
Abstract
Cortical ependymomas are currently not considered a subgroup of supratentorial ependymomas; however, there is a growing body of literature investigating the natural history of these lesions compared to supratentorial ependymomas. We performed a systematic literature review of cortical ependymomas with a focus on the natural history, clinical characteristics, and clinical outcomes of these lesions as compared to supratentorial ependymomas. Our search revealed 153 unique cases of cortical ependymomas. The mean age on presentation was 21.2 years. Males and females comprised 58.8% (90/153) and 41.2% (63/153) of cases, respectively. The most common presenting symptom was seizure activity occurring in 44.4% of the cohort (68/153). The recently recognized C11orf95-RELA fusion was identified in 13.7% of the cohort (21/153) and 95.5% of cases (21/22) reporting molecular characterization. World Health Organization grades 2 and 3 were reported in 52.3% (79/151) and 47.7% (72/151) of cases, respectively. The frontal lobe was involved in the majority of cases (54.9%, 84/153). Gross total resection was achieved in 80.4% of cases (123/153). Tumor recurrence was identified in 27.7% of cases (39/141). Mean clinical follow-up was 41.3 months. Mean overall survival of patients who expired was 27.4 months whereas mean progression-free survival was 15.0 months. Comparatively, cortical ependymomas with C11orf95-RELA fusions and supratentorial ependymomas with C11orf95 RELA fusions exhibited differing clinical outcomes. Further studies with larger sample sizes are necessary to investigate the significance of RELA fusions on survival in cortical ependymomas and to determine whether cortical ependymomas with C11orf95-RELA fusions should be classified as a distinct entity.
Introduction
Ependymomas are a relatively uncommon entity, accounting for approximately 1.8% of all primary central nervous system (CNS) tumors and 6.8% of glial neoplasms.1,2 These tumors arise from ependymal cells lining the ventricular system, choroid plexus, central canal of the spinal cord and filum terminale. 2 Given their embryonic origin, these neoplasms are most commonly found within the ventricular system (e.g. floor of the fourth ventricle, cervicothoracic cord, filum terminale); however, ependymomas can also be found throughout the neuroaxis, even outside the bounds of the ventricular system. 2 Such entities are known as supratentorial extraventricular ependymomas, which exhibit a low incidence and an unclear etiopathogenesis.1,2
New guidelines by the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) committee in 2020 have distinguished ependymal tumors according to methylome profiling data to indicate specific molecular groups based upon anatomic location. 3 These new molecular groups include ependymal tumors of the supratentorial, posterior fossa, and spinal compartments. 3 Specifically, C11orf95 and YAP1 fusion genes are now considered subgroups of supratentorial ependymomas with C11orf95-RELA fusions exhibiting the worst prognosis of all supratentorial ependymomas.3–6 A small subset of supratentorial extraventricular ependymomas, known as cortical ependymomas, selectively involve the cerebral cortex without any ventricular involvement. Cortical ependymomas are currently not considered to be a distinct subgroup of supratentorial ependymomas; however, there is a growing body of literature specifically investigating the natural history and clinical outcomes of these lesions compared to supratentorial ependymomas as a whole.7–48 In fact, several studies have demonstrated cortical ependymomas with high rates of C11orf95-RELA fusions exhibiting favorable outcomes, which is in contradistinction to the natural history of supratentorial ependymomas with C11orf95-RELA fusions.41,43,48 Here, we describe a rare case of a 58-year-old female found to have an ependymoma of the insular cortex. We perform a systematic literature review of cortical ependymomas with a focus on the natural history, clinical characteristics, and clinical outcomes of these lesions as compared to supratentorial ependymomas as a whole.
Systematic literature review
A systematic literature review of cortical ependymomas was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. The PubMed and Web of Science databases were searched through February 2022 using the following search terms: (“cortical ependymoma” [All Fields] OR “supratentorial cortical ependymoma” [All Fields] OR “insula ependymoma” [All Fields] OR “insular ependymoma” [All Fields]). A total of 279 records were returned using these search terms. The bibliographies of these papers were reviewed to search for any additional papers, which yielded five additional records due to the variability of nomenclature used to describe these lesions. These 284 records were screened for original reports of cortical ependymomas including case reports and case series while excluding preclinical or clinical studies, literature reviews, systematic reviews, and meta-analyses. A total of 42 studies met eligibility after applying inclusion and exclusion criteria and were included in the final analysis (Figure 1). These studies encompassed 153 unique cases of cortical ependymomas. Data collection and extraction were performed by one author (AS) with oversight and a second independent review by a second author (JC). Data gathered included: age, sex, gene fusion, tumor grade, location, presence of cystic component, treatment, recurrence, overall survival, progression-free survival, follow-up, and outcome. A summary of the data synthesized is listed in Table 1. PRISMA study selection flowsheet for the systematic review of cortical ependymomas. Systematic review of the literature pertaining to the natural history, clinical features, and treatment strategies of cortical ependymomas. Abbreviations: WHO: World Health Organization; OS: overall survival; PFS: progression-free survival: FU: follow up; F: female; NR: not reported; L: left: GTR: gross total resection; RT: radiotherapy; NA: not available; M: male; R: right; ChT: chemotherapy; STR: subtotal resection. *Overall, the frontal lobe accounted for nine cases, parietooccipital lobe for four cases, frontotemporoparietal region for three cases, and temporal lobe for two cases. Fourteen patients had a cystic appearing tumor.

The average age on presentation was 21.2 years (range: 8–74 years). Males and females constituted 58.8% (90/153) and 41.2% (63/153) of cases, respectively. The most common presenting symptom was seizure activity observed in 44.4% (68/153) of cases. The C11orf95-RELA fusion was observed in 13.7% (21/153) of cases; however, the supratentorial ependymoma C11orf95-RELA fusion subgroup was only recently recognized as a variant by the cIMPACT-NOW committee in 2020. 3 As such, gene fusions were not investigated in the vast majority of cortical ependymomas reported to date. Of cases reporting molecular characterization, 95.5% (21/22) reported the presence of the C11orf95-RELA fusion. World Health Organization (WHO) grades 2 and 3 were reported in 52.3% (79/151) and 47.7% (72/151) of cases, respectively, documenting tumor grades. The most common location was the frontal lobe or at least involvement of the frontal lobe accounting for 54.9% (84/153) of cases. Gross total resection was achieved in 80.4% (123/153) of cases with adjuvant radiotherapy and/or chemotherapy utilized in 43.1% (66/153) and 3.3% (5/153) of cases, respectively. Tumor recurrence occurred in 27.7% (39/141) of cases. Mean clinical follow-up was 41.3 months (range: 2–347 months). Mean overall survival percentage at last known follow-up was 88.3% (128/145). Mean overall survival of patients who expired was 27.4 months (range: 4–72 months). Mean progression-free survival was 15.0 months (range: 4–32 months).
Case illustration
A 58-year-old female presented to the emergency department with three days of mild expressive aphasia and weakness of the right hand. Neurologic examination was otherwise unremarkable. Past medical history was unremarkable. Magnetic resonance imaging (MRI) of the brain demonstrated a heterogeneously enhancing lesion of the left insula with a large cystic component extending superiorly into the left frontal inferior gyrus with significant surrounding vasogenic edema (Figure 2). Metastatic workup was unrevealing. The patient was started on dexamethasone and medically optimized. Given the significant mass effect, symptomatic edema, and need for tissue diagnosis, neurosurgical resection was recommended. Initial MRI of the Brain. (a–c) T1-weighted imaging with a heterogeneously enhancing lesion of the left insular cortex with a large cystic component extending superiorly into the left inferior frontal gyrus with significant surrounding vasogenic edema. (d) Perfusion-weighted imaging with metabolically active tumor within the left insular cortex. (e) T2-weighted imaging with a moderately-sized cystic component extending superiorly into the left inferior frontal gyrus. (f) Fluid-attenuated inversion recovery imaging confirming vasogenic edema surrounding the tumor of the left insular cortex and cystic component.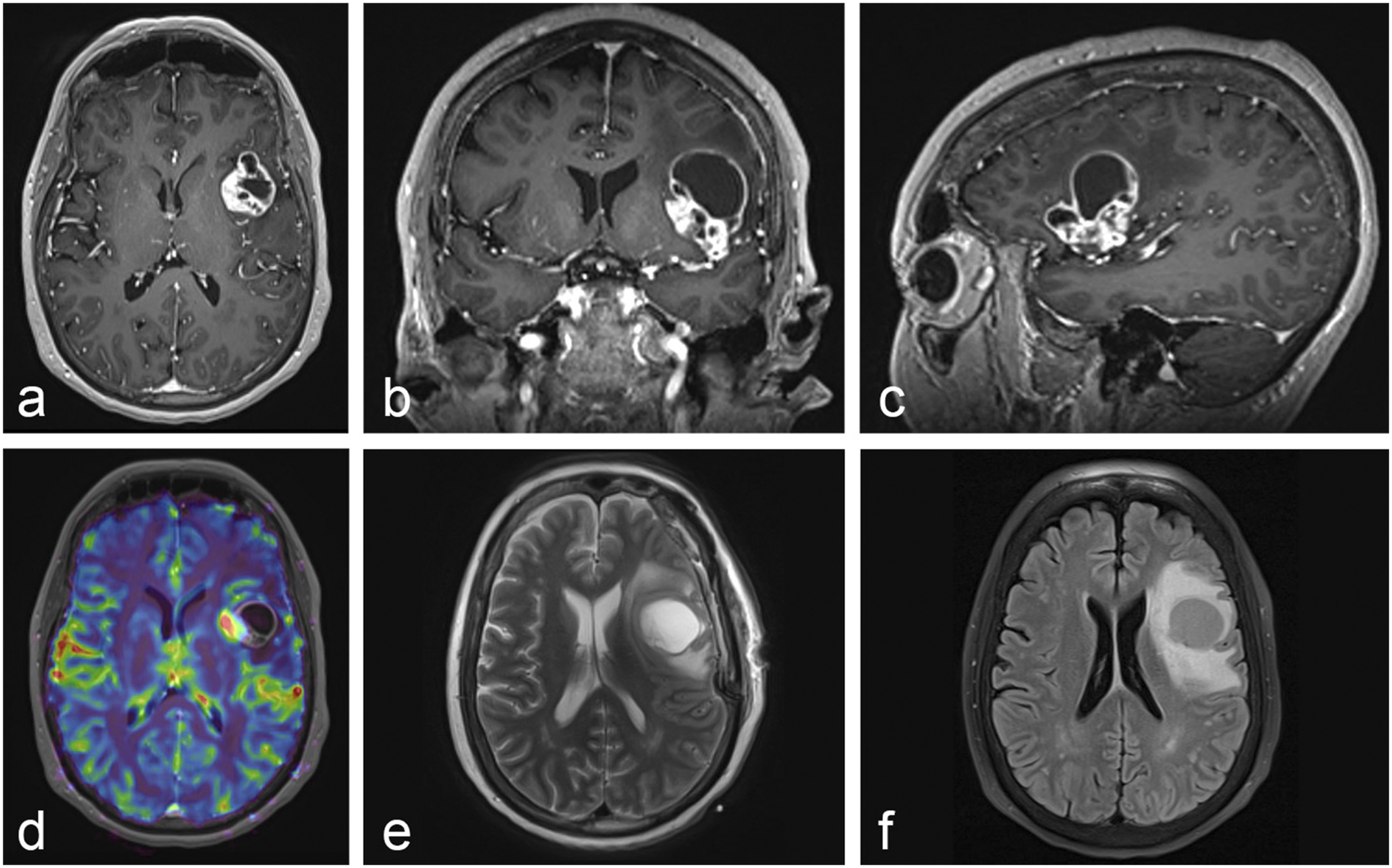
A stereotactic left craniotomy was performed for resection of the left insular lesion and superiorly extending cystic component. A subtotal resection was pursued given that the tumor was found to be encasing the vessels of the Sylvian fissure. The patient tolerated the procedure well and without complication. Post-operative neuroaxis MRI was unremarkable for drop metastases. Histopathology demonstrated cellular neoplastic tissue consisting of cords of relatively monomorphic tumor cells with perivascular pseudorosettes and focal true ependymal rosettes (Figure 3). There was evidence of focal tumor invasion into surrounding brain parenchyma, a feature that can rarely be seen in ependymoma.
15
The Ki-67 proliferative index was variable and focally ranged up to 20% in some areas. Neither necrosis, nor vascular proliferation, nor significant mitotic activity was identified. Tumor cells were strongly and diffusely reactive with glial fibrillary acidic protein (Figure 4). Synaptophysin and neuron-specific enolase highlighted neurons in the infiltrative areas. These stains exhibited limited reactivity in tumor cells; however, the reactivity was patchy and weak compared to the strong and diffuse glial fibrillary acidic protein immunostains. Additional immunostains including Melan-A, SOX10, cytokeratin AE1/AE3, cytokeratin 7, and CD45 were negative in all tumor cells, which ruled out metastatic melanoma, metastatic carcinoma, and lymphoma as potential diagnoses. Neuropathology confirmed a diagnosis of supratentorial ependymoma grade 2 not elsewhere classified according to molecular testing.
3
The patient underwent adjuvant Cyberknife radiosurgical treatment to the resection cavity eight weeks after surgery. The most recent repeat MRI imaging at 15 months revealed a stable disease burden. Histopathology consistent with WHO grade 2 ependymoma. Histopathology demonstrated cellular neoplastic tissue consisting of cords of relatively monomorphic tumor cells with perivascular pseudorosettes and focal true ependymal rosettes. Neither necrosis, nor vascular proliferation, nor significant mitotic activity was identified. (a–b) Hematoxylin and eosin stain at ×40 and ×400 magnification, respectively, showing true ependymal rosettes consisting of columnar cells around central lumens. (c) Hematoxylin and eosin stain at ×200 magnification with pseudorosettes. (d) Hematoxylin and eosin stain at ×100 magnification with focal tumor invasion into surrounding brain parenchyma, a feature that is seen more often in supratentorial ependymomas. Immunohistochemistry consistent with ependymoma. (a–b) GFAP immunostain at ×200 and ×400 magnification, respectively, shows minimal staining in the ependymal cells of these true rosettes. (c–d) GFAP immunostain at ×200 and ×400 magnification, respectively, highlights the ependymal cell processes in pseudorosettes. (e) NSE immunostain at ×100 magnification shows weak staining in pseudorosettes. (f) Synaptophysin immunostain at ×100 magnification shows weak staining in pseudorosettes.

Discussion
Classification of ependymal tumors based upon the anatomical location is a fundamental principle of the recent cIMPACT-NOW guidelines. 3 This reclassification was prompted by molecular profiles to suggest distinct genetic profiles observed in the supratentorial, posterior fossa, and spinal compartments. 3 Specifically, supratentorial ependymomas are now classified according to the genes, C11orf95 and YAP1, which contribute the most significant pathogenic gene fusions in each group with grade defined by morphological criteria. 3 These two groups of supratentorial ependymomas have been distinguished by their clinical characteristics in most studies.3–6 For example, Pajtler et al. demonstrated that supratentorial ependymomas harboring a YAP1 fusion are clinically and molecularly distinct from those with a RELA fusion. 4 Importantly, the authors found that the RELA subgroup exhibited a 10-year overall survival and progression-free survival of 50% and 20%, respectively, whereas patients in the YAP1 subgroup all survived. 4 Comparatively, Merchant et al. found that patients in the RELA subgroup did not have uniformly poor survival when treated with immediate postoperative radiotherapy. 49 In fact, the authors found that the five-year event-free survival differed significantly by tumor grade but not age, location, RELA fusion status, or posterior fossa grouping. 49 Additional fusions genes have been identified in supratentorial ependymomas, such as C11orf95 with MAML2 and YAP1, and YAP1 with FAM118B; however, the clinical significance of these rare gene fusions remains to be elucidated. 3 Lastly, in some cases, supratentorial ependymomas are without a detectable fusion gene. 3
Cortical ependymomas are currently not considered to be a distinct subgroup of supratentorial ependymomas; however, there is a growing body of literature specifically investigating the natural history and clinical outcomes of these lesions compared to supratentorial ependymomas as a whole. Recent studies have demonstrated a higher incidence of C11orf95-RELA fusions in cortical ependymomas (90–100%) compared with supratentorial ependymomas (65.1–70%).4,41,43,48,50,51 Interestingly, Matsumoto et al. found that cortical ependymomas exhibited a comparatively favorable outcome while demonstrating high rates of C11orf95-RELA fusions. 41 Similarly, Wang et al. (2020) found that cortical ependymomas exhibit a higher rate of C11orf95-RELA fusions with 9 of 10 patients being RELA fusion positive. 43 Importantly, the authors found that the nine patients with RELA fusions had favorable outcomes with all patients currently living at last known follow-up. 43 Similar results were published by Wang et al. (2021) in a pediatric population with all RELA fusion positive patients exhibiting favorable outcomes with no deaths at last known follow-up. 48 Some authors have attributed favorable outcomes to the superficial location of cortical ependymomas and achievable gross total resection.41,43,48 Moreover, given these data, some authors have suggested the classification of cortical ependymomas as a new distinct subtype of supratentorial ependymoma.12,41 Further studies with larger sample sizes are necessary to investigate the significance of RELA fusions on survival in cortical ependymomas.
The etiopathogenesis of cortical ependymomas remains ambiguous. In many regards, the clinicopathologic characteristics of cortical ependymomas are similar to ectopic ependymomas, that is, ectopic ependymomas are typically low-grade tumors with indolent behavior.11,21,38 Historically, it has been suggested to group ependymomas based upon their location within the CNS, as natural history, operative mortality, and postoperative survival are known to be closely dependent on tumor location.11,21,38 Nevertheless, cortical ependymomas and ectopic ependymomas have been considered distinct diagnostic entities. Ectopic ependymomas have been reported to involve the trigeminal nerve, neurohypophysis, sella turcica, falx, posterior fossa, and cavernous sinus.52–57 Importantly, all of these sites are devoid of ependymal cells, which suggests that ectopic and cortical ependymomas may originate from a cell line distinct from mature differentiated ependyma.11,21,38 Vernet et al. proposed the etiopathogenesis of ectopic ependymomas to be potentially due to one of several mechanisms including: (i) a migration disorder of the germinal matrix, (ii) primitive neuroectodermal neoplasms differentiating into the ependymal lineage, or (iii) unregulated neoplastic growth of an ependymal cell of ectopic origin. 58 Moreover, Hegyi et al. reported a case of an ectopic retinal ependymoma hypothesized to arise from Muller cells, which would imply that glial cells with progenitor potential may be the origin of these ependymomas rather than matured differentiated ependyma. 59 Extra-axial ependymomas, such as those occurring in ovarian teratomas, further support that these tumors of ectopic origin arise from a progenitor cell line. 21 As such, a progenitor cell line could also be the cell origin of cortical ependymomas. Although several hypotheses have been put forth to explain this phenomenon, the exact pathogenesis of cortical ependymomas has yet to been established. 21
Currently, there is no consensus regarding the treatment of cortical ependymomas as clinical data is based upon limited case series.7–48 Although gross total resection is preferred when feasible, the necessity and efficacy of adjuvant therapy remain debated. Indeed, treatment algorithms for cortical ependymomas remain largely dependent upon clinical studies addressing classical supratentorial intraventricular or infratentorial ependymomas.60–62 There is widespread agreement in the oncology community favoring adjuvant radiotherapy for adults with WHO grade 3 anaplastic ependymomas regardless of the degree of resection and WHO grade 2 ependymomas after subtotal resection.60–62 Conversely, adjuvant radiotherapy for WHO grade 2 ependymomas after gross total resection remains controversial.62,63 Based upon an institutional experience of 13 patients with cortical ependymomas, Wang et al. (2018) advocated for adjuvant radiotherapy for all WHO grade 3 ependymomas regardless of extent of resection and WHO grade 2 ependymomas only in the context of a subtotal resection. 38 In the largest series to date, Wang et al. (2020) performed a comprehensive institutional analysis in 30 patients with cortical ependymomas, in addition to a systematic review of 106 cases previously reported in the literature. 43 The authors found that 69.1% (47/68 cases) of patients with WHO grade 3 cortical anaplastic ependymomas received postoperative adjuvant radiotherapy, which provided a significantly longer overall survival compared to those without irradiation. 43 Importantly, postoperative radiotherapy did not prolong overall survival in patients with WHO grade 2 cortical ependymomas. 43 These data further support widespread favor for adjuvant radiotherapy for WHO grade 3 ependymomas regardless of intracranial location. However, the clinical benefit of adjuvant radiation on disease control in patients with WHO grade 2 cortical ependymomas should be taken with caution, as prospective studies are necessary to validate this finding. 43 Regarding chemotherapeutics, the role of adjuvant chemotherapy, particularly in children, remains unproven despite extensive investigation, as no chemotherapeutic regimen to date has demonstrated significant clinical benefit. 64
Based upon our literature review, there is only one prior report of a cortical ependymoma with insular involvement. 21 Van Gompel et al. described a case of a 43-year-old female who presented with seizures and imaging demonstrating a left frontal tumor with insular involvement. 21 The patient was treated with subtotal resection followed by radiotherapy for the WHO grade 2 lesion. 21 Per report, the patient was alive with a stable disease burden at 60 months 21 Similarly, we report a case of a 58-year-old female with a WHO grade 2 ependymoma of the insular cortex extending into the left frontal cortex. Specifically, our tumor was primarily located in Zone 1 (anterosuperior) of the insula as per the Berger-Sanai classification system.65–67 Our patient also underwent subtotal resection followed by radiotherapy and is currently with a stable disease burden 15 months post-operatively. Although gross total resection is preferred, it may not always be a feasible option, especially when dealing with tumors of eloquent areas, such as the insula. Indeed, the clinical consequences of gross total resection in eloquent areas may supersede disease control when dealing with tumors with more indolent behavior.
Conclusions
Cortical ependymomas are currently not considered to be a distinct subgroup of supratentorial ependymomas; however, there is a growing body of literature specifically investigating the natural history and clinical outcomes of these lesions compared to supratentorial ependymomas as a whole. Preliminary reports describe differing clinical outcomes between cortical ependymomas with C11orf95-RELA fusions and supratentorial ependymomas with C11orf95 RELA fusions. As such, further studies with larger sample sizes are necessary to investigate the significance of RELA fusions on survival in cortical ependymomas and to determine whether cortical ependymomas with C11orf95-RELA fusions should be classified as a distinct entity.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases.
Informed consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article.
Contributorship
JAC and ALO conceived the study idea. MSS, JJE, MRW, and ALO provided guidance and oversight. All authors (JAC, ACS, BMS, MSS, JJE, MRW, ALO) made substantial contributions to study planning and data collection. JAC and ALO drafted the original manuscript. MSS provided pathology images. All authors (JAC, ACS, BMS, MSS, JJE, MRW, ALO) critically reviewed and revised the final manuscript for important intellectual content. All authors (JAC, ACS, BMS, MSS, JJE, MRW, ALO) approval the final version of the manuscript.