
Abstract
Myxoid liposarcoma (MLPS) is the second most prevalent subtype of liposarcoma. It is usually found in the deep tissues of the lower limbs and rarely in gynecologic tract. Herein we present the second case in the English literature of a primary MLPS arising from the broad ligament which was thought to be a borderline ovarian tumor. The aim is to discuss its clinical and pathological characteristics. A 42-year-old woman presented with pelvic pain for the last 6 months. Magnetic resonance imaging was not specific. She underwent a surgical resection of the tumor mass, and pathological examination confirmed the diagnosis of MLPS deriving from the broad ligament. She received radiotherapy and the patient is doing well at 3 months follow-up. The clinical aspects, pathological diagnosis, prognosis, and therapy approach of broad ligament MLPS are all poorly understood. Complete surgical resection with or without radiotherapy is the mainstay of treatment in located MLPS.
Introduction
Gynecologic sarcomas account for 13% of all sarcomas and 3%–4% of all gynecologic malignancies. The uterus is the most common primary site (83%), followed by the ovary (8%), vulva and vagina (5%), and other gynecological organs (2%). 1
Myxoid liposarcoma (MLPS) constitutes approximately 5% of all adult soft tissue sarcomas and 20–30% of all liposarcomas. 2 More than half of cases are seen in the thigh muscles, where it is typically found in the deep soft tissue of the extremities. 3
Primary tumors of the broad ligament are rare and diverse as benign (leiomyoma, epithelial tumors of Mullerian type like Paratubal/paraovarian broad ligament cysts, Wolffian tumor/adnexal tumor, papillary cystadenoma, fibroma, endometriosis, Müllerianosis) and malignant tumors (leiomyosarcoma). 4 Broad ligament MLPS is very uncommon with only one case reported in the English literature. 5
In this report, we present a case of a primary broad ligament MLPS in a 42-year-old female and aim through a review of the literature to discuss the clinicopathological characteristics of this uncommon entity in this site.
Case presentation
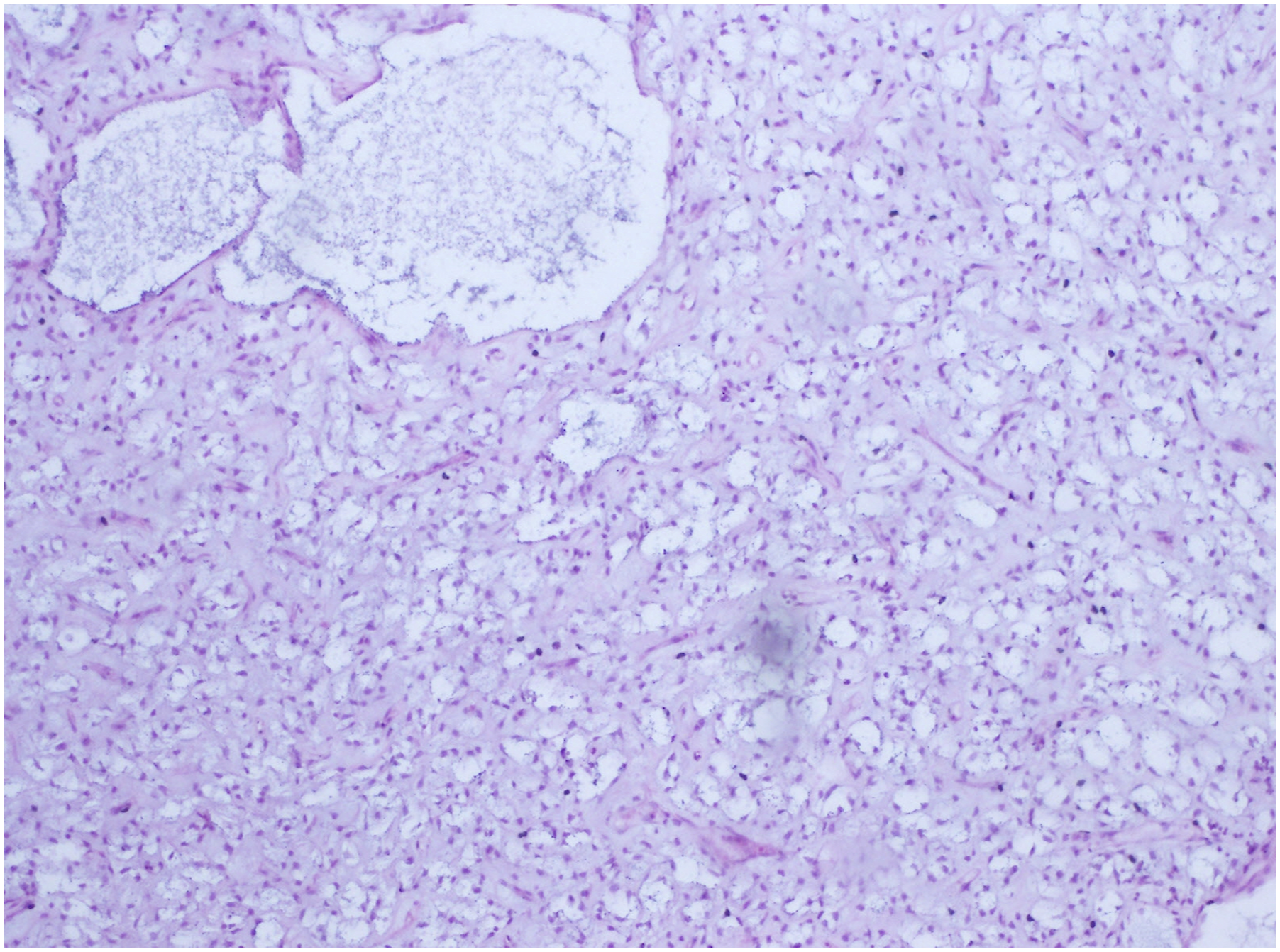
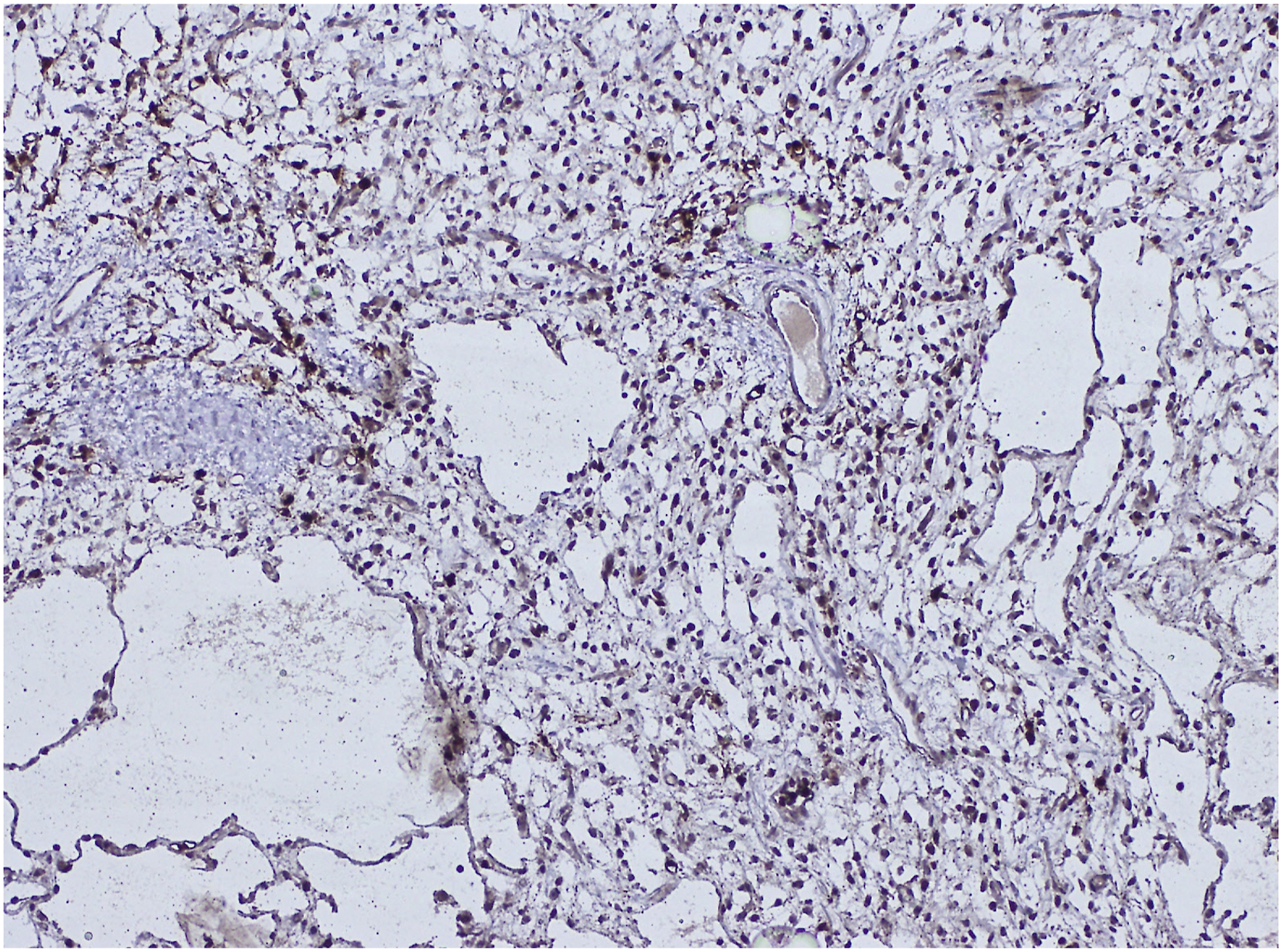
A 42-year-old gravida 4, para 2 woman with no comorbidities presented with the complaint of pelvic pain associated to a sensation of heaviness and then an increase in abdominal volume without digestive or urinary signs present for the last 6 months. The patient had a good general condition. On palpation, she had a pelvic well defined and mobile mass lateralized to the right extending beyond the umbilicus of 20 cm. There was no inguinal adenopathy. Blood work and tumor markers found elevated CA 125: 56.27 U/mL [0–35 U/mL] and normal CEA: 1.18 μg/L [0–5 μg/L]. All the routine blood investigations were performed and were normal. The abdomino-pelvic magnetic resonance imaging (MRI) showed a large multiloculated irregular cystic lesion of 211×199 × 106 mm arising from the right ovary with thickened wall and some endocystic vegetations, seat of irregular and thickened partitions as well as vegetations (Figure 1). There wasn’t lombo-aortic or ilio-femoral lymphadenopathy. There was a small intraperitoneal effusion with no evidence of carcinosis. This radiologic morphology was reminiscent of an ovarian borderline tumor at first glance. Exploratory laparotomy was carried out revealing a voluminous cystic thin-walled abdominal-pelvic mass of 25 cm without extracystic vegetation developing from the right ovary and the broad ligament and extending to the retroperitoneal space in the right pelvis where it spontaneously ruptured with gelatinous contents. The left adnexa and uterus were intact. Frozen section examination concluded to a peritoneal pseudomyxoma and the examination of the appendix was mandatory. A total hysterectomy, bilateral salpingo-oophorectomy, appendectomy, and omentectomy were performed. Gross examination revealed a complex cystic lesion developing from the broad ligament with a smooth outer surface. The cross-sections of the tumor showed large number of nodular areas showing a gelatinous cut surface (Figure 2). At low magnification, the histopathological analysis revealed a myxoid, moderately cellular tumor with a smooth border. Tumor cells had indistinct cytoplasmic borders and oval, stellate-shaped nuclei (Figure 3). There were areas of elevated cellularity. Mitotic count was estimated at <2 mitosis/10 high power fields. The myxoid stroma of the tumor was composed of numerous thin vascular structures with branching resembling chicken wire and large thick-walled veins. Mucin pools were seen in both macrocystic and microcystic areas. Since some areas of the myxoid stroma exhibited microvacuolization, which gave those areas a lipoblast-like appearance, it was difficult to distinguish between lipoblasts and nonlipogenic cells that displayed vacuolar degeneration. Necrosis was absent. On immunohistochemical staining, tumor cells were positive for S-100 (Figure 4), and negative for CK, CK7, EMA, and HMB45. Therefore, the final diagnosis of a low-grade primary broad ligament MLPS of histoprognostic grade 1 according to FNCLCC was retained. The omentum, left and right fallopian tubes, ovaries, uterus and appendix did not contain neoplasm. The margins of resection were free of tumor. Postoperative course was uneventful. A general assessment by a whole-body MRI was performed and reveals negative. The patient received radiation therapy 1 month after surgery because the tumor was large in size. No chemotherapy was administered because the mass was considered to be adequately removed. At 3 months follow-up, the patient remains free of clinical disease. Radiological control was performed by a whole-body MRI which showed no tumor residue. The patient will be followed every 6 months for 4 years with radiographic controls and a complete clinical examination. Magnetic resonance imaging of the lesion: A/Coronal T2 weighted B/Axial T1 weighted images showing a large well-defined multiloculated irregular cystic lesion of 211 × 199 × 106 mm arising from the right ovary with hyperintense septal fat. Macroscopic findings of the specimen. The tumor is arising from the broad ligament. The mass is composed of a yellowish lobulated glistening gelatinous cut-surface. Histological findings of low-grade myxoid liposarcoma. H&E, original magnification ×40. The tumor is composed of an abundant myxoid matrix, lymphangioma-like cystic spaces with component of mature fat tissue. Immunohistochemical staining of low-grade myxoid liposarcoma. Diffuse nuclear and cytoplasmic expression of S100 protein in tumor cells.

Discussion
The present case is the second reported case of primary broad ligament MLPS in the English literature emphasizing how gynecologic pathologists need to be aware of this neoplasm and be able to tell it apart from other soft tissue tumors.
The first case of primary broad ligament MLPS was described in 1992 by Singh et al 5 occurring in a 54 year-old woman who presented with a mass protruding from the left side of her abdomen. It was surgically resected and the patient was free of disease after 2 years of follow-up.
MLPS is the second most common liposarcoma subtype of children and adolescents but has a peak incidence in the fourth to fifth decades with no sex predilection. 2 Another characteristic that sets apart MLPSs from other liposarcomas is their propensity to metastasis in uncommon places, which is associated with a worse prognosis. These atypical locations include the trunk, extremities, bone, retroperitoneal location, chest wall, pleura, and pericardium. 6
MLPS is characterized by the recurrent translocation t (12; 16) or rarely t (12; 22) on the 12q13.3 locus resulting in FUS-DDIT3 and EWSR1-DDIT3 gene fusion transcripts, respectively. 7
MLPSs usually present as large and painless masses. They have no specific symptoms. 2 Clinical symptoms are usually dependent by where the MLPS arises. In the present case, the patient presented with a pelvic pain with a sensation of heaviness. In a case of a dedifferentiated liposarcoma arising from the vagina, the patient complained from vaginal discomfort and pain. 8
Before resection surgery, accurate diagnosis is quite challenging. The only way to diagnose MLPS is through postoperative pathology, yet it has been shown that a significant percentage of MLPS patients had an initial pathological misdiagnosis. An incorrect diagnosis could lead to a delayed or ineffective treatment regimen. 8 In fact, in our case, the tumor was thought to be radiologically, a borderline ovarian tumor and in frozen section a pseudomyxoma peritonei.
On macroscopic examination, MLPSs are typically large (>10 cm), well-defined, multinodular neoplasms. The cut surface is smooth, gelatinous and glistening, like our case. Higher-grade MLPSs show a firmer, fleshy tan surface. 2 Necrosis is not common. Adequate sampling is required to estimate the level of hypercellularity, which is a crucial prognostic factor. 2
Microscopically and at low magnification, MLPSs are moderately cellular, lobulated tumors with increasing peripheral cellularity, composed of patternless arrays of homogeneous, tiny, ovoid cells lacking morphological adipocytic differentiation, with varied numbers of small lipoblasts. The tumors have a dense, mildly basophilic, myxoid stroma with a remarkable plexiform, delicately arborizing capillary network similar to a chicken wire, lymphangioma-like cystic spaces, which neoplastic cells frequently cluster around. Extracellular mucin frequently creates huge pools of mucin, akin to microcystic lymphangioma or pulmonary edema. 9 Except for the presence of large lipoblasts, the tumor in our case has all these characteristics stated. No signs of necrosis, hypercellularity, or primitive round cells that would indicate high grade MLPS were present.
The differential diagnosis in the present case was made with tumors that exhibit myxoid matrix. It includes aggressive angiomyxoma, pseudomyxoma peritonei, myxoid leiomyoma and myxoma. These tumors may share comparable gross characteristics, such as large tumor size. Pseudomyxoma peritonei usually presents as loculated collections along peritoneal surfaces of pouch of Douglas and rectovesical space, with a scalloped appearance. 10 The appendix was totally normal in our case, so this diagnosis was ruled out. The less conspicuous vascular pattern and the presence of smooth muscle fibers inside the myxoid matrix help to identify the myxoid leiomyoma. The aggressive angiomyxoma is distinguished by its extensive vasculature and lack of discernible lipoblasts and its positivity for Desmin and estrogen receptors. 11
Although it has little impact on MLPS diagnosis, immunohistochemistry may help distinguish high-grade MLPS from other round cell sarcomas. Preoperative chemotherapy or radiotherapy frequently results in a significant decrease in cellularity, with only scattered ovoid cells, significant stromal hyalinization, and sometimes maturation into white adipose tissue. 2
In terms of treatment, the role of chemotherapy in patients with soft tissue sarcomas has been thoroughly researched as MLPS is radiosensitive. 12 Surgical excision when the tumor is resectable with or without radiotherapy is the mainstay of treatment in located MLPS. 13 Because broad ligament MLPS is so rare, there is no universally consensual treatment. In the present case, the tumor was surgically resected and the patient had radiotherapy.
Local recurrence occurs in 12–25% of cases and distant metastases develop in approximately 30–60%, sometimes years after initial diagnosis, and may progress slowly. Unlike most sarcomas, MLPSs often metastasize to other soft tissue sites and can metastasize to bone (particularly spine), in preference to lung. 2 At 3 months follow-up which is a short period and considered a limitation of our study, the present patient remains free of clinical disease and will be followed every 6 months for 4 years with radiographic controls and a complete clinical examination.
Tumors with a high histological grade (>5% hypercellularity) have a statistically significant greater rate of metastasis or death from disease. 2 Necrosis, as well as TP53 and CDKN2A mutations, have been attributed to a poor prognosis. Less is known about the prognostic importance of transitional regions with more limited hypercellularity. FUS-DDIT3 transcript isoforms have no correlation with either grade or prognosis.2,14
Conclusion
Although MLPS are frequently found in adults’ extremities, they are very uncommon in the mesorectum. Patients frequently present with nonspecific symptoms, and by the time a diagnosis is established, surgical resection is required. The mainstay of treatment is still surgical excision with clear margins. The role of chemotherapy or radiotherapy is still unknown.
Footnotes
Author contributions
All the authors read and approved the final version of the manuscript. Farah Sassi (MD): conception, acquisition of data, literature research and preparing the manuscript. Ghada Sahraoui (MD): conception, acquisition of clinical data, and revising the manuscript. Lamia Charfi (MD): revising the manuscript. Zemni Ines (MD): Supervision and acquisition of clinical data. Karima Mrad (MD): revising the manuscript critically. Raoudha Doghri (MD): manuscript editing and revising the manuscript critically.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Salah Azaiez Institute does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Guarantor
Sassi Farah
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).