
Abstract
Objective
To investigate the role and characterize the molecular mechanisms regulating apoptosis and autophagy in nitric oxide (NO)–induced chondrocyte cell death.
Design
Cell apoptosis and autophagy were evaluated in chondrocytes treated with sodium nitroprusside (SNP) combined with the presence or absence of interleukin-1 beta (IL-1β) and nutrient-deprived conditions. The concentration of nitrite was determined by Griess reaction. Activation of apoptosis and autophagy were determined by immunocytochemistry, Western blot, and quantitative real-time polymerase chain reaction (qPCR) analysis. Flow cytometry and MTT assay were used to assess cell viability.
Results
Cotreatment of chondrocytes with SNP and IL-1β under nutrient-deprived condition potentially enhanced the effect of NO-induced cell death. Immunocytochemistry, Western blot, and qPCR analysis indicated that treatment of chondrocytes with SNP significantly reduced autophagic activity, autophagic flux, and multiple autophagy-related (Atg) genes expression. These findings were associated with an increase in ERK, Akt, and mTOR phosphorylation, whereas autophagy induction through mTOR/p70S6K inhibition by rapamycin significantly suppressed NO-induced cell apoptosis. Furthermore, the cleavage of poly(ADP-ribose) polymerase (PARP) and caspase-3 activation in response to apoptosis was weakly detected. These results corresponded with a significant increase in apoptosis-inducing factor (AIF) expression, suggesting the involvement of the caspase-independent pathway.
Conclusions
These results demonstrate that in chondrocyte cultures with cells induced into an osteoarthritis state, NO inhibits autophagy and induces chondrocyte apoptosis mainly, but not completely through the caspase-independent pathway. Our data suggest that autophagy is a protective mechanism in the pathogenesis of osteoarthritis and could be proposed as a therapeutic target for degenerative joint diseases.
Introduction
Osteoarthritis (OA) is the most common cartilage degenerative disease, which primarily affects joints and progressively causes impaired mobility, disability, and chronic pain, especially in the aging population worldwide.1,2 Despite the huge economic impact and high prevalence of OA in humans and animals, there are no current effective therapeutic approaches that could completely restore degraded cartilage or prevent the progression of the disease.2-5 Because of the complexity and multifactorial nature of the disease, the underlying molecular mechanisms regulating the pathogenesis of OA have not been fully understood yet.5,6 Several biochemical mediators are currently proposed to be involved in the initiation and progression of OA through the articular cartilage metabolism.2,7 In the disease process, the chondrocytes, as well as synoviocytes, actively produce large quantities of inflammatory mediators.2,6-9 Among numerous inflammatory mediators associated with the pathological process of OA and rheumatoid arthritis, nitric oxide (NO) is a predominant mediator that is primarily responsible for the catabolic activity of chondrocytes and typically observed in the synovial fluid of arthritis patients.9-12
Although NO is implicated in several cellular functions, including chondrocyte proliferation, differentiation, and even in protection against oxidative stress,13-16 it has been well demonstrated that excessive production of NO resulting from the upregulation of inducible nitric oxide synthase (iNOS) in OA chondrocytes contributes to the development and maintenance of OA by its involvement in the inflammatory condition, cartilage destruction, mitochondrial dysfunction, and induction of apoptotic cell death.10,15-18 To date, several studies have suggested that the death of chondrocytes during the progression of OA is a central feature of advanced stages in osteoarthritic cartilage, which contributes to the progressive cartilage destruction.15,19-21 Although chondrocyte cell death could occur through a variety of molecular pathways, it has been shown that a strong correlation exists between the cytotoxicity of NO and apoptotic cell death.15,22-24 However, the molecular mechanisms of NO-induced chondrocyte cell death remains unresolved due to inconsistence in results outcome between each experimental conditions previously evaluated.15,22 On the other hand, another form of programmed cell death also known as a self-digesting mechanism termed autophagy has been considered as the protective mechanism in the pathogenesis of OA.21,25-27 While excessive autophagy could lead to cell death, it is still recognized as an essential degradation mechanism that occurs to maintain cellular homeostasis by degrading organelles and proteins, which enables cells to survive through unfavorable conditions. 21 Autophagy can be upregulated in response to avoid cell death triggered by a variety of cellular signals such as nutrient deprivation, hypoxia, and endoplasmic reticulum stress. 15 Despite the fact that the effects of NO on regulation of autophagy in chondrocytes have been previously reported, the results are still unclear and further studies are required to confirm the role of autophagy in NO-induced chondrocyte cell death.28-30
Both apoptosis and autophagy are important cellular degradation mechanisms that influence the homeostasis and survivability of chondrocytes. Further investigations on the mutual relationship and interaction between these 2 pathways will undoubtedly provide valuable information on the pathophysiology and therapeutic approaches for OA. The objective of this study was to investigate the roles and characterize the molecular mechanisms regulating apoptosis and autophagy in NO-induced articular chondrocyte cell death using sodium nitroprusside (SNP) as the exogenous NO donor compound.
Materials and Methods
Chondrocytes Isolation and Culture
The use of animal samples was in accordance with Hokkaido University Institutional Animal Care and Use Committee guidelines. Healthy articular cartilage tissue samples from the femoral head were obtained from 3 beagle dogs (3-4 years old) that were euthanized at the end of an experimental study not related to the musculoskeletal system. Briefly, cartilage was harvested and dissected into small sections, then digested in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 0.3% collagenase I (Wako Pure Chemicals Industries, Osaka, Japan) for 12 hours at 37 °C in a 5% CO2 incubator. Chondrocytes were separated by filtering through a nylon filter and primary chondrocytes were seeded at 15,000 cells/cm2. Following the above procedure, the second passage chondrocytes were used for any further experiments. The culture media used in this study was DMEM containing 10 mM HEPES (Dojindo, Kumamoto, Japan), 25 mM NaHCO3 (Wako), 100 U/mL penicillin G potassium (Wako) and 73 U/mL streptomycin sulphate (Wako).
Chondrocytes Treatment
Unless specified otherwise, all cultured chondrocytes were seeded in 60 mm culture dishes (Corning, Lowell, MA, USA) in DMEM containing 10% fetal bovine serum (FBS; Nichirei Biosciences Inc., Tokyo, Japan) and incubated for 24 hours at 37 °C in a 5% CO2 incubator. Thereafter, the culture media was removed, and chondrocytes were either not treated or treated with 10 ng/ml recombinant canine interleukin-1 beta (IL-1β; Kingfisher Biotech, Saint Paul, MN, USA) in DMEM containing 10% FBS. After 72 hours, the culture media was changed to either DMEM containing 10% FBS or serum-free DMEM with the presence or absence of 100 µM SNP (Wako) and incubated for a further 8 hours. The effects of autophagy on cultured chondrocytes were evaluated by treating with 10 µM rapamycin (Invivogen, San Diego, CA, USA) during this last 8 hours of treatment. In addition, to assess the autophagic flux, 100 nM bafilomycin (Cell Signaling Technology, Danvers, MA, USA) was added at 4 hours before the end of the treatment.
Cell Viability Assay
Chondrocytes were seeded into 96-well plates (Corning) and treated as described above with various concentrations of SNP (0, 25, 50, 100 and 200 µM) for 24 hours. Cell viability was evaluated by 3-(4,5-dimehylthiazolyl-2)2,5-diphenyltetrazolium bromide (MTT; Dojindo) colorimetric assay. Briefly, the MTT solution was added into each well and culture plates were incubated for 4 hours. The solution was then removed and MTT formazan crystals that formed were dissolved by dimethyl sulfoxide (Wako). The absorbance was measured with a microplate reader (Thermo Scientific, Vantaa, Finland) at 570 nm.
Cell Apoptosis Assay
Chondrocyte apoptosis was evaluated with annexin V and propidium iodide (PI) stain using FITC annexin V apoptosis detection kit I (BD Bioscience, Heidelberg, Germany) in accordance with the manufacturer’s protocol. In order to evaluate the roles of autophagy in cultured chondrocytes, the cells were incubated with the presence or absence of autophagy inducer (10 µM rapamycin) or inhibitor (100 nM bafilomycin) during the last 8 hours of treatment, together with SNP. After treatment, chondrocytes were harvested, washed with phosphate-buffered saline (PBS) and resuspended in binding buffer. FITC-annexin V and PI were added into the cell suspension, and then incubated for 15 min in darkness at room temperature. The samples were analyzed using a FACS Verse flow cytometer (BD Biosciences).
Caspase-3 Activity Assay
Caspase-3 activity was determined using a caspase-3 assay kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions. After treatment, chondrocytes were harvested, washed with PBS and resuspended in chilled lysis buffer. The protein concentration of each sample was measured with protein quantification assay kit (Macherey-Nagel, Dürren, Germany) and adjusted, then DTT and DEVD-pNA substrate were added. Samples were incubated for 2 hours and the absorbance was measured with a microplate reader at 405 nm.
Quantification of Nitrite
The concentration of nitrite that accumulated in the cell culture supernatants was determined by Griess reaction with sodium nitrite as standard. In brief, 50 µL of cell culture supernatants were mixed with an equal amount of Griess reagent containing 0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride (Sigma-Aldrich, St. Louis, MO, USA) and 1% sulfanilamide (Sigma-Aldrich) in 5% H3PO4 (Wako), then incubated for 10 minutes. The absorbance was measured with a microplate reader at 540 nm.
Protein Isolation and Western Blot Analysis
The following antibodies were used: antibodies against microtubule-associated protein light chain 3 (LC3; Cat. #: NB100-2331) and sequestosome-1 (p62/SQSTM1; Cat. #: NBP1-48320) were purchased from Novus Biologicals (Littleton, CO, USA); antibodies against β-actin (Cat. #: 4970), apoptosis-inducing factor (AIF; Cat. #: 5318), cleaved caspase-3 (Cat. #: 9661), poly (ADP-ribose) polymerase (PARP; Cat. #: 9542), ERK1/2 (Cat. #: 4695), phospho-ERK1/2 (Cat. #: 4370), Akt (Cat. #: 9272), phospho-Akt (Cat. #: 9271), mammalian target of rapamycin (mTOR; Cat. #: 2983), phospho-mTOR (Cat. #: 5536), p70 ribosomal protein S6 kinase (p70S6K; Cat. #: 9205) and HRP conjugated anti-rabbit IgG (Cat. #: 7074) were purchased from Cell Signaling Technology; antibodies against phospho-p70S6K (Cat. #: SC-230) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Total proteins were extracted from cultured chondrocytes using chilled radioimmunoprecipitation assay (RIPA; Sigma-Aldrich) buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich) and cell debris was removed by centrifugation at 12,000 rpm for 20 minutes at 4 °C. The protein concentration was measured with protein quantification assay kit, adjusted and 4 µg of protein from each sample was separated on 4% to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) then transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 10% skim milk in PBS supplemented with 0.1% Tween 20 (PBST) for 1 hour and incubated with primary antibodies in 1:1,500 (1:4,000 for β-actin) dilution overnight at 4 °C. Subsequently, membranes were incubated with HRP conjugated anti-rabbit IgG at a dilution of 1:3,000 (1:8,000 for β-actin) for 1 hour then incubated in Western Blot Ultra-Sensitive HRP substrate (Takara Bio Inc., Otsu, Japan) for 5 minutes. The results of Western blot were visualized using Image Quant LAS 4000 (GE Healthcare, Buckinghamshire, UK) and the intensity of each protein bands was quantified using ImageJ software (NIH, Bethesda, MD, USA). Quantified values were normalized by β-actin as a loading control, while phosphorylated proteins were normalized by the corresponding total proteins.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Total RNA was extracted from cultured chondrocytes using TRIZol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The RNA concentration was measured, adjusted and 1 µg of RNA from each sample was reverse transcribed into cDNA using M-MLV RT kit (Invitrogen). The synthesized cDNA was then used to perform qPCR analysis using KAPA SYBR FAST qPCR kit (KAPA Biosystems, Woburn, MA, USA). The relative mRNA expression levels of each gene were quantified by delta-delta Ct method and normalized against the reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Sequence of primers used in the experiment were designed using BLAST programs on the National Center for Biotechnology Information (NCBI) website, whereas the specificity of primers was validated by a single peak in the melting curve analysis. The sequence, amplicon length and accession number for each of the primers are indicated in (Table 1).
Primer Sequences Used for the Quantitative Real-Time Polymerase Chain Reaction Analysis.
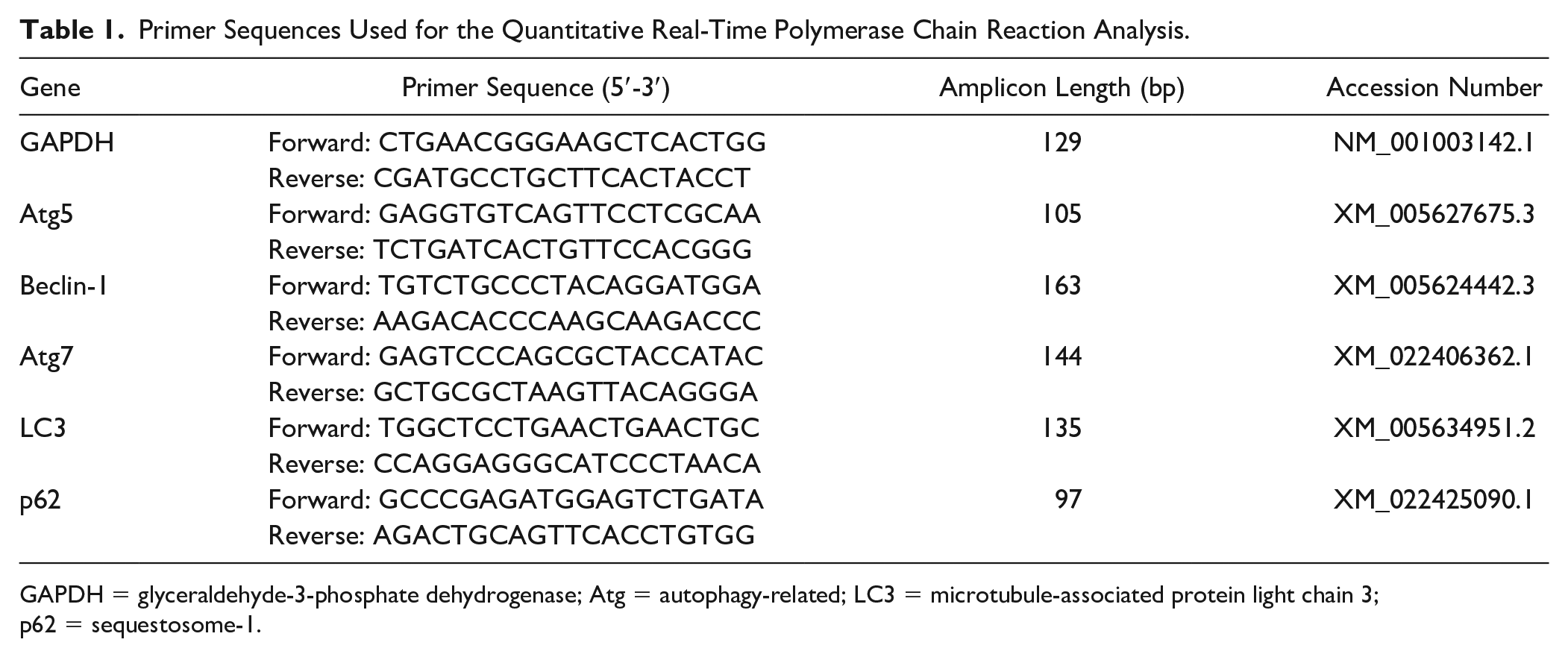
GAPDH = glyceraldehyde-3-phosphate dehydrogenase; Atg = autophagy-related; LC3 = microtubule-associated protein light chain 3; p62 = sequestosome-1.
Immunocytochemistry Assay
Immunocytochemistry was performed on chondrocytes grown on 8-well chamber slides (Iwaki, Tokyo, Japan). After treatment, chondrocytes were washed with PBS and fixed in 4% paraformaldehyde (Wako) for 10 minutes, incubated with 0.1 M glycine (Wako) in PBS for 10 minutes, permeabilized with 0.05% digitonin (Sigma-Aldrich) in PBS for 5 minutes and blocked with 5% FBS in PBS for 30 minutes. The fixed cells were incubated with LC3 antibody (MBL, Nagoya, Japan; Cat. #: PM036) in 1:500 dilution overnight at 4 °C. Subsequently, cells were incubated with FITC conjugated anti-rabbit IgG (Invitrogen; Cat. #: A-11034) at a dilution of 1:1,000 for 1 hour. NucBlue Fixed Cell ReadyProbes (Invitrogen) and ProLong Gold Antifade reagent (Invitrogen) were used to stain the nucleus and mount the slides, respectively. The fluorescent images were captured using LSM 700 laser scanning confocal microscope and ZEN software (Carl Zeiss, Jena, Germany).
Statistical Analysis
Quantitative data analysis was performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA). All quantitative results are presented as mean ± standard error (SE). Statistical comparisons were performed using analysis of variance (ANOVA), with a Dunnett’s multiple comparison test to compare between groups. P < 0.05 was considered significant.
Results
IL-1β and Starvation Condition Enhance the Effect of NO-Induced Cell Death
The viability of chondrocytes treated with various concentrations of SNP was assessed by MTT assay. As shown in Figure 1 , a decrease in chondrocyte viability was observed with a higher concentration of SNP at 200 µM. However, the viability of chondrocytes preincubated with IL-1β and treated with SNP under serum starvation was decreased even with 4-fold lower concentration of SNP.
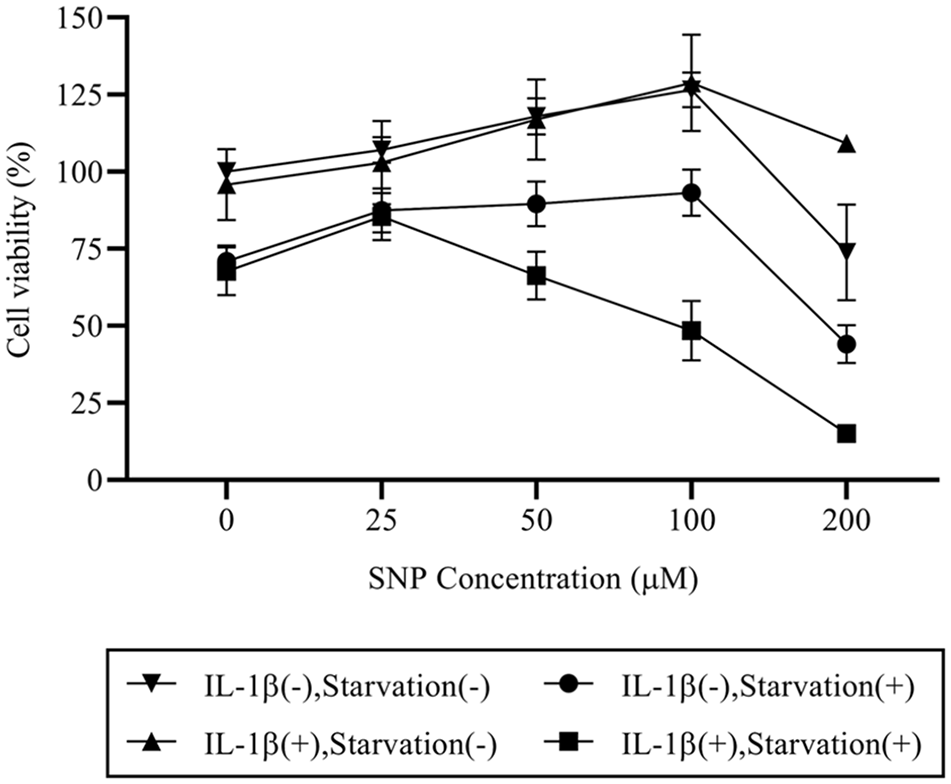
Cotreatment with sodium nitroprusside (SNP) and interleukin-1 beta (IL-1β) under nutrient-deprived condition enhances the effect of nitric oxide (NO)–induced cell death. Chondrocytes were either not treated or treated with 10 ng/mL IL-1β for 72 hours prior to the stimulation with various concentrations of SNP (0, 25, 50, 100, and 200 µM) in the culture media with the presence or absence of serum supplement for 24 hours. Reduction in cell viability as evaluated by MTT assay was observed at 200 µM SNP in 3 groups, except the chondrocytes that were pretreated with IL-1β and stimulated with SNP in culture media without serum supplement in which cell viability reduction commenced with the lower concentration of SNP at 50 µM.
Cotreatment with SNP and IL-1β under Nutrient-Deprived Condition Exhibits Low Cell Viability but Expresses High Autophagosome Formation, as Detected by LC3-II
To investigate the autophagic activity of SNP treated chondrocytes, Western blot analysis was performed to evaluate the conversion of LC3-I to LC3-II (LC3 lipidation) as a marker of autophagosome formation ( Fig. 2A ). The concentration of SNP at 100 µM, which was the highest concentration that did not reduce cell viability in 3 other groups, was used ( Fig. 1 ). The results indicated that LC3-II protein expression level of serum-starved chondrocytes was increased in all groups, although the significant increase was only observed in chondrocytes preincubated with IL-1β and treated with SNP (P < 0.001), compared to untreated nonstarved chondrocytes ( Fig. 2B ). In addition, p62 protein expression level was significantly decreased only in untreated serum-starved chondrocytes (P = 0.041) ( Fig. 2C ).
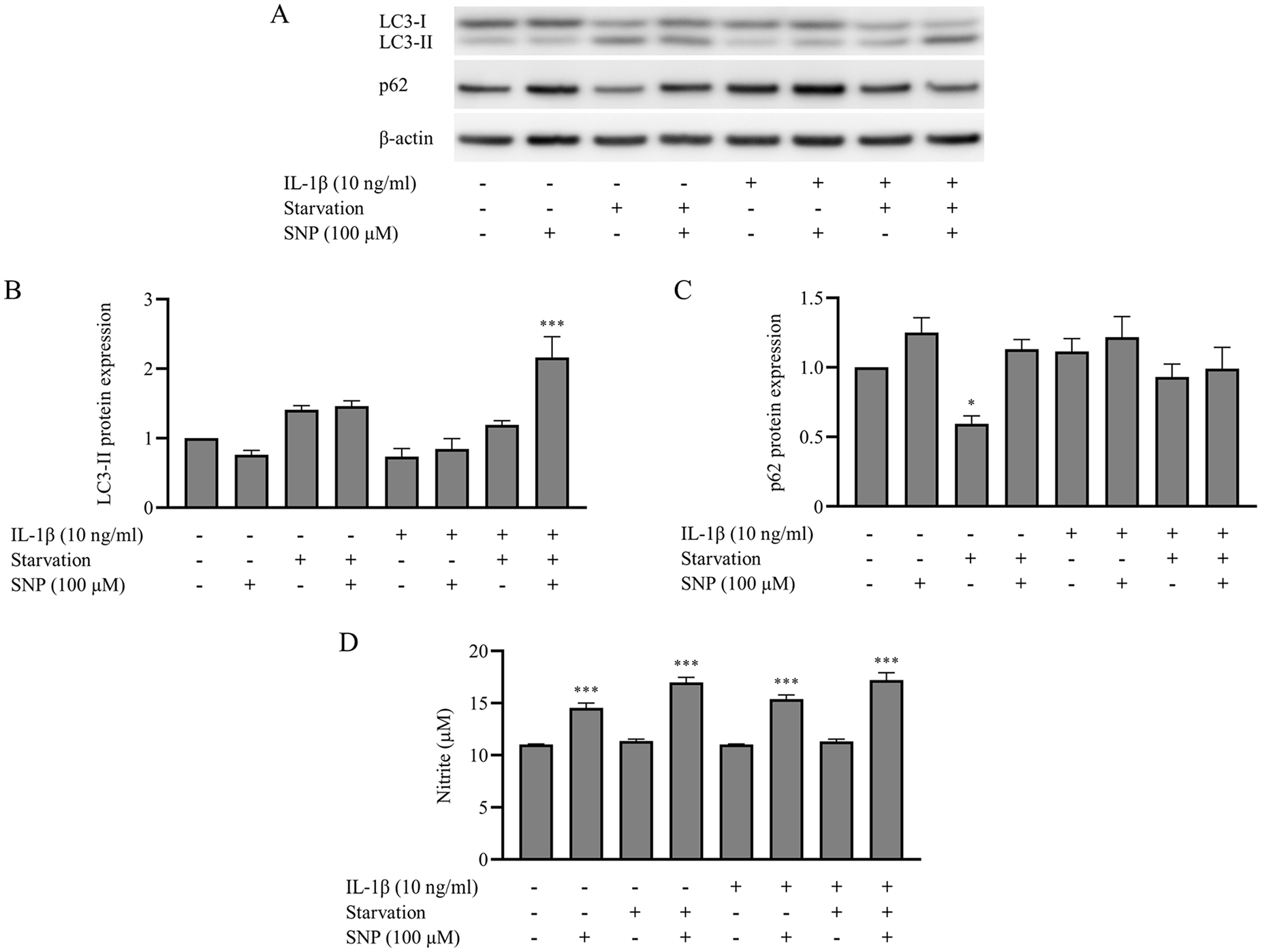
Autophagosome formation was increased in the chondrocytes that were pretreated with interleukin-1 beta (IL-1β) and stimulated with sodium nitroprusside (SNP) under nutrient-deprived condition. Chondrocytes were either not treated or treated with 10 ng/mL IL-1β for 72 hours prior to the stimulation with 100 µM SNP in the culture media in the presence or absence of serum supplement for 8 hours. (
Activation of Autophagy by Rapamycin Reduces Chondrocyte Apoptosis
The effect of NO on chondrocyte apoptosis was evaluated by flow cytometry analysis with annexin V and PI staining. The autophagy inducer, rapamycin, and inhibitor, bafilomycin were cotreated with SNP to characterize the interaction between autophagy and apoptosis, and to exclude the possibility of autophagic cell death. The results from flow cytometry analysis indicated that chondrocyte apoptosis was induced by SNP treatment ( Fig. 3A ). As shown in ( Fig. 3B ), the proportion of live cells were significantly reduced (P < 0.001) in all SNP-treated chondrocytes. However, SNP-treated chondrocytes that were cotreated with rapamycin showed a significant increase in the proportion of live cells (P < 0.001) compared with either SNP alone or a combination of SNP and bafilomycin. However, the proportion of live cells between SNP and bafilomycin co-treated chondrocytes and chondrocytes treated with SNP alone showed no significant difference (P = 0.666).
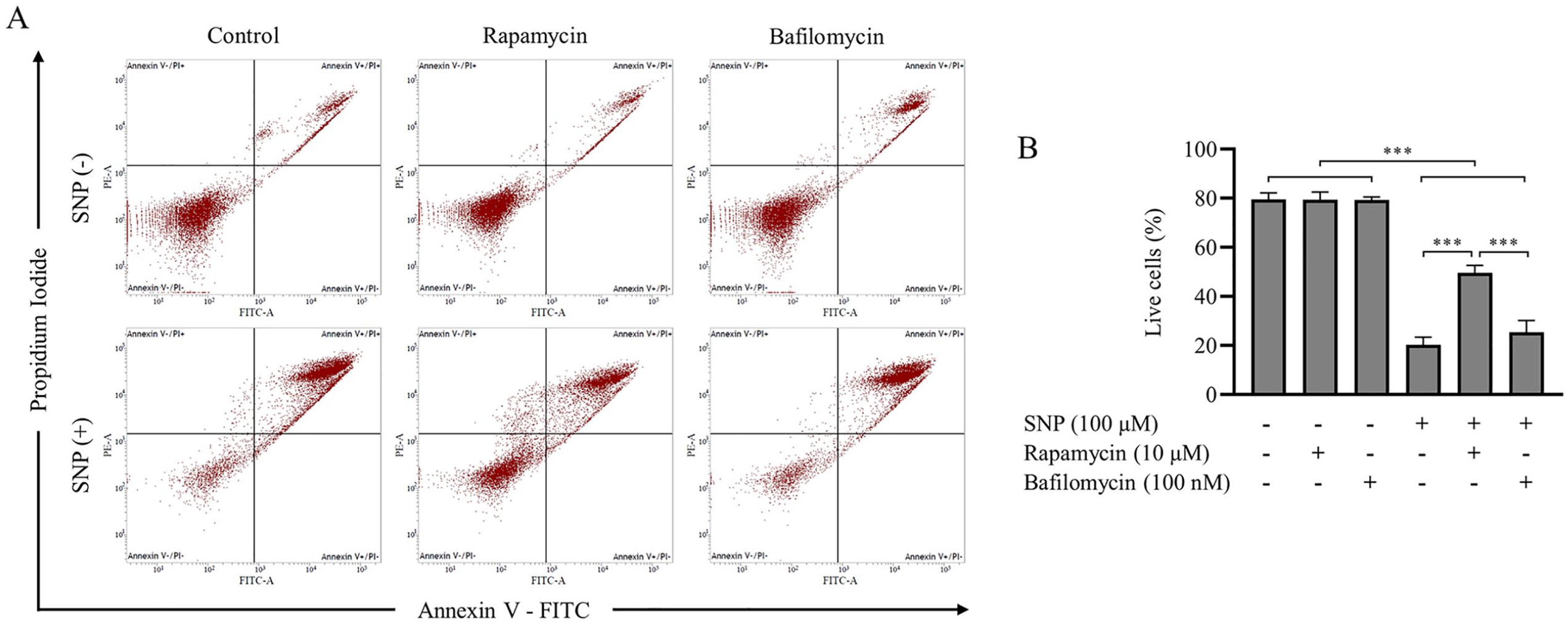
Activation of autophagy by rapamycin reduced chondrocyte apoptosis, whereas the inhibition of autophagy by bafilomycin had no effect on cell apoptosis. Chondrocytes were preincubated with interleukin-1 beta (IL-1β) and either not treated or treated with 100 µM sodium nitroprusside (SNP) under nutrient-deprived condition for 8 hours. Together with SNP treatment, the chondrocytes were incubated in the presence or absence of autophagy inducer (10 µM rapamycin) or inhibitor (100 nM bafilomycin). (
NO Reduces Activation of Autophagy, Autophagic Flux, and Autophagy-Related (Atg) Genes mRNA Expression
Increased autophagosome formation in SNP-treated chondrocytes was further distinguished between high autophagic activity and inhibition of subsequent events by blocking the fusion of autophagosomes and lysosomes. Bafilomycin, the V-ATPase inhibitor was added at 4 hours prior to the end of the treatment to inhibit autophagosome-lysosome fusion and LC3 conversion was evaluated by Western blot analysis ( Fig. 4A and D ). In the absence of bafilomycin, our results demonstrated that LC3-II protein expression level was significantly increased in chondrocytes treated with rapamycin (P = 0.006), SNP (P = 0.008), and a combination of SNP and rapamycin (P < 0.001), compared to untreated chondrocytes ( Fig. 4B ). The protein expression level of p62 was significantly increased by the treatment with rapamycin (P = 0.003) and a combination of SNP and rapamycin (P = 0.039) ( Fig. 4C ). However, the LC3-II protein expression level of SNP treated chondrocytes was significantly decreased (P = 0.008) in the presence of bafilomycin ( Fig. 4E ). In contrast, the LC3 conversion remained significantly increased in chondrocytes treated with either rapamycin alone (P = 0.016) and a combination of SNP and rapamycin (P = 0.006) ( Fig. 4E ). The protein expression level of p62 was significantly increased only by the treatment with a combination of SNP and rapamycin (P = 0.032) ( Fig. 4F ). Furthermore, the cytoplasmic LC3 puncta formation in chondrocytes was assessed by immunocytochemistry assay. Consistent with Western blot analysis, in the absence of bafilomycin, the lowest amount of accumulated autophagosome was observed in untreated chondrocytes, whereas an accumulation of autophagosome was reduced by the treatment with SNP under the presence of bafilomycin ( Fig. 5 ). Further investigations were performed to verify the effects of NO on autophagy at the gene transcription level using qPCR analysis. There was a significant decrease in beclin-1 (P < 0.001), Atg7 (P < 0.001), and LC3 (P = 0.003) mRNA expression in chondrocytes treated with SNP alone ( Fig. 4H-J ), compared with untreated chondrocytes. Interestingly, the cotreatment with rapamycin had no effect on these Atg genes. The mRNA expression level of beclin-1 (P < 0.001), Atg7 (P < 0.001), and LC3 (P < 0.001) remained significantly decreased ( Fig. 4H-J ) in chondrocytes co-treated with SNP and rapamycin similar to chondrocytes treated with SNP alone. The expression of Atg5 mRNA was not affected by any treatment condition ( Fig. 4G ). In addition, the mRNA expression level of p62 was significantly increased by the treatment with rapamycin (P = 0.021) and a combination of SNP and rapamycin (P < 0.001) ( Fig. 4K ).
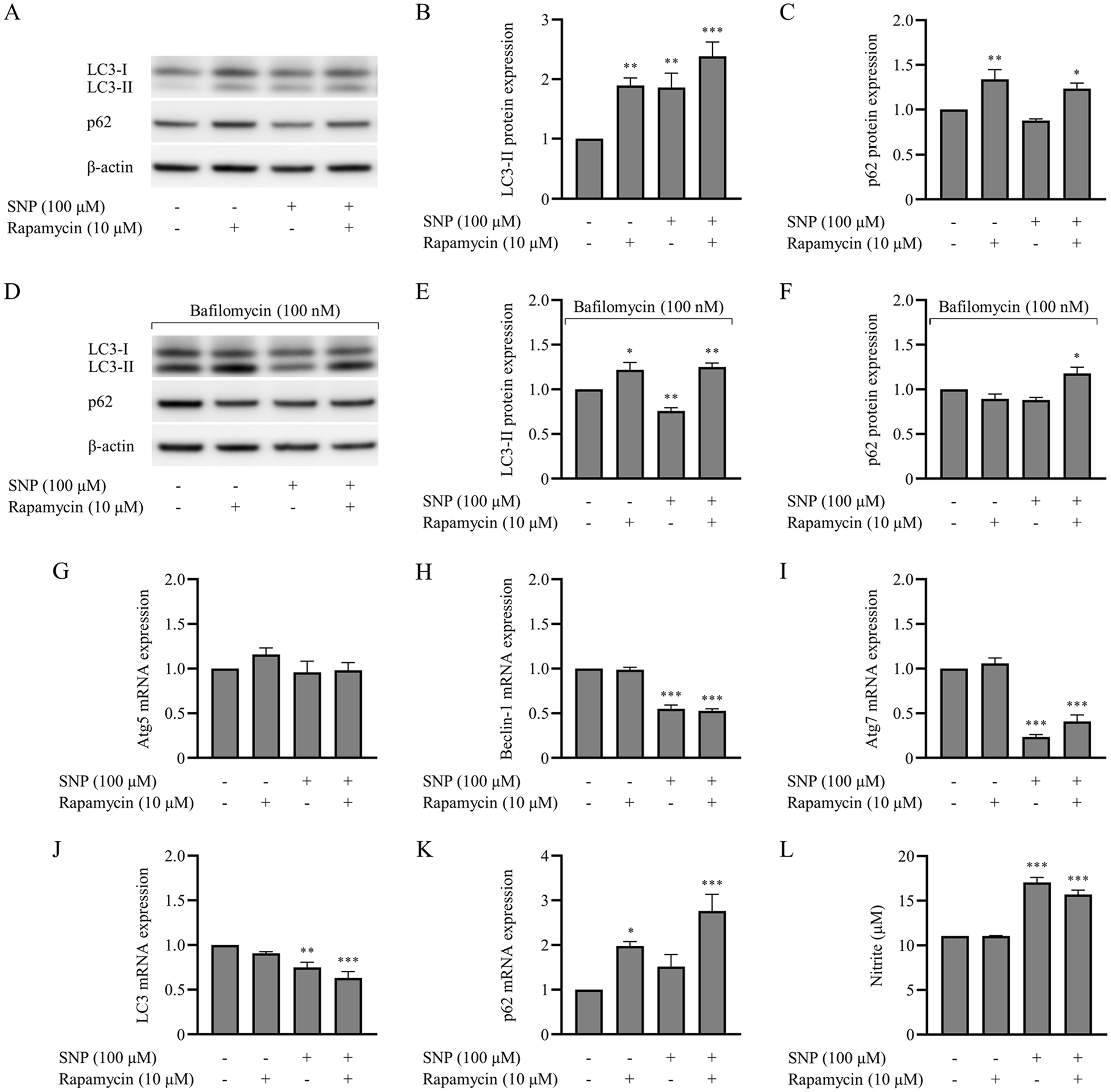
Treatment with sodium nitroprusside (SNP) resulted in reduction in autophagic activity, autophagic flux, and autophagy-related (Atg) genes mRNA expression in chondrocytes. Chondrocytes were preincubated with interleukin-1 beta (IL-1β) and either not treated or treated with 100 µM SNP under nutrient-deprived condition for 8 hours. Together with SNP treatment, the chondrocytes were incubated with the presence or absence of 10 µM rapamycin. In addition, 100 nM bafilomycin was added at 4 h before the end of the treatment to assess the autophagic flux. (A and D) Total protein was analyzed by Western blot, using an antibody for microtubule-associated protein light chain 3 (LC3), sequestosome-1 (p62), and β-actin. (
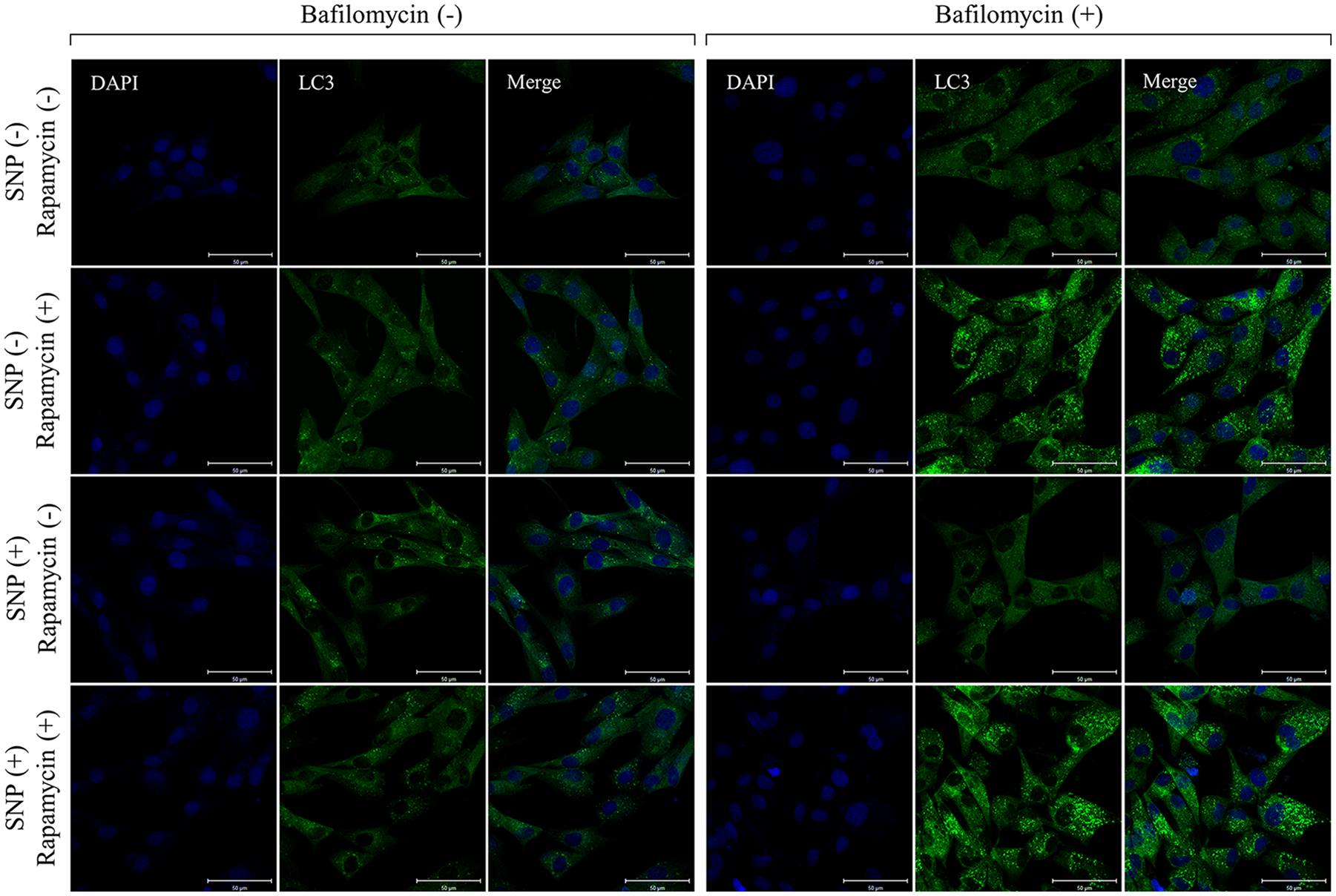
Treatment with sodium nitroprusside (SNP) resulted in reduction in cytoplasmic microtubule-associated protein light chain 3 (LC3) puncta under the presence of bafilomycin. Chondrocytes were preincubated with interleukin-1 beta (IL-1β) and either not treated or treated with 100 µM SNP under nutrient-deprived condition for 8 hours. Together with SNP treatment, the chondrocytes were incubated with the presence or absence of 10 µM rapamycin. In addition, 100 nM bafilomycin was added at 4 hours before the end of the treatment to assess the autophagic flux. The cytoplasmic LC3 puncta formation were detected by immunocytochemistry, using an antibody for LC3 (green). Nuclei were stained with DAPI (blue). Scale bars: 50 µm.
NO Generated by SNP Is Not Directly Related to Cell Death
Nitrite quantification by Griess reaction was used to confirm NO production from SNP-treated chondrocytes. As shown in ( Figs. 2D and 4L ), the NO donor compound SNP significantly increased the production of NO in all groups (P < 0.001) regardless of the presence or absence of IL-1β, rapamycin, or starvation condition. However, only with the combination of IL-1β and nutrient-deprived condition was there potentially reduced chondrocyte viability under the high concentration of NO.
NO Reduces Autophagy via Promotion of the ERK, Akt, and mTOR Pathways
To investigate the molecular mechanism of NO on autophagy, we focused on the ERK and Akt pathway of chondrocytes. The results from Western blot analysis ( Fig. 6A ) revealed that compared to untreated chondrocytes, treatment with SNP significantly promotes (P < 0.001) the phosphorylation of ERK, Akt, mTOR, and p70S6K, a downstream substrate of the mTOR signaling pathway ( Fig. 6B-E ). While the cotreatment of chondrocytes with SNP and rapamycin inhibited the phosphorylation of mTOR signaling, it had no effect on the phosphorylation of ERK and Akt ( Fig. 6B and C ).
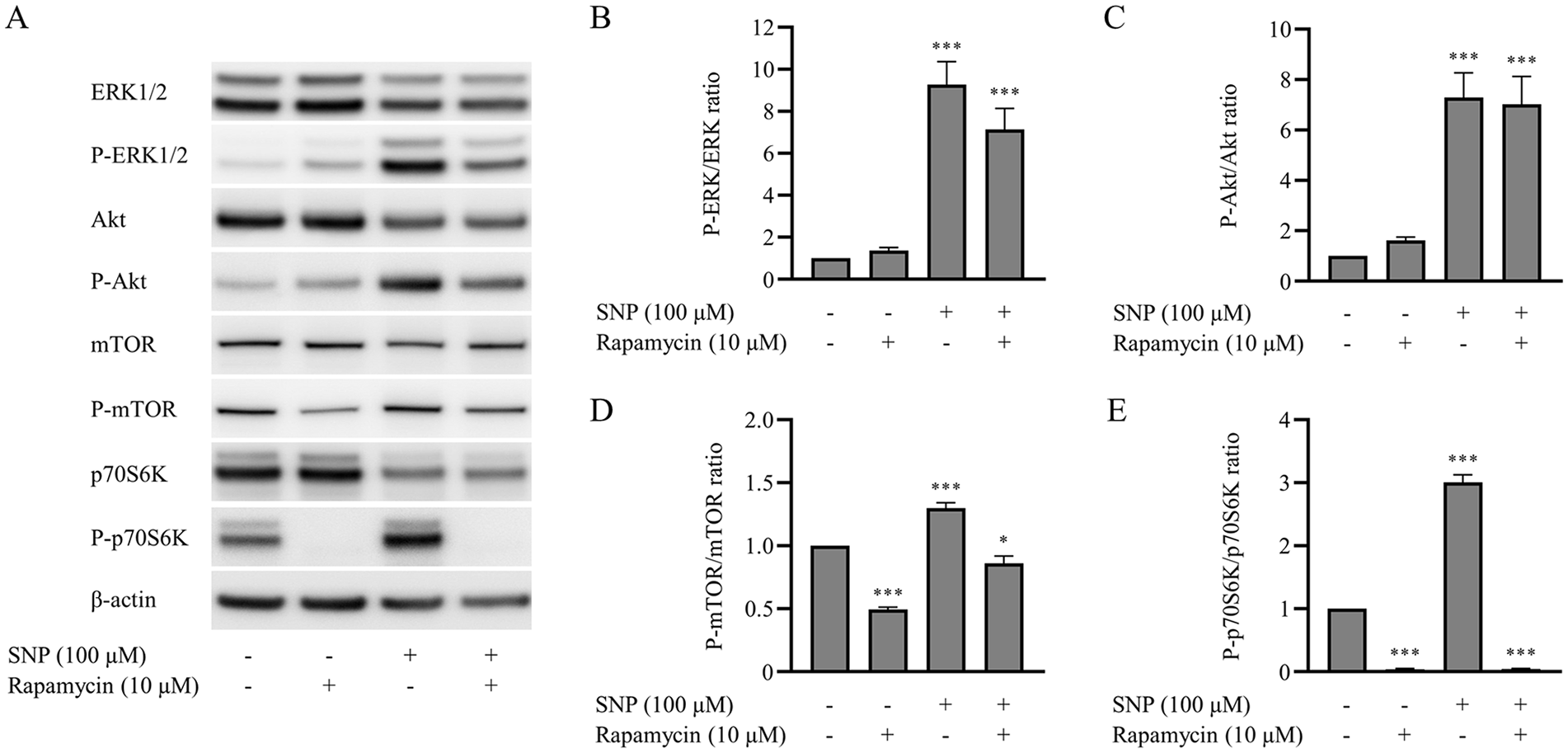
The phosphorylation of ERK, Akt, mammalian target of rapamycin (mTOR), and p70 ribosomal protein S6 kinase (p70S6K) were promoted in sodium nitroprusside (SNP)–treated chondrocytes. Chondrocytes were preincubated with interleukin-1 beta (IL-1β) and either not treated or treated with 100 µM SNP under nutrient-deprived condition for 8 hours. Together with SNP treatment, the chondrocytes were incubated with the presence or absence of 10 µM rapamycin. (
Inhibition of mTOR/p70S6K Pathway by Rapamycin Impairs NO-Induced Cell Apoptosis
The effect of rapamycin on mTOR inhibition was demonstrated by Western blot analysis. As shown in ( Fig. 6D and E ), rapamycin treatment alone significantly (P < 0.001) inhibits the phosphorylation of mTOR and p70S6K compared with untreated chondrocytes. Similar results were consistently observed under SNP and rapamycin cotreatment conditions, in which the mTOR phosphorylation was promoted by SNP through the ERK and Akt. Although the cotreatment with SNP attenuated the inhibition effect of rapamycin on mTOR, the significant reduction (P = 0.035) in mTOR phosphorylation was still detected ( Fig. 6D ). In addition, the phosphorylation of p70S6K remained significantly inhibited (P < 0.001) despite the presence of SNP ( Fig. 6E ).
NO-Induced Cell Apoptosis Is Predominantly Through the Caspase-Independent Pathway
To characterize the molecular mechanism of NO-induced cell apoptosis, we investigated the activation of caspase-3 using Western blot analysis and caspase-3 assay kit. The results surprisingly demonstrated low activation of caspase-3, as evidenced by the low albeit detectable caspase-3 cleavage and PARP cleavage protein expression levels in all treatment conditions (
Fig. 7A
). These results were supported by caspase-3 assay kit, which showed rather low and no significant difference (P > 0.05) in caspase-3 activity between each treatment condition (
Fig. 7B
). For further investigation, an important caspase-independent death regulator, AIF protein expression was evaluated by Western blot analysis. As shown in
Figure 7C
and
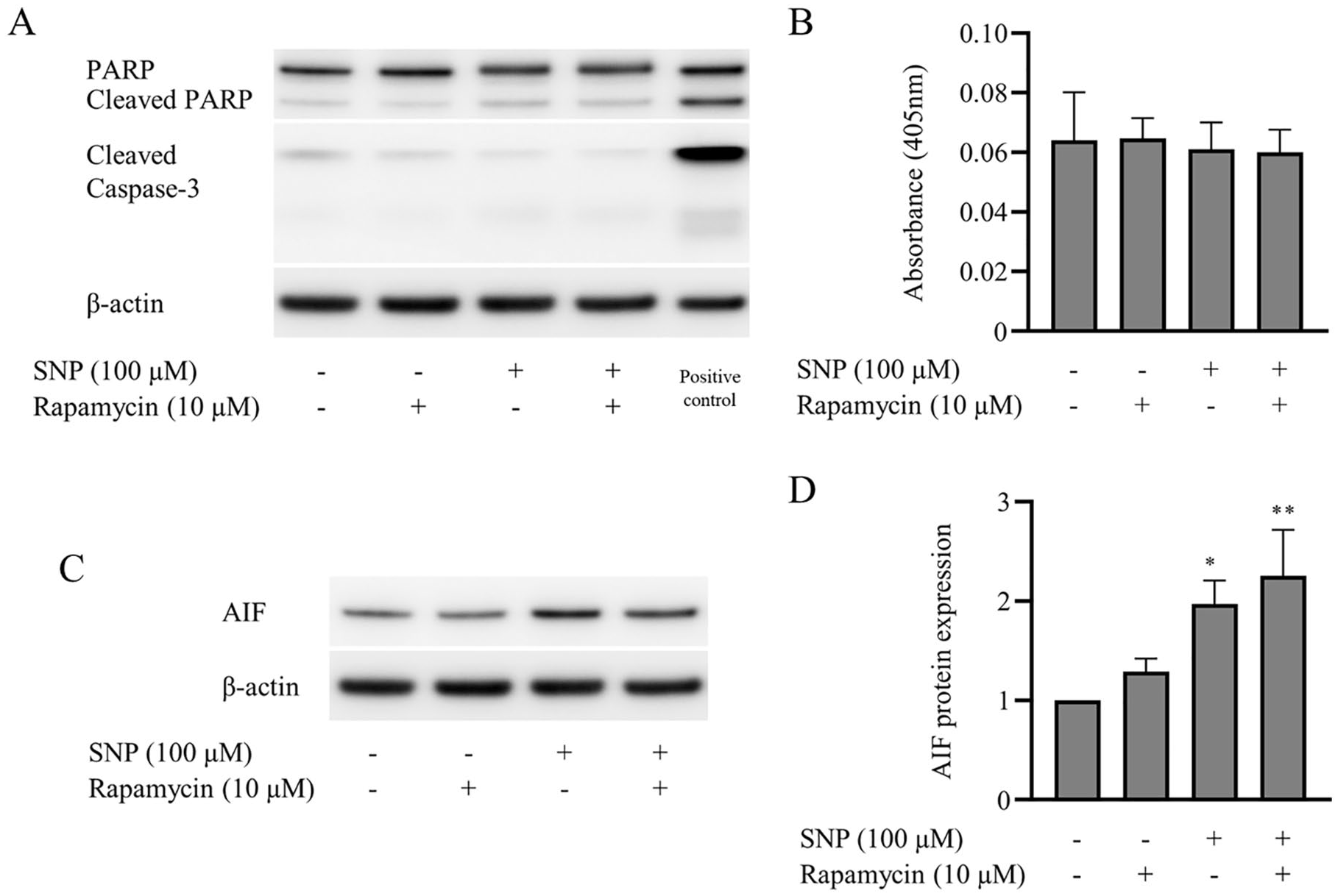
The cleavage of poly(ADP-ribose) polymerase (PARP) and caspase-3 activation in response to apoptosis was weakly detected, while apoptosis-inducing factor (AIF) was highly expressed in sodium nitroprusside (SNP)–treated chondrocytes. Chondrocytes were preincubated with interleukin-1 beta (IL-1β) and either not treated or treated with 100 µM SNP under nutrient-deprived condition for 8 hours. Together with SNP treatment, the chondrocytes were incubated with the presence or absence of 10 µM rapamycin. (
Discussion
It cannot be denied that NO is an important cellular signaling molecule that regulates several cellular functions. Some previous studies have reported a protective role of NO under certain conditions and have shown that NO alone is not cytotoxic to cultured chondrocytes.6,16,31 Conversely, several studies have shown the catabolic effects of NO on articular cartilage and chondrocyte functions, particularly the cytotoxicity of NO that mediates chondrocyte apoptosis.6,11,23,32 Interestingly, although a wide range of pharmaceutical agents have shown a decent effect to suppress the expression of iNOS and subsequent NO production on both in vitro and in vivo studies, neither of them could provide promising results under the clinical trials. 33 On the other hand, it has been well demonstrated that chondrocyte apoptosis in the articular cartilage evidently increases with age and is positively correlated with the severity of OA. 34 While apoptosis is considered as the main cause of OA, it seems that another form of programmed cell death; autophagy is necessary to maintain normal cellular metabolism, especially in low regenerative capability cells such as chondrocytes. Numerous studies have demonstrated the relationship between autophagy and pathogenesis of OA, which suggest that a low level of autophagy could be observed in senescent chondrocytes and associated with OA cartilage.25,26,35,36 Furthermore, the severity of experimental OA is related to the activation and inhibition of autophagy.27,37 Since the biological responses in arthritis involves multiple mediators and a variety of features of cell death, understanding these complex events will provide us a new aspect on joint diseases. However, it should be noted that the in vitro model of OA using chondrocytes-induced into an OA state might differ from the naturally occurring disease due to the multifactorial nature of OA. Thus, further studies conducted on naturally occurring OA cartilage are required to provide more evidence and enhance our understanding of the cellular responses in different biological aspects. In this study, we demonstrated that NO-induced chondrocyte cell death is associated with both apoptosis and autophagy features, as the treatment with NO donor resulted in apoptotic cell death and significantly reduced the autophagic activity ( Fig. 8 ).
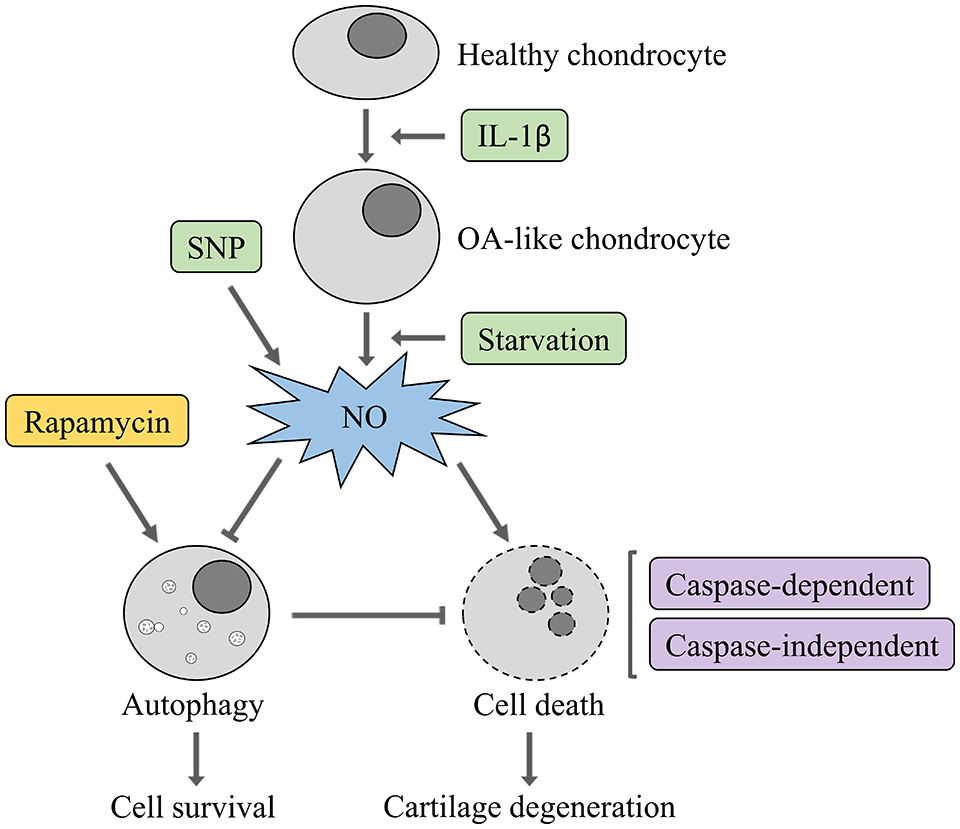
Schematic diagram illustrating the effect of nitric oxide (NO) on the interrelation between autophagy and cell death in osteoarthritis (OA)-like chondrocyte.
Pro-inflammatory cytokine, IL-1β has been extensively reported as a potent inducer of iNOS that promotes NO production in chondrocytes, but does not induce cell death.10,15,22,23 Contrary to previous findings, an increase in NO production occurred regardless of the presence of IL-1β, as it was observed in chondrocytes treated with SNP alone or a combination of IL-1β with SNP. Our results indicate that NO generated by SNP treatment is not directly related to cell death. Nevertheless, with the same concentration of SNP, cell death only occurred in the presence of IL-1β and nutrient-deprived conditions. We suggested that treatment with IL-1β under nutrient-deprived conditions could potentially enhance the effect of NO-induced cell death. In agreement with previous findings by colleagues from our study group, 38 it appears the induction of iNOS and production of NO, which leads to cytotoxicity in canine articular chondrocytes is dependent on the involvement of other mediators, while the observed cell death may be related to an impaired autophagic activity.
Despite several differences in study methodology,28-30 our results suggest that NO inhibits autophagy in chondrocytes. The treatment of chondrocytes with NO donor resulted in the lowest cell viability but high autophagosome formation. Agreeably, further investigations will be necessary to clarify whether autophagosome accumulation is due to increase in synthesis or decrease in degradation. 39 The present study demonstrated that the effects of NO on autophagosome formation can be reduced by addition of V-ATPase inhibitor as shown by the significant reduction in autophagic flux in bafilomycin treated chondrocytes. Consistent with Western blot and immunocytochemistry analysis, the mRNA expression of Atg genes; beclin-1, Atg7, and LC3 were downregulated. Taken together, our finding indicates that the activation of autophagy, autophagic flux and multiple Atg genes expression in chondrocytes is negatively regulated by the presence of NO. This inhibition of autophagy seems to depend on the promotion of mTOR, the key regulator of autophagy in mammalian cells.40,41 In agreement with findings elsewhere, these findings suggest that NO positively regulates the phosphorylation of ERK and Akt,42-44 whereas the activation of these pathways acts synergistically to promote mTOR/p70S6K signaling. 45 These results indicate that the reduction of autophagy in SNP treated chondrocytes is associated with the promotion of ERK, Akt and mTOR signaling pathways. Our results further reveal that rapamycin could attenuate NO-induced cell apoptosis by inhibition of mTOR signaling and reducing phosphorylation of p70S6K, which results in activation of autophagy as indicated by increase in LC3-II expression. This finding suggests that the activation of autophagy through the inhibition of mTOR/p70S6K pathway by rapamycin plays a protective role against NO-induced cell apoptosis.
Apoptosis is an active form of programmed cell death, which is mediated by caspases. This form of cell death is characterized by nuclear fragmentation, chromatin condensation, and membrane blebbing. 46 Previous studies have described that SNP induces chondrocyte apoptosis through the classical pathway, with activation of caspase and the presence of DNA fragmentation.15,22,44,47 In contrast, our results suggest that NO-induced cell apoptosis may be predominantly through the caspase-independent pathway as shown by an unexpectedly low level of caspase-3 and PARP cleavage protein expression, which corresponded with the result from the caspase-3 assay kit. This finding was further supported by the observed upregulation of AIF, a mitochondrion-localized flavoprotein and a key mediator in caspase-independent death pathway suggesting that the mechanism of canine chondrocyte apoptosis induced by SNP may be distinct from the classical apoptosis pathway.24,48,49 Hence the pathways of chondrocyte cell death are diverse and may possibly occur as a combination of multiple pathways. Indeed, according to various outcomes from several studies, we also believe that the mechanisms of NO donor-induced chondrocyte cell death could vary among experimental conditions. 22
In conclusion, our results demonstrate that in chondrocyte cultures with cells induced into an OA-state, NO inhibits autophagy and induces chondrocyte apoptosis mainly, but not completely through the caspase-independent pathway. Furthermore, the cytotoxicity of NO on chondrocytes could be enhanced by the involvement of other inflammatory mediators and experimental conditions. These findings suggest that autophagy is a protective mechanism in the pathogenesis of OA and could be proposed as a valuable therapeutic target for degenerative joint diseases, particularly to reduce the toxicity of NO.
Footnotes
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors state that the first author, Ekkapol Akaraphutiporn has a scholarship (Scholarship ID: MEXT 163007) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the government of Japan. All the authors declare that the provider of the scholarship for the first author had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval for this study was obtained from Hokkaido University Institutional Animal Care and Use Committee (approval number 12-0059).
Animal Welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.