
Abstract
Idelalisib is a first in class, delta isoform specific, PI3-kinase inhibitor. Based on its high level of efficacy and acceptable safety profile, this oral drug has been approved by the US Food and Drug Administration as a single agent for the treatment of relapsed or refractory small lymphocytic lymphoma, and follicular non-Hodgkin lymphoma, and in combination with rituximab for patients with chronic lymphocytic leukemia. Adverse effects of particular concern include diarrhea, pneumonitis, and transient elevations of hepatic transaminase levels. Efforts to improve on the activity of this drug have included combinations with standard chemotherapy agents, such as bendamustine, and other targeted therapies, including checkpoint inhibitors. However, other combinations have been associated with life-threatening and fatal toxicities. Thus, the development of such regimens should be conducted carefully in the context of a clinical research study. Idelalisib has a vital role as second-line therapy for chronic lymphocytic leukemia, especially for patients with high-risk disease and multiple comorbidities, and studies are exploring the use of this agent as front-line therapy to improve the outcome of patients with indolent B-cell malignancies.
Keywords
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the western world accounting for 30% of all leukemia cases [Siegel et al. 2015]. The disease is characterized by a clonal proliferation of mature, yet functionally inept lymphocytes expressing CD5, CD19, CD23 and dim levels of CD20. CLL exhibits a heterogeneous course ranging from asymptomatic patients who can be monitored closely without treatment, to those with rapidly increasing lymphocytosis, progressive lymphadenopathy or splenomegaly, constitutional symptoms, or standard treatment resistant cytopenias, who require intervention [Hallek et al. 2008]. Staging, according to either the Rai or Binet classification, is useful in predicting the prognosis of patients and guiding clinical management [Binet et al. 1981; Rai et al. 1975]. The presence of genetic abnormalities such as unmutated immunoglobulin variable heavy chain (IgVH), deletion (del) 17p and 11q and mutations in tumor protein 53 (TP53) and NOTCH1 indicate a poorer prognosis [Mertens and Stilgenbauer, 2014].
Front-line CLL therapy has evolved over the years from akylator-based regimens, to nucleosides, to combination chemoimmunotherapy. Depending on patient age, comorbidities, and lifestyle preferences, fludarabine, bendamustine or chlorambucil in combination with immune therapy such as rituximab or obinutuzumab, all currently serve as reasonable options for treatment naïve CLL [Eichhorst et al. 2014; Fischer et al. 2012; Goede et al. 2014; Hallek et al. 2010]. Despite high response rates, CLL remains an incurable disease with patients inevitably experiencing a relapse. Owing to the adverse effects seen with standard chemoimmunotherapy, elderly patients or those with comorbidities often tolerate treatment poorly and may have lingering toxicities making retreatment challenging. In addition, patients with poor prognostic features such as del 17p or mutations in TP53 or NOTCH1 have an inherent resistance to standard chemoimmunotherapy [Nabhan et al. 2015].
Until recently, the treatment options for relapsed or refractory CLL were limited and included either retreatment with a prior regimen or agents such as alemtuzumab, ofatumumab, bendamustine, or rituximab [Fischer et al. 2011; Keating et al. 2002; Wierda et al. 2010]. Fortunately, the treatment paradigm for relapsed or refractory CLL has now expanded with the availability of an increasing number of small molecules targeting the tumor microenvironment, and pathways associated with the B-cell receptor (BCR), or apoptosis. Idelalisib is a first in its class phosphatidylinositol 3-kinase delta (PI3Kδ) inhibitor, which demonstrates impressive activity in CLL, with manageable toxicities. This review focuses on the role of idelalisib in the treatment of relapsed or refractory CLL.
Biology of CLL
B-cell receptor
The BCR pathway is aberrantly activated in malignant B cells, playing an integral role in the pathogenesis of CLL. In a normal mature B cell, antigen binding to the BCR complex composed of membrane immunoglobulin (IgM) bound to intracellular CD79a/CD79b, recruits tyrosine kinase Lyn and spleen tyrosine kinase Syk, which initiates the signaling cascade [Woyach et al. 2012]. Through the activation of additional kinases and adaptor proteins, 3 main downstream pathways are activated: Bruton tyrosine kinase (BTK), phospholipase C-γ2 (PLC-γ2) and PI3K. This signal transduction leads to B-cell maturation, survival and antibody producing ability. In CLL, BCR signaling differs from normal B cells and is characterized by a variable response to antigen stimulation, decreased levels of IgM expression and constitutive activation of specific kinases such as Lyn and Syk leading to tonic activation of anti-apoptotic pathways. The constant BCR pathway stimulation also increases the production of CCL3 and CLL4, chemokines responsible for promoting lymphocyte survival and tissue homing [Burger et al. 2009].
PI3K pathway
PI3Kδ expression is restricted to hematopoietic cells and plays a central role in B- cell homeostasis and function [Vanhaesebroeck et al. 1997]. PI3Ks are lipid kinases composed of 3 classes [Herman et al. 2010]. Class 1 consists of 4 isomers; alpha (α) beta (β), gamma (γ) and delta (δ), and each is composed of a catalytic subunit p110 and regulatory subunit p85. Lyn and Syk activate the PI3K pathway through the phosphorylation of B-cell adaptor protein (BCAP) and CD19, leading to the recruitment of p85/p110 complex [Aiba et al. 2008]. This activated complex phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which excites downstream signaling pathways such as protein kinase B (AKT) contributing to cellular growth [Engelman et al. 2006]. Other stimulators of PI3K pathway include Toll-like receptors, sphingosine 1-phosphate, the chemokine CXCL13, and cytokines such as interleukin (IL)-4 and B-cell activating factor (BAFF) [Bilancio et al. 2006; Durand et al. 2009; Henley et al. 2008; Patke et al. 2006]. Mouse models harboring mutations in p85α, p110δ and CD19/BCAP subsequently have impaired immunity and inability to develop mature, functional B cells demonstrating the importance of the PI3K pathway [Aiba et al. 2008; Fruman et al. 1999; Okkenhaug et al. 2002; Suzuki et al. 1999].
In B-cell malignancies, the PI3K pathway is hyperactive, stimulating the tumor environment to aid in the growth, survival, and migration of CLL cells [Herman et al. 2010]. Due to this vital role, inhibition of this pathway serves as an attractive target for CLL therapy.
Pharmacology
Mechanism of action
Idelalisib (GS-1101 or CAL-101) is a reversible, highly selective inhibitor of the delta isoform of PI3K [Gilead Sciences, 2014]. The molecular formula is C22H18FN7O and chemical name is 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]quinazolin-4(3H)-one (Figure 1).
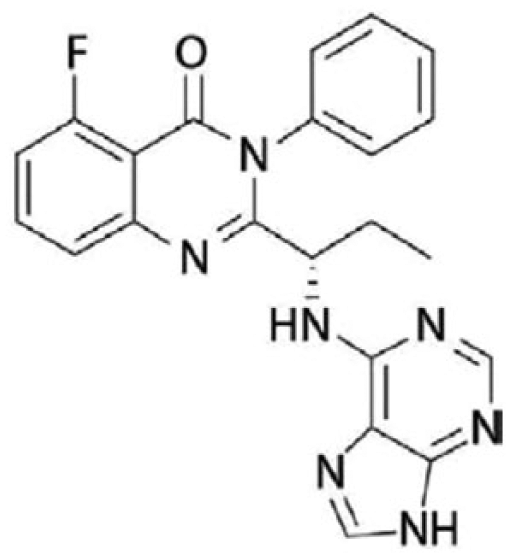
Structure of idelalisib [Gilead Sciences, 2014].
Idelalisib targets malignant B-cell proliferation, survival, migration and homing to lymphoid tissues through multiple mechanisms. There is an interference with BCR signaling through a decrease in phosphorylation of downstream pathways such as AKT and mitogen-activated protein kinase (MAPK) [Lannutti et al. 2011]. In a dose- and time-dependent manner, idelalisib promotes caspase-dependent apoptosis, which is independent of CLL prognostic factors such as del 17p and 11q [Herman et al. 2010; Lannutti et al. 2011]. Idelalisib also targets the tumor microenvironment by hindering the secretion of cytokines and chemokines, which disrupts the communication between CLL cells and monocyte derived nurse-like cells (NLCs) thereby reducing chemo taxis [Hoellenriegel et al. 2011]. Similarly, idelalisib specifically inhibits survival signals from stromal derived factors CD40 ligand, BAFF, tumor necrosis factor, and fibronectin [Herman et al. 2010; Hoellenriegel et al. 2011]. In vivo, T-cell and natural killer cell viability are not affected by idelalisib; however, their production of inflammatory and anti-apoptotic cytokines such as IL-4, IL-6, IL-10 and interferon gamma (IFNγ) is diminished [Herman et al. 2010].
Pharmacokinetics
Idelalisib was found to be well-tolerated at doses of up to 400 mg daily in healthy volunteers, without reaching a maximal tolerated dose [Webb et al. 2010]. However, the current recommended dose is 150 mg twice daily due to a lack of a substantial increase in plasma concentration with higher doses and a stable, steady-state level reached by day 8 with twice daily rather than daily dosing [Brown et al. 2014; Webb et al. 2010]. Idelalisib can be taken irrespective of food and reaches its peak within 1.5 and 4.5 hours when ingested with and without food, respectively. More than 80% of it is bound to plasma proteins with a mean volume of distribution of 23 liters [Gilead Sciences, 2014]. The majority of idelalisib is metabolized by aldehyde oxidase and cytochrome P4503A (CYP3A) to its major, inactive metabolite, GS-563117 and minimally metabolized by UGT1A4. The half-life is 8.2 hours with most of its metabolites being excreted in feces and to lesser extent, urine. Idelalisib can be administered safely to patients with severe renal impairment [creatinine clearance (CrCl) 15–29 ml/min] without increasing toxicity. On the other hand, caution is advised in patients with moderate hepatic impairment [aspartate aminotransferase (AST) or alanine aminotransferase (ALT) greater than 2.5 times the upper limit of normal (ULN) or bilirubin greater than 1.5 ULN] due to a potential increase in systemic exposure. However, in a study with 32 patients with moderate to severe hepatic impairment, no clinically relevant changes in either idelalisib or GS-563117 exposures were observed when compared with matched healthy subjects [Jin et al. 2014a]. Concurrent administration of idelalisib with strong CYP3A and P-glycoprotein (P-gp) inducers should be avoided due to the possibility of decreased drug levels [Jin et al. 2014b]. Conversely, although idelalisib is metabolized by CYP3A, it is not a sensitive substrate and can be given with CYP3A and P-gp inhibitors with watchfulness [Gilead Sciences, 2014; Webb et al. 2010]. GS-563117 is a moderate inhibitor of CYP3A advising caution when administering idelalisib with other drugs utilizing CYP3A metabolism [Jin et al. 2014b]. Foods such as grapefruit, pomegranates, passion fruit, and Seville oranges are restricted due to an effect on drug metabolism.
Clinical studies
Idelalisib as a single agent
Given the molecular role of idelalisib and pharmacologic profile, the first phase I, multicenter study evaluating the clinical efficacy of idelalisib as a single agent in patients with relapsed or refractory disease was conducted [Brown et al. 2014]. Idelalisib 50–350 mg once or twice daily was given to 54 patients with relapsed or refractory CLL (median of 5 prior therapies). High-risk disease features included unmutated IgVH (91%), refractory disease (70%), del 11q (28%), del 17p and/or TP53 mutation (24%) and NOTCH1 mutation (17%). The overall response rate (ORR) was 72% with a complete response (CR) rate of 39% according to the updated response criteria [Cheson et al. 2012]. Treatment resulted in a reduction of lymph node size (82%), resolution of hepatomegaly (100%), and splenomegaly (70%) and normalization of cytopenias: anemia (68%), thrombocytopenia (79%), and neutropenia (100%). The median progression-free survival (PFS) was 15.8 months and the 3-year overall survival (OS) was 75%. Patients treated with idelalisib 150 mg or greater twice daily (n = 28) had a median PFS of 32 months compared with 7 months for those treated at lower doses (n = 26). Unlike standard chemoimmunotherapy, notable responses were seen in patients with high-risk disease. The ORR was 54% in patients with del 17p and/or TP53 mutation with a median PFS of 3 months among all dose groups and 5 months for those treated with higher doses. The 9 patients with a NOTCH1 mutation had an ORR of 67%, with a duration of response (DOR) of 7.8 months. There were no reported dose-limiting toxicities and the most common grade 3 or 4 adverse events included pneumonia (20%), neutropenic fever (11%), diarrhea (6%), pyrexia (4%), cough (4%), fatigue (2%), and transaminitis (2%). A total of 23 patients (43%) were enrolled into a phase I extension study with results still pending [ClinicalTrials.gov identifier: NCT01090414]. Idelalisib has also been explored as front-line therapy for elderly patients with CLL producing an ORR of 81% [Zelenetz et al. 2014].
Idelalisib, especially when administered as a single agent, is associated with a transient increase in absolute lymphocyte count (ALC). Rather than representing disease progression, this observed lymphocytosis is secondary to the interference of lymphocyte homing by idelalisib. The reduced interaction between NLCs and BCR-induced chemokines, CXCL12 and CXCL13 and CLL cell chemokine receptors, CXCR4 and CXCR5, results in decreased migration of malignant cells to lymphoid tissues [Hoellenriegel et al. 2011]. The circulating CLL cells lack prosurvival signals from the tissue microenvironment making them more prone to death, which makes the observed ALC elevation often transient. Typically, the lymphocytosis is maximal within the first 8 weeks of treatment; however, can persist above baseline for up to 24 weeks [Brown et al. 2014]. Currently, lymphocytosis alone does not define disease progression, but rather the development of signs or symptoms of progressive CLL such as cytopenias, or lymphadenopathy, are considered to be indicators of true progression [Cheson et al. 2012].
Idelalisib and chemotherapy, chemoimmunotherapy and immune therapy
Early phase studies
Given the impressive results seen with idelalisib monotherapy, especially in high-risk disease, a pharmaceutical sponsored, 7-arm, phase Ib study was conducted in relapsed and refractory CLL [Barrientos et al. 2015a]. Of 114 patients enrolled, high-risk features included unmutated IgVH (79%), refractory disease (51%), del 17p/TP53 mutation (29%), NOTCH1 mutation (28%), and del 11q (13%). The population was heavily pretreated with a median of 3 prior therapies (range 1–9). To discover the most efficacious and least toxic regimen, idelalisib was investigated with various doublet and triplet combinations consisting of chemotherapy [bendamustine (B), fludarabine (F), chlorambucil (Chl)], chemoimmunotherapy [bendamustine plus rituximab (BR), chlorambucil plus rituximab (ChlR)], and monoclonal antibodies [rituximab (R), ofatumumab (Ofa)]. The following section will discuss the details and results of the 7 arms.
Promising preclinical data have demonstrated a sensitization to bendamustine and fludarabine in idelalisib-treated CLL cells [Hoellenriegel et al. 2011]. To explore the clinical application of this finding, idelalisib 150 mg (100 mg for n = 4) twice daily was given continuously with either B 90 mg/m2 days (D) 1 and 2 for 6, 28-day cycles (n = 15), F 40 mg/m2 D1–5 for 6, 28-day cycles (n = 12) or Chl 10 mg/m2 D1–7 for 3–12, 28-day cycles (n = 18) as part of the multi-arm, phase Ib study [De Vos et al. 2013]. The population was high-risk: IgVH unmutated (84%), refractory disease (58%), del 17p/TP53 mutation (42%) and NOTCH1 mutation (26%). For the entire group, the ORR was 78% and among each cohort was idelalisib-B 78% (CR 0%), idelalisib-F 92% (CR 0%) and idelalisib-Chl 67% (CR 7%). The combinations demonstrated activity in patients with del 17p or TP53 mutation (ORR 70%). The median PFS was 28.5 months overall, 19.9 months for the idelalisib-B cohort and not reached for both idelalisib-F and idelalisib-Chl groups. The most common grade 3 or 4 adverse events among all groups were mostly hematologic and included neutropenia (67%), thrombocytopenia (24%), and anemia (22%). Nonhematologic grade 3 or 4 side effects were febrile neutropenia (22%), diarrhea (16%), and transaminitis (16%). The addition of idelalisib to traditional chemotherapy was able to overcome the chemotherapy resistance seen with high-risk disease producing significant clinical activity; however with appreciable hematologic toxicity.
Triplet therapy was also explored in relapsed and refractory CLL as part of the 7-arm, phase Ib study. A total of 29 patients received idelalisib 150 mg twice daily continuously with either B 70 or 90 mg/m2 D1+2 plus R 375 mg/m2 D1 for 6, 28-day cycles (n = 15) or Chl 10 mg/m2 D1–7 for 12 cycles plus R 375 mg/m2 D1 for 6, 28-day cycles (n = 14) [Barrientos et al. 2013]. High-risk features included rituximab-refractory disease in 55% of patients. The ORR was 87% (CR 7%) and 93% (CR 14%) for the idelalisib-BR and idelalisib-ChlR groups, respectively. At 18 months of follow up, the median PFS of the idelalisib-BR group was not reached with a 1-year PFS of 87% [Coutre et al. 2012]. At 8 months of follow up, the median PFS for the idelalisib-ChlR group was not reached [Barrientos et al. 2013]. The most common grade 3 or 4 adverse events in the idelalisib-BR and idelalisib-ChlR groups were pyrexia (0% versus 7%), diarrhea (13% versus 7%), fatigue (0% versus 21%), transaminitis (0% versus 21%), neutropenia (60% versus 43%), thrombocytopenia (7% versus 21%), and anemia (13% versus 14%). As opposed to idelalisib-B, the idelalisib-BR group had fewer reports of grade 3 of 4 hematologic toxicity, pneumonia/pneumonitis (29% versus 0%), transaminitis (18% versus 0%), infections (18% versus 0%), and diarrhea (12% versus 7%) [Coutre et al. 2012]. In comparison to idelalisib plus cytotoxic therapy, idelalisib in combination with chemoimmunotherapy had comparable clinical activity with a better toxicity profile, warranting further exploration.
Rituximab in combination with cytotoxic therapy has shown to be quite efficacious in the treatment of CLL [Fischer et al. 2011, 2012; Hallek et al. 2010]. Ofatumumab is a second-generation, fully humanized IgG anti-CD20 monoclonal antibody, which has demonstrated single agent activity in relapsed or refractory CLL [Wierda et al. 2010]. As part of the 7-arm, phase Ib study, idelalisib 150 mg twice daily was given in combination with R 375 mg/m2 weekly for 8 infusions (n = 19) or Ofa 300 mg D1+2 of week 1, then 1000 mg weekly for 7 weeks followed by 1000 mg every 4 weeks for 4 doses for a total of 12 infusions (n = 21) [Furman et al. 2014a]. Of 40 patients enrolled, 98% had received prior rituximab and 55% had rituximab-refractory disease. The ORR for both cohorts was 83% (CR 5%) with a median PFS of 24 months. The median OS was not reached at 24 months with an estimated 2-year OS of 80%. Among the idelalisib-R group, the ORR was 78% (CR 0%) with a 1-year PFS of 74% [Coutre et al. 2012]. The 28% of patients with del 17p and/or TP53 mutation demonstrated an ORR of 73% and median PFS of 20 months [Furman et al. 2014a]. The most common grade 3 or 4 adverse events amongst both arms included diarrhea/colitis (23%), pneumonia (18%), transaminitis (10%), and pneumonitis (5%). Of note, transaminitis did not recur upon re-exposure to idelalisib after resolution with the exception of 1 case. Secondary malignancies including breast cancer, colon cancer and acute myeloid leukemia (AML), were observed in 3 patients, but deemed unrelated to treatment. Immune therapy in combination with idelalisib had similar efficacy to that observed with cytotoxic doublet therapy demonstrating activity in high-risk, refractory disease. Despite the addition of a monoclonal antibody, the toxicity profile was similar to that observed with idelalisib monotherapy.
Barrientos and colleagues [Barrientos et al. 2015a] reported long-term follow up of the 7-cohort (idelalisib plus B, F, Chl, BR, ChlR, R, or Ofa,), phase Ib study. The ORR was 83% with a response rate of 70% in patients with del 17p/TP53 mutation. The time to response was rapid and seen after a median of 1.9 months. The median PFS was 26.1 months for all 7 cohorts and 20.3 months for patients with del17p/TP53 mutation. The 3-year OS was 73% overall and lower at 57% for patients with del 17p/TP53 mutation. Owing to durable responses, 54% of patients enrolled in an extension study evaluating the role of idelalisib maintenance and 34% were still on study at 3 years with final results pending [ClinicalTrials.gov identifier: NCT01090414]. The combinations of idelalisib with BR, R and Ofa were successful demonstrating rapid and appreciable clinical activity in poor prognostic, refractory and heavily pretreated CLL with manageable toxicities. In particular, the efficacy and safety profile seen with idelalisib-BR appeared to be superior to the other combinations; however, additional comparative studies are necessary to explore the validity of this observation.
Late phase studies
Due to promising clinical activity and acceptable safety profile observed in phase I studies, idelalisib-R was further evaluated in Study 116, a pharmaceutical sponsored, multicenter phase III trial [Furman et al. 2014b]. Patients were eligible if they had disease progression within 24 months of their last treatment and were unfit to receive cytotoxic therapy due to severe myelosuppression from previous therapy, reduced kidney function (CrCl < 60 ml/min) or a Cumulative Illness Rating Scale (CIRS) score greater than 6. The median number or prior therapies was 3 (range 1–14) and must have included either a CD20 antibody or at least 2 prior cytotoxic regimens. A total of 220 patients were stratified according to the presence of del 17p and/or TP53 mutation (n = 95) or unmutated IgVH (n = 184) and then randomized to receive R 375 mg/m2 for 1 dose, followed by 500 mg/m2 every 2 weeks for 4 doses and then every 4 weeks for 3 doses, for a total of 8 infusions with either idelalisib 150 mg (n = 110) or placebo (n = 107) twice daily. The study was terminated at an interim analysis due to a significantly longer median PFS in the idelalisib-R group compared with placebo-R group (not reached versus 5.5 months, p < 0.0001). Based on these data, idelalisib-R was approved by the US Food and Drug Administration (FDA) for relapsed or refractory CLL in July 2014. During the second interim analysis at the end of the blinded phase, the median PFS was still not reached in the idelalisib-R group with a 1-year PFS of 66% compared with 13% in the placebo arm [Sharman et al. 2014]. Similarly, the PFS observed in high-risk patients with del 17p and/or TP53 mutation (not reached versus 4 months) or unmutated IgVH (not reached versus 5.5 months) was better with idelalisib-R. Secondary endpoints including ORR (77% versus 15%), and reduction in lymph node size (93% versus 4%) favored the idelalisib-R group. Although, median OS was not reached in both groups, it appeared to favor the idelalisib-R group. Furthermore, the addition of rituximab blunted and shortened the lymphocytosis often seen with idelalisib monotherapy. The most common grade 3 or 4 adverse events in the idelalisib-R arm versus placebo-R group were neutropenia (37% versus 27%), thrombocytopenia (11% versus 18%), anemia (7% versus 17%), pneumonia (8% versus 9%), transaminitis (8% versus 1%), diarrhea/colitis (5% versus 0%), and pneumonitis (4% versus 1%). Patient quality of life in terms of emotional, social and functional well-being, and leukemia-specific concerns was significantly better in the idelalisib-R compared to the placebo-R group [Ghia et al. 2014]. Patients who had an ongoing response were offered participation in a 2-arm (idelalisib 150 or 300 mg twice daily), double blind, parallel group extension trial, Study 117, which has completed accrual with results pending [ClinicalTrials.gov identifier: NCT01539291]. In addition, at the time of disease progression, patients could enroll in Study 117 and receive idelalisib (placebo-R group) or idelalisib at a higher dose of 300 mg twice daily (idelalisib-R group). Idelalisib-R is a reasonable second-line option especially for patients with poor prognostic features or who are intolerable of traditional chemotherapy. A pharmaceutical sponsored, phase II study evaluated the combination as front-line therapy in elderly patients and found an ORR of 97% (CR 19%) and 2-year PFS of 91% [O’Brien, Lamanna et al. 2014]. A similar trial evaluating idelalisib-R in treatment naive patients with del 17p is currently recruiting participants [ClinicalTrials.gov identifier: NCT02044822].
Similar to idelalisib-R, the combination of idelalisib-Ofa was further evaluated in a pharmaceutical sponsored, open-label, phase III trial, Study 119, given the efficacy and safety profile observed in earlier studies [Jones et al. 2015]. The trial included 261 patients who had progressed within 2 years from their last therapy and received 2 or more cycles of a purine analogue or bendamustine. Patients were stratified on the basis of relapsed or refractory CLL, IgVH mutation status and the presence of del 17p and/or TP53 mutation. Patients were then randomized to either the control arm: Ofa 300 mg IV for 1 week followed by 2 g IV weekly for 7 weeks and then every 4 weeks for 4 doses (n = 174) or the experimental arm: idelalisib 150 mg twice daily continuously with Ofa 300 mg IV for 1 week followed by 1 g IV weekly for 7 weeks and then every 4 weeks for 4 doses (n = 87). The ORR was significantly higher in the idelalisib-Ofa group in comparison with the Ofa-only group (75% versus 18%, p < 0.0001). Similarly, the median PFS was longer in the idelalisib-Ofa group versus Ofa-only group (16.3 months versus 8 months, p < 0.0001), which held true in patients with del 17p and/or TP53 mutation (13.7 months versus 5.8 months, p < 0.0001). However, there was no difference observed in median OS (20.9 months versus 19.4 months, p = 0.27). The most common grade 3 or 4 adverse events reported in the idelalisib-Ofa cohort were diarrhea/colitis (20%), pneumonia (13%), and febrile neutropenia (12%). Based on efficacy and safety, the combination of idelalisib-Ofa is a reasonable treatment option for patients with relapsed or refractory CLL; however, further studies are necessary to determine its use in comparison to idelalisib-R. The Dana-Farber Cancer Institute is currently recruiting patients for a phase II study evaluating idelalisib-Ofa in treatment naïve CLL [ClinicalTrials.gov identifier: NCT02135133].
Owing to the early phase results seen with idelalisib-BR, the regimen is being investigated further in 2 phase III, randomized, double blind trials in recurrent and previously untreated CLL [ClinicalTrials.gov identifiers: NCT01569295, NCT01980888] [Eradat et al. 2014; Salles et al. 2014]. Both studies have similar designs with patients receiving BR with or without idelalisib and have completed accrual with results pending.
A summary of completed clinical trials with idelalisib is given in Table 1.
Summary of clinical trials with idelalisib.

CR, complete response; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
Idelalisib and other novel agents
The significant antitumor activity and manageable toxicities enable idelalisib to be an attractive partner for other novel therapies. Obinutuzumab, a fully humanized, glycoengineered type II, CD20 antibody, promotes high levels of apoptosis and antibody-dependent cellular cytotoxicity [Robak, 2009]. Based on the results of the phase III, CLL11 study, the FDA approved obinutuzumab in combination with chlorambucil for previously untreated CLL in 2013 [Goede et al. 2014]. Obinutuzumab and chlorambucil had a superior median PFS (26.7 months) when compared to chlorambucil alone (11.1 months) or chlorambucil plus rituximab (16.3 months). Unlike ibrutinib, an irreversible inhibitor of BTK, idelalisib had minimal impact on the immune effects of obinutuzumab and rituximab in preclinical models [Herter et al. 2014]. The German CLL Study Group is conducting the CLL2-BCG study to evaluate the role of idelalisib plus obinutuzumab as induction and maintenance therapy following debulking with bendamustine in both treatment naïve and pretreated CLL [ClinicalTrials.gov identifier: NCT02445131]. The combination is also being explored in untreated CLL in a phase III, pharmaceutical sponsored study [ClinicalTrials.gov identifier: NCT01980875]. Other potential immune therapy combinations include BI 836826, which is an IgG1 chimerized and glycoengineered, anti-CD37 monoclonal antibody [Stephens et al. 2014]. In vitro, BI 836826 demonstrated enhanced direct cytotoxicity with PI3K inhibition against the highly resistant, p53 mutant CLL cells making it an attractive agent for future combination studies.
Idelalisib has been combined with other small molecules targeting pathways downstream from the BCR. Entospletinib (GS-9973), an adenosine triphosphate competitive inhibitor of Syk, has demonstrated greater selectivity than its predecessor, fostamatinib [Currie et al. 2014]. In a phase II study of 41 patients with relapsed or refractory CLL, entospletinib monotherapy produced an ORR of 61% and median PFS of 13.8 months [Sharman et al. 2015]. Owing to in vitro synergy with PI3K inhibition leading to enhanced growth inhibition, further interruption of chemokine signaling and decreased CLL survival, entospletinib was studied in combination with idelalisib in a phase II study [Burke et al. 2014]. A total of 66 patients with relapsed or refractory CLL (n = 35) or non-Hodgkin lymphoma (NHL) (n = 31) received escalating doses of entospletinib (400–800 mg) with idelalisib (100–150 mg) twice daily [Barr et al. 2015]. Among the CLL cohort, 60% of patients achieved a partial response and 20% had stable disease. However, the study was terminated early due to the development of severe, pneumonitis in 12 patients (2 deaths) attributed to excessive mammalian target of rapamycin (mTOR) like activity. Combinations with biologic agents targeting other components of the BCR signaling pathway, such as GS-4059, a second-generation BTK inhibitor, are underway [ClinicalTrials.gov identifier: NCT02457598]. Idelalisib in combination with CX-4945, a selective inhibitor of casein kinase 2 (CK2), which is a fundamental protein that aids in CLL survival, serves as potential regimen for future studies due to synergism observed in pre-clinical models [Prins et al. 2013]. Similarly, panobinostat and vorinostat, both histone deacetylase inhibitors (HDI), have demonstrated in vitro synergism with idelalisib [Bodo et al. 2013]. In CLL lines, PI3K inhibition led to a dramatic increase in HDI-mediated apoptosis through AKT dependent and independent mechanisms and disruption of MAPK pathway. Lenalidomide, a modulator of the tumor microenvironment, has demonstrated anti-tumor activity in CLL both as monotherapy and in combination with rituximab [Chanan-Khan et al. 2006; Badoux et al. 2013]. The combination of lenalidomide and idelalisib is yet to be studied in CLL; however phase I studies exploring idelalisib, rituximab and lenalidomide in both relapsed follicular and mantle cell lymphoma were modified to exclude rituximab due to a high rate of toxicity concerning for cytokine release syndrome [Smith et al. 2014].
Toxicity
Idelalisib has a tolerable safety profile that enables administration to elderly patients and those with complex comorbidities such as renal failure. Although caution is advised, idelalisib has been given safely to patients with hepatic dysfunction [Jin et al. 2014a]. Transaminase elevations are common, occurring in up to 50% of patients (Grade 3/4 14%) [Gilead Sciences, 2014]. The rise typically occurs within the first 12 weeks of therapy and is reversible in most patients with temporary drug cessation and dose titration if necessary [Brown et al. 2014; Gilead Sciences, 2014; Jin et al. 2014a; Webb et al. 2010]. Diarrhea is frequent and has 2 peaks of occurrence. A lower grade diarrhea can be experienced by greater than 30% of patients in the first few weeks of therapy, whereas a more severe colitis can occur 6–12 months into treatment in 9% of patients [Coutre et al. 2015b]. The late onset diarrhea tends to respond poorly to anti-motility agents lasting 1 week to 1 month following temporary interruption in treatment and in some cases nonabsorbable steroids [Coutre et al. 2015a]. Severe and sometimes fatal pneumonitis can be triggered by idelalisib treatment in 2–4% of patients. Further investigation is necessary at any signs of pulmonary dysfunction and if idelalisib induced pneumonitis is suspected, therapy should be stopped and steroids and antibiotics implemented [Coutre et al. 2015a]. Maculopapular skin rash occurring in 10–13% of patients is typically low grade and responsive to steroids [Hou et al. 2015]. Grade 3 or 4 neutropenia is common occurring in greater than 30% of patients; however, the rate of infection is not significantly increased with idelalisib use [Coutre et al. 2015b]. Thrombocytopenia has been reported in 30% of patients and tends to be mild to moderate in severity with no increase in bleeding even in patients on anticoagulant therapy [Barrientos et al. 2015b]. Tumor lysis syndrome is uncommon with no reported cases in the approval study of idelalisib [Brown et al. 2014]. The management of the most common toxicities is depicted in Table 2.
Management of common toxicities [Coutre et al. 2015a].

ALT, alanine transaminase; AST, aspartate transaminase; ULN, upper limit of normal.
Other PI3K inhibitors
In addition to idelalisib, there are several other PI3K inhibitors being explored in CLL (Table 3). Pilaralisib, a pan-PI3K inhibitor, was studied in a phase I trial of relapsed or refractory CLL and lymphoma [Brown et al. 2015]. All grades of diarrhea (92%) were more common with pilaralisib than idelalisib. However, grade 3 or 4 diarrhea (20%) occurred at a similar rate to that of idelalisib, which suggests a role for delta inhibition in the development of severe colitis. Unlike idelalisib, pilaralisib was associated with hyperglycemia (28%) and higher rates of rash (32%), which may be toxicities related to alpha inhibition. In a phase IIa study of relapsed or refractory CLL and NHL, copanlisib, another pan-PI3K inhibitor most potent against α and δ subunits, was investigated [Dreyling et al. 2014]. As opposed to idelalisib, the most common grade 3 or 4 toxicities of copanlisib included hypertension (49%), hyperglycemia (30%) and neutropenia (30%). Despite being a dual inhibitor of PI3K γ and δ, duvelisib has a comparable toxicity profile to that of idelalisib. The most frequent grade 3 or 4 adverse effects experienced in 2 phase I studies with treatment naive and pre-treated CLL included the following: neutropenia (31%), diarrhea (22%), transaminitis (17%), febrile neutropenia (15%), thrombocytopenia (11%), rash (11%) and pneumonia (11%) [O’Brien, Patel et al. 2014; Patel et al. 2015]. TGR-1202, an inhibitor of PI3K δ, was first explored in a phase I study of relapsed or refractory CLL and NHL. Similar to idelalisib, all grade adverse events reported included diarrhea (34%), fatigue (31%), nausea (29%), and cough (26%) [Burris et al. 2015]. However, in contrast to idelalisib, no TGR-1202 related hepatotoxicity or colitis have been observed to date. Although, comparative studies are necessary, the unique toxicity profiles of each PI3K inhibitor will undoubtedly play a role in therapeutic decision-making in CLL, allowing for more individualized treatment.
Studies in CLL with other PI3K inhibitors.

BTK, Bruton tyrosine kinase; CLL, chronic lymphocytic leukemia; NHL, non-Hodgkin lymphoma; PI3K, phosphatidylinositol 3-kinase; SLL, small lymphocytic lymphoma.
Clinical practice
Idelalisib-R is an attractive treatment option for relapsed or refractory CLL. The regimen has demonstrated an acceptable safety profile making it optimal for treatment of older patients or those with confounding comorbidities, which tends to be the limitation of standard chemoimmunotherapy. However, patients should be monitored closely for the development of severe transaminitis, colitis or pneumonitis, which can prompt permanent termination of idelalisib therapy. The antitumor activity of idelalisib is broad, targeting patients with refractory, heavily pre-treated disease and poor prognostic features such as del 17p and TP53 mutation. Similarly, ibrutinib has demonstrated considerable activity in CLL leading to its approval for relapsed or refractory disease and as front-line therapy for del 17p [Byrd et al. 2014]. The once-daily administration of ibrutinib offers greater convenience to patients over the twice-daily dosing of idelalisib. While the approval study for idelalisib included patients with reduced renal function, poor bone marrow reserve and CIRS score greater than 6, the approval study for ibrutinib, the Resonate trial, excluded or had a minimal number of such patients. Given the respective study populations, idelalisib and rituximab is the appropriate choice of therapy for patients with confounding comorbidities such as renal and hepatic dysfunction and persistent toxicities related to prior treatment. Also, the use of ibrutinib is limited by an increased risk for bleeding and atrial fibrillation, observed in 44% and 7% of patients, respectively [Byrd et al. 2014]. On the other hand, idelalisib has not been associated with cardiac dysfunction and can be administered safely with concurrent anticoagulant therapy [Barrientos et al. 2015b]. With our current understanding, patients should continue on idelalisib therapy indefinitely until disease progression. Despite the high frequency of durable remissions observed with idelalisib, complete responses are few and patients often require salvage therapy. A clinical trial is the preferred option in such settings as there are limited data on effective treatment post idelalisib. Venetoclax (ABT-199), a highly potent BCL-2 inhibitor, has yielded promising results in relapsed and refractory CLL producing an ORR of 79% (CR 22%) and median DOR of 20.5 months [Seymour et al. 2014]. A pharmaceutical sponsored, phase II study evaluating the role of venetoclax in CLL after failure of a BCR pathway inhibitor is currently recruiting patients [ClinicalTrials.gov identifier: NCT02141282]. Furthermore, similar to idelalisib and ibrutinib, venetoclax demonstrated efficacy in del 17p (ORR 78%), making it an attractive option for salvage therapy and possibly as front-line therapy in the future. A phase II study exploring venetoclax in untreated and previously treated CLL with del 17p has completed accrual with results pending [ClinicalTrials.gov identifier: NCT01889186].
Conclusion
The treatment paradigm of CLL has been revolutionized by an increasing number of novel targeted agents. Idelalisib has a vital role as second-line therapy for CLL, especially for patients with high-risk disease and multiple comorbidities and the use of idelalisib as front-line therapy will likely emerge. Ongoing clinical trials such as the phase III study investigating the efficacy of idelalisib-BR in both treatment naïve and previously treated CLL will help address the question of the optimal timing and composition of combination regimens. Critical data regarding long-term safety with prolong use of idelalisib, ibrutinib, and venetoclax are lacking and will hopefully become available with time. Although more trials are encouraged, caution is advised for future studies combining small biologic molecules due to unforeseen toxicities observed with idelalisib in combination with entospletinib in CLL and lenalidomide in previously treated follicular or mantle cell lymphoma [Barr et al. 2015; Smith et al. 2014]. The emergence of drug resistance CLL is of concern with the inhibition of such intricate pathways and its effect on salvage therapy is unknown [Woyach and Johnson, 2015]. Methods such as next-generation sequencing and in vitro models will help gain a deeper understanding of the complexities driving CLL. As more data become available, the algorithm for CLL therapy will continue to evolve shifting towards more individualized treatment.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
Bruce Cheson, MD is a consultant for Gilead Sciences, AbbVie, Pharmacyclics LLC, Celgene, Astellas Pharma, and Roche-Genentech.