patient. 13 A study published in 1993 focused on the use of Hyate:C® in patients with AHA. 13 Data were collected from 47 centres in Europe and North America pertaining to 74 bleeding episodes in 65 patients. The median anti-pFVIII antibody levels at presentation were 1 BU/ml (range 0–15) compared to anti-hFVIII antibody titres of 38 BU/ml (1.2–1024). Objective clinical responses were rated as good or excellent in 78% of cases. Unlike patients with congenital haemophilia, the response to treatment was not predicted by the FVIII level or the inhibitor titre at presentation (human or porcine) or the initial dose of pd-pFVIII or subsequent dose alterations. The mean initial dose of pd-pFVIII was 84 IU/kg, which increased the plasma FVIII activity by 0.85 IU/ml. Side effects were uncommon. The median level of anti-pFVIII antibody after treatment was unchanged, with a mean of 8.5 treatment days and an average of 11 infusions. Of note, treatment resulted in the development of anti-pFVIII antibody in two-thirds of patients, with one-third showing no change over the study. In the former, about half stopped responding to treatment and the remainder continued to respond to treatment despite an increase in anti-pFVIII inhibitor levels.
Recombinant porcine FVIII concentrate (rpFVIII): OBI-1
The manufacturing of pd-pFVIII was ceased in 2004 due to concerns relating to porcine parvovirus. However, this left a gap in the treatment armamentarium available to clinicians looking after patients with AHA, and thus potential for a recombinant product. As a result, OBI-1 (Obizur®, susoctocog alfa) a recombinant, glycosylated B-domain deleted pFVIII molecule (rpFVIII) was developed. It is produced in a baby hamster kidney (BHK) cell line with a manufacturing process that involves two viral inactivation steps – solvent/detergent and nano-filtration.11,14 OBI-1 circulates as a hetero-dimer with only 2% in single-chain form. It binds to human VWF and is activated by thrombin. Pre-clinical safety studies including animal studies involving cynomolgus monkeys, mice and dogs along with in vitro studies demonstrated similar efficacy, recovery rates, immunogenicity and half-life (t1/2) as Hyate:C®.8,11 OBI-1 is a recombinant protein and the preparations contain >99% rpFVIII in contrast to Hyate:C® that contained only 1% pFVIII. As it does not contain pVWF, the risk of treatment-associated thrombocytopaenia is not present.
OBI-1 (rpFVIII): phase I trial in patients with congenital haemophilia A and inhibitors
The first trial with rpFVIII was in patients with congenital haemophilia A with inhibitors. This multi-centre, randomized, double blind, placebo-controlled phase I trial was conducted to assess the safety and pharmacokinetic profile of OBI-1 (100 U/kg) or placebo with Hyate:C® (100 U/kg) in the non-bleeding state. 11 The study had to be terminated early due to the lack of availability of Hyate:C®, thus data were only collected on nine patients. All patients underwent pharmacokinetic assessment with samples taken up to 48 hours post-dose. Patients were stratified into two groups as per their anti-pFVIII inhibitor titres during screening [low-titre (0–10 BU/ml) versus high-titre (10.1–20 BU/ml)]. Safety assessments included infusion-related events and inhibitor formation at day 29.
OBI-1 appeared to have a higher recovery and a shorter t max than Hyate:C®, with an incremental recovery of 0.8–2.6 and t1/2 of around 11 h. However, the time taken to infuse Hyate:C® and small sample size may have had an influence. Three patients with baseline anti-pFVIII titres of >5 BU/ml did not have a measurable increase in FVIII levels after infusion and further these patients had a marked increase in the titres of anti-pFVIII and anti-hFVIII inhibitors. One subject who was negative for anti-pFVIII inhibitors at baseline developed a low-titre anti-pFVIII inhibitor at day 29 with a concurrent increase in the anti-hFVIII inhibitor titre.
OBI-1 (rpFVIII): phase II dose-finding study in patients with congenital haemophilia A and inhibitors
Following on from the findings of the phase I safety study, a phase II study was initiated to evaluate the haemostatic efficacy of rpFVIII in treating non-life-/non-limb-threatening bleeds in patients with congenital haemophilia A and inhibitors. 15 This international, prospective, multi-centre, open-label, non-comparative study was conducted across 14 sites. Of 28 patients screened, 24 were eligible; of these, nine experienced at least one bleed and received rpFVIII for a total of 25 bleeding episodes. Seven of these nine patients completed the study – one withdrew consent and one was discontinued by the sponsor. The peak historical anti-hFVIII inhibitor titre was >10 BU/ml for eight of nine patients; the remaining patient had an inhibitor titre of ⩽5 BU/ml. At the time of the first bleed after enrolment in the study, the anti-hFVIII inhibitor titre ranged from <0.8 BU/ml to 15.7 BU/ml and the anti-pFVIII titres from <0.8 BU/ml to 6.2 BU/ml.
The study was terminated early after review of the positive safety and efficacy data for the 25 episodes in which rpFVIII was used. In the nine patients who received rpFVIII, all 25 episodes of bleeding were effectively controlled with a median single dose of rpFVIII, even in the presence of a high-titre anti-hFVIII inhibitor. A median dose of 200.8 U/kg was sufficient to treat bleeding in 80% of patients. As a result of this finding, a dose of 200 U/kg was set for use in latter studies.
OBI-1 (rpFVIII): phase II/III study in patients with AHA
The findings of a phase II/III study in patients with AHA were published in 2015. 14 This was a prospective, multi-centre, international, open-label clinical trial that demonstrated the safety and haemostatic efficacy of OBI-1 for treatment of serious bleeds in patients with AHA. The primary efficacy endpoint was haemostatic control assessed at 24 h on a four-point scale. Other endpoints included assessment of cross-reactivity to rpFVIII at baseline and development of anti-pFVIII antibodies post-treatment, in addition to other safety parameters and utilization of treatment.
Patient population
Male and female patients with a diagnosis of AHA aged ⩾18 and suffering from a serious bleed (as deemed by the investigator) were eligible, and exclusion criteria included the presence of an anti-pFVIII inhibitor titre >20 BU/ml and a platelet count <100 × 109/l. Bleeding events threatening vital organ function, requiring a blood transfusion, compromising muscle viability or neurovascular integrity or impacting a major joint, resulting in a large muscle or soft tissue haematomas, or planned surgery or bleeding from surgical sites were included as these enabled assessments of haemostatic control. Patients who had received prior treatment with rFVIIa or aPCC could participate, providing there was an adequate washout period (3 or 6 h respectively) and there was requirement of ongoing therapy that enabled assessment of haemostatic control. Patients were also excluded if they were haemodynamically unstable or if the bleeding episode was likely to resolve on its own if left untreated. Twenty-nine patients were enrolled and treated with OBI-1 (19 male, 10 female) and had a median age of 70 years. Thirteen of 29 had a significant associated comorbidity such as malignancy or autoimmune disease. The remaining 16 appeared to have idiopathic disease. At screening the median FVIII level was 3% (range <1–30) and the median anti-hFVIII inhibitor titre was 46 BU/ml. One patient was excluded as the local results could not be confirmed by the central laboratory. All patients were already receiving immunosuppressive therapy. Eleven of 29 had received haemostatic therapy in the previous month consisting of BPA or tranexamic acid (TXA).
Dosing and efficacy assessment
A fixed dose of 200 U/kg was selected on the basis of the phase II study findings in patients with congenital haemophilia with inhibitors. 15 Subsequent dosing was based on the clinical status of the patient and FVIII levels measured either by one-stage or chromogenic assay. Trough levels of >80% were targeted for serious bleeds of concern and >50% for less serious bleeds. Bleed assessment was done at two time points; an initial haemostatic assessment was undertaken at 24 h using the four-point scale described in Table 1. Investigators assessed the control of bleeding based on multiple observations, including but not limited to obvious blood loss (external blood loss and bodily fluids), haemoglobin results, red cell transfusion and blood component requirements, physical examination of the bleeding site, examination for organ failure due to haematoma and imaging studies. In terms of efficacy, the response at 24 h was used to predict eventual bleed control.
Efficacy assessment criteria used in the phase II/III study. 14

At 24 h, all 28 patients included in the final analysis had a positive response to treatment as defined by the protocol (95% CI: 88.1–100) and of these 24 were recorded as effective and 4 as partially effective. Control of the primary bleed at the time of the final dose of OBI-1 was considered to have been achieved in 24/28 patients. FVIII activity levels demonstrated the ability to monitor replacement therapy effectively and titrate the dose accordingly to maintain haemostatic levels. Ten subjects discontinued after the primary efficacy endpoint was reached but before the end of the 90-day follow-up period, and reasons for discontinuation include the development of anti-pFVIII inhibitor (2), death due to intracranial haemorrhage (2), death due to sepsis (2), renal failure (1), lack of efficacy (1), lost to follow up (1) and discontinued due to noncompliance (1).
Impact of cross-reacting antibodies on OBI-1 (rpFVIII) use
The presence of cross-reacting antibody had significant impact on the recovery. Based on the level of anti-pFVIII inhibitor levels and the response to treatment, three groups were identifiable. The first encompassed patients with no cross-reacting antibody; the second included those with low level titres (<5 BU/ml) and the remainder had titres between 10 BU/ml and 30 BU/ml (the latter being a patient who responded to OB1-1 prior to the antibody titre being available and was allowed to continue in the study even though one of the exclusion criteria was a titre >20 BU/ml). The antibody titres, FVIII levels, consumption and response assessment are presented in Table 2. High levels of cross-reacting antibody were associated in the initial 24 h with an increased number of infusions, increased consumption of concentrate and lower FVIII levels after the first dose and at 24 h. The absence of cross-reacting antibody not surprisingly was associated with supra-therapeutic FVIII levels, longer intervals between infusions and lower consumption of concentrate. The responses to rpFVIII treatment across the three groups are presented in Figures 1 and 2.
Data from phase II/III study in AHA; rpFVIII usage in relation to the anti-pFVIII inhibitor from phase II/III study in AHA.

Results presented as median (range) or as number of patients; patients excluded if p-FVIII titre >20 BU/ml (one exception made). 14

Post treatment factor FVIII levels in relation to anti-pFVIII inhibitor titre.

rpFVIII usage in relation to anti-pFVIII antibody titre. 14
Antibody assessment and monitoring
Neutralizing anti-hFVIII and anti-pFVIII antibodies were measured at baseline and every 5 days during treatment and subsequently at every follow-up visit in order to monitor for any change in the titres and identify the development of de novo anti-pFVIII antibodies. No specific laboratory standard is required for measurement of OBI-1 levels in the patient’s plasma. 8 However, for detection and measurement of porcine antibodies it is necessary for laboratories to use OBI-1 as the substrate in the Nijmegen modified Bethesda assay. 16
Of the 28 patients, 10 had baseline anti-pFVIII antibodies with titres ranging between 0.8 BU/ml and 29 BU/ml. All patients with baseline anti-pFVIII inhibitors achieved a rise in FVIII levels to >100% during the initial 24 h period in at least one sample and had a positive response to treatment. Over the course of the study the anti-pFVIII antibodies disappeared in 8 of 10 patients and in the remaining 2 patients the titres increased. Of the 18 patients who did not have anti-pFVIII antibodies at baseline, 5 developed de novo antibodies that were neutralizing and precluded further treatment with OB-1. It is worth noting that the presence of cross-reacting anti-pFVIII antibodies at baseline was associated with both clinical response and measurable factor VIII, while the de novo antibodies were neutralizing and were not associated with any increase in factor VIII levels post-infusion. As definitive management of AHA includes inhibitor eradication with immunosuppression, the long-term impact of de novo cross-reacting neutralizing antibodies is difficult to assess.
Adverse events
Seven deaths occurred during the study, including three secondary to sepsis and one due to worsening of renal failure in a patient with a history of renal insufficiency. Three deaths were secondary to haemorrhage; in all instances it was not considered to be due to a failure of treatment with the study drug (one patient had treatment withdrawn due to futility and had a fatal intracranial bleed 1 day after the last OBI-1 dose; two other patients had fatal events 4 and 7 days after the last dose of OBI-1). No thrombotic events were reported. Of note, there were no reported hypersensitivity reactions or any instances of thrombocytopaenia as seen with pd-pFVIII.
OBI-1, susoctocog alfa in clinical practice
OBI-1 is now licensed by the European Medicines Agency (EMA) and the Food & Drugs Administration (FDA) for the treatment of bleeding episodes in adults with AHA. It has been shown to be effective and FVIII levels can be used to assess adequacy of replacement therapy. There does not appear to be a correlation between baseline anti-pFVIII antibodies and response to treatment,13,14,17–19 and this is reflected in the summary of the product characteristics where the presence of anti-pFVIII antibody levels of >30 BU/ml were not considered a contraindication to treatment. Instead, emphasis is laid on the need for use by an experienced clinician and the absolute need for monitoring replacement therapy and clinical response in the context of inter-individual variability in rise of factor VIII levels that is considerably influenced by the presence of cross-reacting anti-pFVIII antibodies.
Real-world experience
Clinical experience of treating bleeding episodes in AHA with OBI-1 is entering the literature.17–19 Although the numbers of patients are small, they also confirm the efficacy of the product. However, there is a common concern regarding the supra-physiological levels of FVIII seen following the initial 200 U/kg loading dose. One retrospective study looked at the use of rpFVIII as a rescue agent following failed responses to BPAs. 19 In six of seven patients, 100 U/kg was used as a loading dose, and one patient received a loading dose of 200 U/kg. Six of seven patients had a factor VIII level after the first infusion >100%, and for one patient there was no measurable response. rpFVIII was effective in five patients, ineffective in one patient and doubtfully effective in one patient who continued to bleed despite normal levels of FVIII. Subsequent dosing was based on FVIII levels and clinical response and the median doses per infusion was lower than the registration trial at 30–100 U/kg; similarly the total dose per patient was lower than the registration trial. Importantly, results were not available for the presence of cross-reacting anti-pFVIII inhibitor. In another study, four patients were treated with a loading dose of 100 U/kg. Three of the four patients had detectable anti-pFVIII inhibitors at baseline (0.6–8 BU/ml). The patient with the highest anti-pFVIII inhibitor failed to respond to rpFVIII, with lack of bleeding control and had an anamnestic rise in the anti-pFVIII antibodies. One patient was diagnosed with a distal deep vein thrombosis (DVT) 10 days after the 45th dose (when the FVIII level was <1 IU/dl). 17 Based on their experience, the authors propose a treatment algorithm based on an initial loading dose of 100 U/kg and subsequent treatment guided by peak and trough FVIII levels. Another article documents the use of the lower loading dose in two patients, chosen in light of known data using pd-pFVIII, using FVIII level to guide further dosing (irrespective of anti-pFVIII or anti-hFVIII inhibitor levels). 18 Treatment was successful in both patients.
In addition to potential adverse events, there remains the concern about cross-reactivity between anti-hFVIII and anti-pFVIII antibodies, response to treatment and the development of anti-pFVIII inhibitors. Roughly one-third of patients have detectable anti-pFVIII antibodies at baseline (35.7%; similar to studies with pd-pFVIII). 14 Although there is a degree of sequence discrepancy between human and porcine FVIII, this is not an unexpected finding. Previous studies and the most recent study in AHA demonstrate that FVIII levels do not rise as rapidly in the first 24 h in patients with a pre-existing anti-pFVIII antibody as in those without, but therapeutic levels could still be achieved. It would therefore appear that if incremental FVIII activity is present and the clinical response is good, treatment with rpFVIII can still be continued even if anti-pFVIII antibodies persist.
Proposed algorithm for the potential first-line use of OBI-1
Patients with AHA can be clinically challenging to treat, particularly the elderly with multiple comorbidities including thrombotic risk factors. Given the clinical challenge and the laboratory requirement for specialist tests, this cohort require tertiary-level care overseen by clinicians with experience in bleeding disorders. Given the demonstrable efficacy of rpFVIII, the question arises as to the role of this agent in this disorder. The majority of patients are likely to be initially treated with BPA and currently OBI-1 is being used as second-line therapy.
A potential haemostatic algorithm for the initial management of bleeding episodes in AHA that includes rpFVIII as a first-line option is presented in Figure 3. During the development of the algorithm, the following factors were taken into consideration. First, the clinical trial and subsequent case series demonstrated an association in the initial 24 h between high titres of cross-reacting antibody (≥10 BU/ml) and higher number of infusions, higher concentrate utilization and lower FVIII levels after the first dose and at 24 hours. This increased treatment intensity increases cost of care with potential for treatment failure. The second factor was the severity of presenting bleed and need for immediate management as life or limb-threatening bleeds in the absence of information about cross-reacting antibodies are ideally treated with BPAs. These are readily available and, importantly, clinicians are familiar with their use and efficacy. The third issue considered was the availability of laboratory monitoring for FVIII levels and inhibitor results as both influence the choice of treatment. The presence of cross-reacting antibody will dictate the initial loading dose as higher inhibitor titres require higher doses of treatment. Further factor VIII levels measured post rpFVIII can be used to determine initial response and subsequent titration of dose and frequency of infusions.17–19 The fourth factor considered was the desired target FVIII levels, as levels beyond 100% of normal are not required for haemostasis and potentially can be associated with thrombosis in this patient group. A dosing regimen aimed to achieve a peak FVIII level of 80–150% of normal with troughs of around 70–80% for severe bleeds is appropriate, and this can be achieved by stratifying the dose according to anti-pFVIII inhibitor levels with a dose of 100 U/kg achieving haemostatic levels in most instances, as demonstrated in the registration trial and subsequent publications.14,17–19 The fifth and final factor was related to the considerable variability in response to rpFVIII treatment, which reinforces the importance of monitoring of peak and trough levels to ensure treatment is cost-effective without exposing the patient to excess risk of thrombosis or treatment failure.
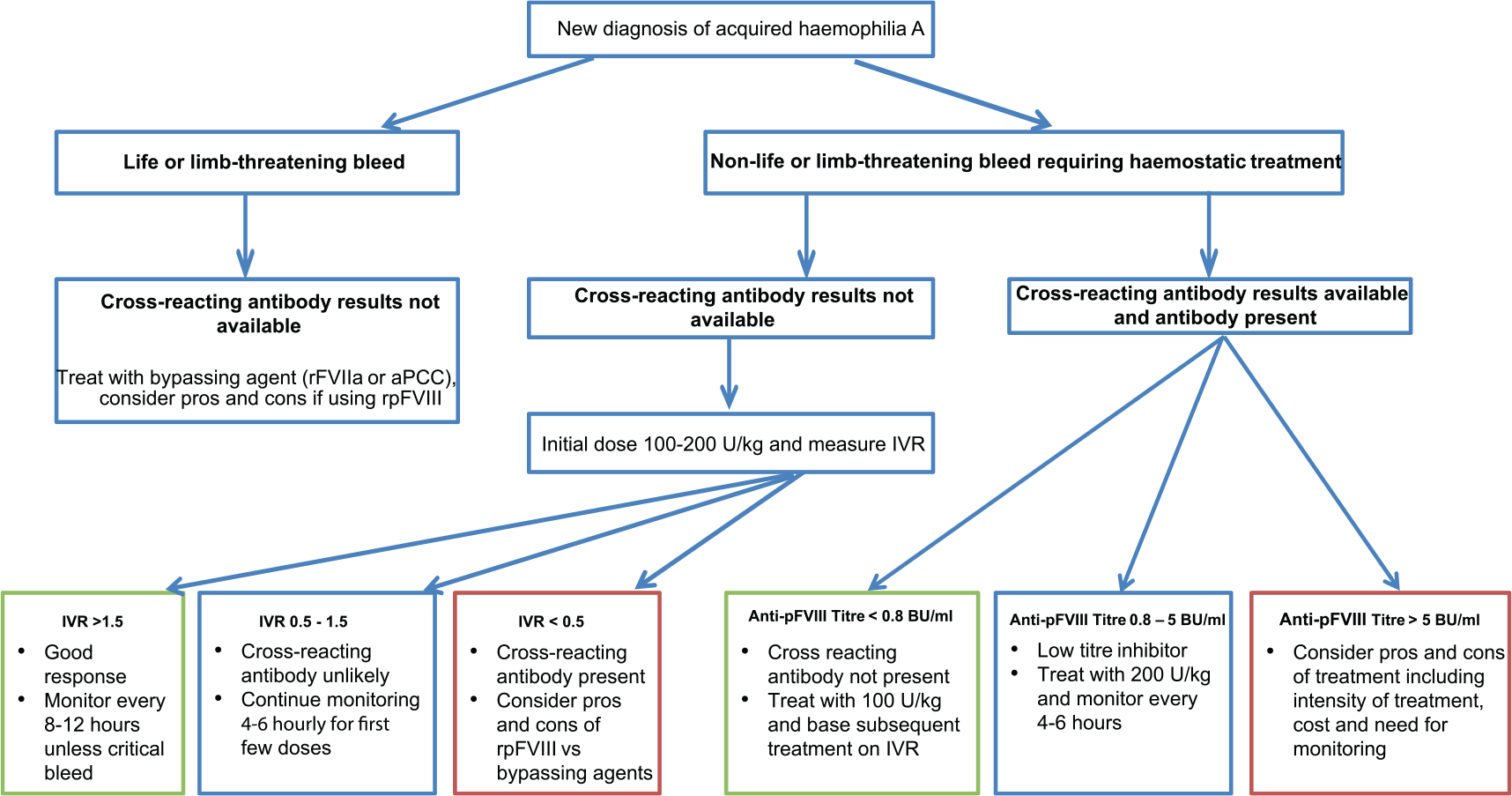
Proposed treatment algorithm for first-line use of rpFVIII in AHA.
Conclusion
In summary, rpFVIII is an effective treatment option in AHA that can be monitored using routine laboratory assays. The response to treatment is unique to each individual patient and importantly this response can be influenced by the presence and development of anti-pFVIII antibodies. As the number of patients treated with this product thus far is small, there is need for ongoing data collection to define and stratify patients based on clinical and laboratory criteria to enable the development of more robust algorithms.
Footnotes
Acknowledgements
We would like to thank the staff of the KD Haemophilia and Thrombosis Centre for their active participation in the pivotal phase II/III study of OB-1.
EF and PC have reviewed the literature and written the manuscript. AD and AR have critically reviewed and revised the manuscript. All authors have reviewed and authorized the final version.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflict of interest statement
P Chowdary, A Drebes and A Riddell were part of the OB-1 pivotal study.
PC has served on advisory boards for Baxalta/Shire, Biogen Idec, CSL Behring, NovoNordisk, Pfizer and Sobi and has received research funding from CSL Behring, Novo Nordisk, Pfizer and Sobi.