
Abstract
Objective
Previously we showed that genetic deletion of Fgfr1 in chondrocytes protected mice from progression of osteoarthritis (OA). The aim of this study is to evaluate the effect of PD166866, a potent selective inhibitor of Fgfr1, on cartilage degeneration induced by interleukin-1β (IL-1β) and to clarify underlying global gene expression pattern.
Design
Cartilage explants and primary rat chondrocytes were stimulated with IL-1β to establish an inflammatory OA in vitro model. The effects of PD166866 were determined by measuring the release of glycosaminoglycans (GAG) in cartilage explants and primary rat chondrocytes, and the underlying molecular mechanism was analyzed by microarray and RT-PCR analysis in primary chondrocytes.
Results
In cartilage explants, PD166866 significantly counteracts IL-β stimulated GAG release. In addition, PD166866 impede IL-1β-stimulated nuclear translocation of p65 in rat chondrocytes. Based on microarray analysis, a total of 67 and 132 genes with more than 1.5-fold changes were identified in IL-1β-treated versus control and PD166866 cotreatment versus IL-1β treatment alone, respectively. Only 19 thereof were coregulated by IL-1β and PD166866 simultaneously. GO and KEGG pathway analysis showed that some pathways, including “cytokine–cytokine receptor interaction,” “chemokine signaling pathway,” and “complement and coagulation cascades,” as well as some key genes like chemokines, complement, and matrix metalloproteinases may relevant for therapeutic application of Fgfr1 blockade in IL-1β-stimulated chondrocytes.
Conclusion
Our results clearly demonstrated that blockade of Fgfr1 with PD166866 could effectively suppress the catabolic effects induced by IL-1β, and elucidated whole genomic targets of Fgfr1 inhibition responsible for the therapeutic effects of Fgfr1 blockade against inflammatory OA.
Introduction
Osteoarthritis (OA) is the most common form of arthritis, affecting more than 240 million people worldwide. 1 However, no disease-modifying drug is currently available for OA therapy, which is primarily associated with a poor understanding of the pathogenesis. 2
Remarkable progress has been made in the identification of signal pathways that regulate cartilage development and homeostasis maintenance rencently. 3 The role of fibroblast growth factors (FGFs) and their receptors in the cartilage metabolism and OA development has caught special attention.4,5 Fgfr1 and Fgfr3 are primary receptors expressed in human chondrocytes, and their function seems opposite in maintaining homeostasis of articular cartilage. 5 Chondrocyte-specific Fgfr3 knockout mouse accelerated cartilage degeneration and increased expression of matrix-degrading enzymes. 6 Intra-articular injection of FGF-18, through binding with FGFR3, could prevent cartilage destruction and enhance the regeneration of damaged cartilage in a surgical rat OA model. 7 Currently, the recombinant human FGF-18 protein, named sprifermin, is undergoing phase II clinical trials for OA treatment. Primary 3-year data showed that sprifermin is effective in increasing cartilage thickness in a dose-dependent manner and has an acceptable safety profile in OA patients. 8 With regard to Fgfr1, we have previously demonstrated the upregulation of Fgfr1 in degenerative cartilage chondrocytes, and found that conditional deletion of Fgfr1 alleviated the cartilage destruction with decreased matrix metalloproteinase-13 (MMP-13) expression. 9 It has been reported that FGF2 mainly binds with Fgfr1 to promotes cartilage catabolism through MAPK-extracellular signal-regulated kinase (ERK) 1/2 pathway in human articular chondrocytes. 10 In addition, blockade of Fgfr1 with inhibitor or peptide could attenuate the degeneration of articular cartilage in surgically-induced mouse OA models.11,12 These results suggested that Fgfr1 may serve as potential therapeutic targets to prevent the onset of OA and delay disease progression. Despite of those early findings, whether manipulation of Fgfr1 with pharmacological inhibition could suppress the inflammatory catabolic response in cartilage requires further study, and the specific underlying molecular mechanisms are still poorly understood.
The research on intra-articular injection of therapeutics targeting FGF signaling is still in its infancy as regards the development of drugs for OA. 4 By contrast, a variety of small-molecule compounds are under development as pro- or anti-FGF signaling therapeutics for human diseases.13,14 In this study, we examined whether PD166866, a selective FGFR1 inhibitor, have a therapeutic effect against catabolic effects induced by interleukin-1β (IL-1β) in the ex vivo model of cartilage explants. Moreover, we performed gene expression analyses using microarrays in an effort to determine how PD166866 treatment affects the global gene expression profile of articular chondrocytes based on IL-1β-stimulated inflammatory OA in vitro model.
Methods
This study was conducted according to protocols was approved by the Experimental Animals Center of the Fourth Medical Centre of Chinese PLA General Hospital, Beijing, China.
Glycosaminoglycan Release in Joint Explants
Porcine cartilage explants were aseptically isolated from the patellar groove of fresh porcine knees that obtained were from the slaughterhouse. The cartilage was cut into a uniform cube and placed into 96-well plates with Dulbecco’s modified Eagle medium (DMEM). After 48 hours of culture, explants were stimulated with 25 ng/mL IL-1 to induce glycosaminoglycan release for 72 hours in the presence of increasing concentrations of PD166866 (100 nM, 1 μM, 10 μM). To quantify the amount of GAGs, culture media and the papain-digested cartilage explants were mixed with dimethyl methylene blue in formate buffer, and absorbance at 525 nm was determined. The ratio between the secreted GAG (medium) and total GAG (the retained GAG in digested explants plus the secreted GAG) was used to calculate the percent release of GAG.
Isolation and Culture of Rat Chondrocytes
To further investigate the effects of Fgfr1 inhibition on chondrocytes at a cellular and molecular level, primary rat articular chondrocytes were isolated and stimulated with IL-1β to establish an inflammatory OA in vitro model. To obtain rat primary chondrocytes, we harvested the knee joints from 5-day-old newborn rats and digested the joints with 0.1% collagenase (Gibco) overnight. The use of laboratory rats and experimental animal procedures was approved by the Experimental Animals Center of the Fourth Medical Centre of Chinese PLA General Hospital. After filtration to remove tissue debris, chondrocytes were maintained in a 100 mm culture dish in complete DMEM (supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2. After confluence, chondrocytes were trypsinized, pooled, and seeded into 24-well plates and 60-mm culture dishes. On day 3, cultures were pretreated with 100 nM PD166866 for 30 minutes and then stimulated with 10 ng/ml IL-1β (PD166866 was still present during the culture period after exposure to IL-1β). The cultures were divided into 3 groups, including control (no IL-1β and PD166866), IL-1β treated group, and PD166866 + IL-1β group. After 24-hour stimulation, chondrocytes in 24-well plates were fixed and performed with toluidine blue staining to determine proteoglycan content, while cultures in 60-mm culture dishes were used to isolate RNA. For GAG quantification, culture media and the papain-digested rat chondrocytes in 24-well plates (n = 6 each group) were collected and analyzed according to the method described above.
Immunofluorescence of P65
Immunofluorescence analysis of nuclear factor-κB (NF-κB) p65 subunit was performed on 50% confluent rat primary chondrocytes. The cells were either untreated or treated with IL-1β (10 ng/mL) in the presence and absence of PD166866 (100 nM). After 12-hour stimulation, the chondrocytes were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.1% Triton X-100, deprived endogenous peroxidase activity with 3% H2O2, and blocked with normal goat serum for 30 minutes at room temperature. The chondrocytes subsequently incubated with anti-p65 antibody (ab16502) at a 1/500 dilution for 1 hour. After rinsing with phosphate buffered saline (PBS), FITC-conjugated goat anti-mouse secondary antibody was applied, and images of cells were acquired with a confocal microscope (Nikon, Japan) and Nikon digital imaging system.
Total RNA Isolation and Real-Time Polymerase Chain Reaction Analysis
Total RNA was extracted from rat chondrocytes using the TRIzol reagent according to the manufacturer’s instructions (Invitrogen, USA). For real-time polymerase chain reaction (PCR) analysis, the isolated RNA was treated by DNase I to remove traces of contaminating DNA and was then reverse-transcribed to cDNA using All-in-One cDNA Synthesis SuperMix (Bimake, USA). Amplification by PCR was carried out in a 25-μL reaction volume using an SYBR Green UltraSYBR Mixture (Cwbio, China). Relative expression levels were normalized to GAPDH and calculated by QuantStudio Design & Analysis desktop software. After the comparison of the data by analysis of variance, the different groups were compared using Fisher’s t test (n = 3; P < 0.05). The annealing temperature was 57 °C and primer sequences were provided under the requirement.
Microarray Hybridization and Analysis
For microarray analysis, a total of 9 RNA samples (3 repetitions in each group) were extracted using TRIzol reagent and further purified by Qiagen RNeasy Minikit. Overall RNA quality was examined by 1% agarose gels. Microarray hybridization and data analyses were performed by the GMINIX Informatics Ltd. Co. (Shanghai, China) according to the standard Affymetrix protocol. Briefly, 5.5 μg of biotin-labeled cDNA from each sample was hybridized for 16 hours at 45 °C to Affymetrix GeneChip Rat Gene 2.0 ST Array (Affymetrix). Posthybridization staining and washing were performed following standard protocols, and each array was scanned, quantified, and exported to .CEL file format using the GeneChip Command Console Software. Data were analyzed with a robust multi average method (RMA workflow) using Affymetrix Expression Console Software (version 1.3.1). The median summarization of transcript expressions was calculated.
Bioinformatics Analysis
CEL-files of the raw data, which obtained from Affymetrix GeneChip Command Console Software, were uploaded to the website of Gminix-Cloud Biotechnology Information (GCBI, http://www.gcbi.com.cn/gclib/html/ index, Genminix Informatics Co. Ltd., Shanghai, China) for further analysis, including GO/pathway enrichment analysis, serial test of cluster (STC) analysis, and gene coexpression net analysis. GCBI is a platform that integrates a variety of research findings, genetic information, sample information, data algorithms, and bioinformatics to create a “gene knowledge base,” which include biology, medicine, informatics, computer science, mathematics, graphics, and other disciplines. Before analyzing data for biological variation, GCBI performed data processing (which included data normalization and filtering of flagged data) and quality control. The random-variance model (RVM, which is commonly used for comparison of more than 2 groups) F test was applied to filter the differentially expressed genes for the control and experiment group because the RVM F-test can raise degrees of freedom effectively in the cases of small samples. After the significant analysis and the false discovery rate (FDR) analysis, we selected the differentially expressed genes with a fold expression change >1.5 at cutoff values Q < 0.05 and P < 0.05. Gene ontology (GO) analysis was applied to analyze the main function of the differential expression genes according to Gene Ontology, which is the key functional classification of NCBI (National Center of Biotechnology Information) and can organize genes into hierarchical categories and uncover the gene regulatory network on the basis of biological process and molecular function. Pathway analysis was used to find out the significant pathway of the differential genes, according to Kyoto Encyclopedia of Genes and Genomes (KEGG; P < 0.05). Based on the interaction relationship from KEGG, we built gene regulatory networks based on GCBI. Within one network, core regulatory factors connect with adjacent genes and have the most considerable degrees.
Statistical Analysis
Results were reported as the mean ± standard deviation. Unpaired Student t test was used to determine the statistical difference between groups. P values <0.05 were considered significant.
Results
Blockade of Fgfr1 with PD166866 Counteracts IL-β-Stimulated GAGs Release in Cartilage Explants
The effects of increasing concentration of PD166866, a selective Fgfr1 inhibitor, on cartilage catabolism under inflammatory conditions in joint explants were evaluated by measuring the amount of GAG released into the culture medium and the fraction of secreted GAG in media. Consistent with previous studies, IL-1β treatment dramatically increased the amount of GAG in the culture medium from 13.14 ± 2.34 up to 29.09 ± 3.98 μg after 72 hours ( Fig. 1A ). GAG release of explants was sharply grown to 29.08% ± 3.98% on IL-1β treatment compared with that of 13.1% ± 2.34% in controls ( Fig. 1B ). However, PD166866 exposure, even at a concentration of 100 nM, could significantly decrease the IL-1β- induced release of GAGs. At the concentration of 1 μM and 10 μM of PD166866, the amount of GAG content decreased to the level close to the baseline ( Fig. 1A ). The GAG release of explants was reduced to 24.42% ± 3.57% and 16.99% ± 2.79%, respectively, when cotreated with PD166866 at 100 nM and 1 μM ( Fig. 1B ). No difference was observed between 1 μM and 10 μM concentrations.
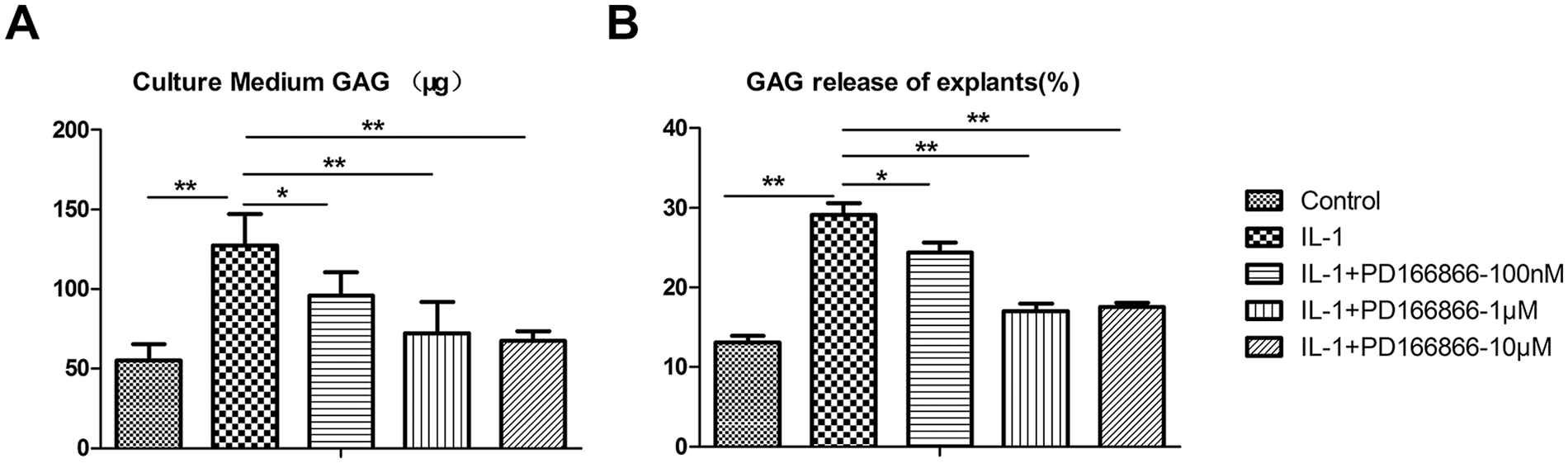
Protection of cartilage explants from the catabolic effects of IL-1β with PD166866. (
Blockade of Fgfr1 with PD166866 Impedes the Nuclear Translocation of p65 in Rat Chondrocytes Stimulated by IL-1β
Decreased toluidine blue staining was observed in the IL-1β stimulated chondrocytes, while PD166866 exposure greatly reduced the loss of proteoglycan induced by IL-1β ( Fig. 2A ). GAG amount in the culture medium and chondrocytes following treatment were also quantified. IL-1β treatment significantly increased GAG in the culture medium, which was remarkable decreased after being cotreated with PD166866 ( Fig. 2B ). Consistent with toluidine blue staining, IL-1β treatment led to decreased GAG and PD166866 cotreatment significantly increased the GAG of chondrocytes ( Fig. 2B ).
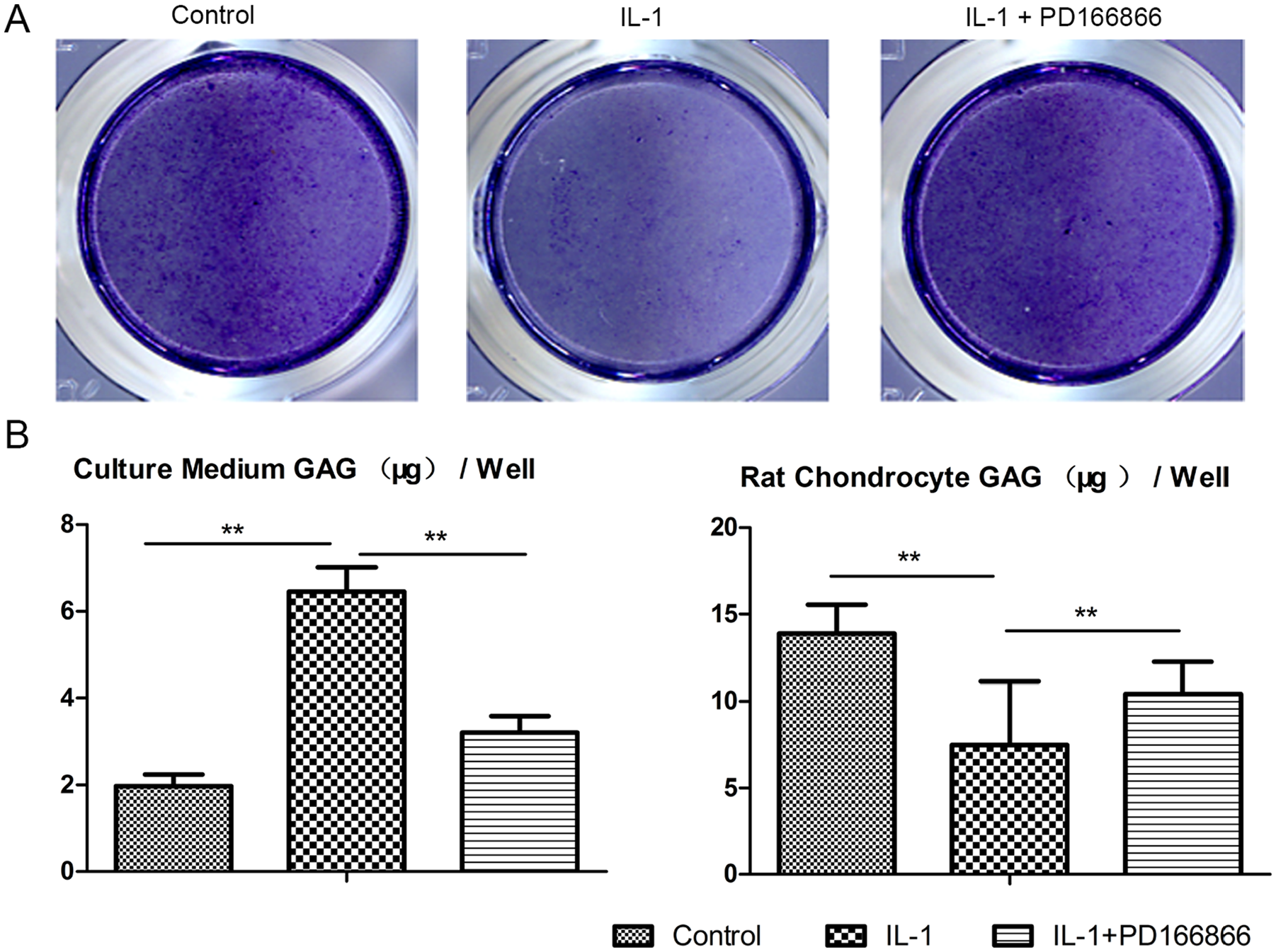
PD166866 treatment inhibited loss of proteoglycan in rat chondrocytes stimulated by IL-1β. (
To determine whether blockade of Fgfr1 affects the activation of the NF-κB pathway, we measured the translocation of the p65 NF-κB subunit from the cytoplasm to the nucleus on IL-1β stimulation in the presence and absence of PD166866. Consistent with previous studies, IL-1β treatment led to the localization of p65 protein in the nucleus of almost all chondrocytes. However, the nuclear translocation of p65 proteins was almost completely inhibited in chondrocytes cultured in the presence of 100 nM PD166866 ( Fig. 3A and B ).
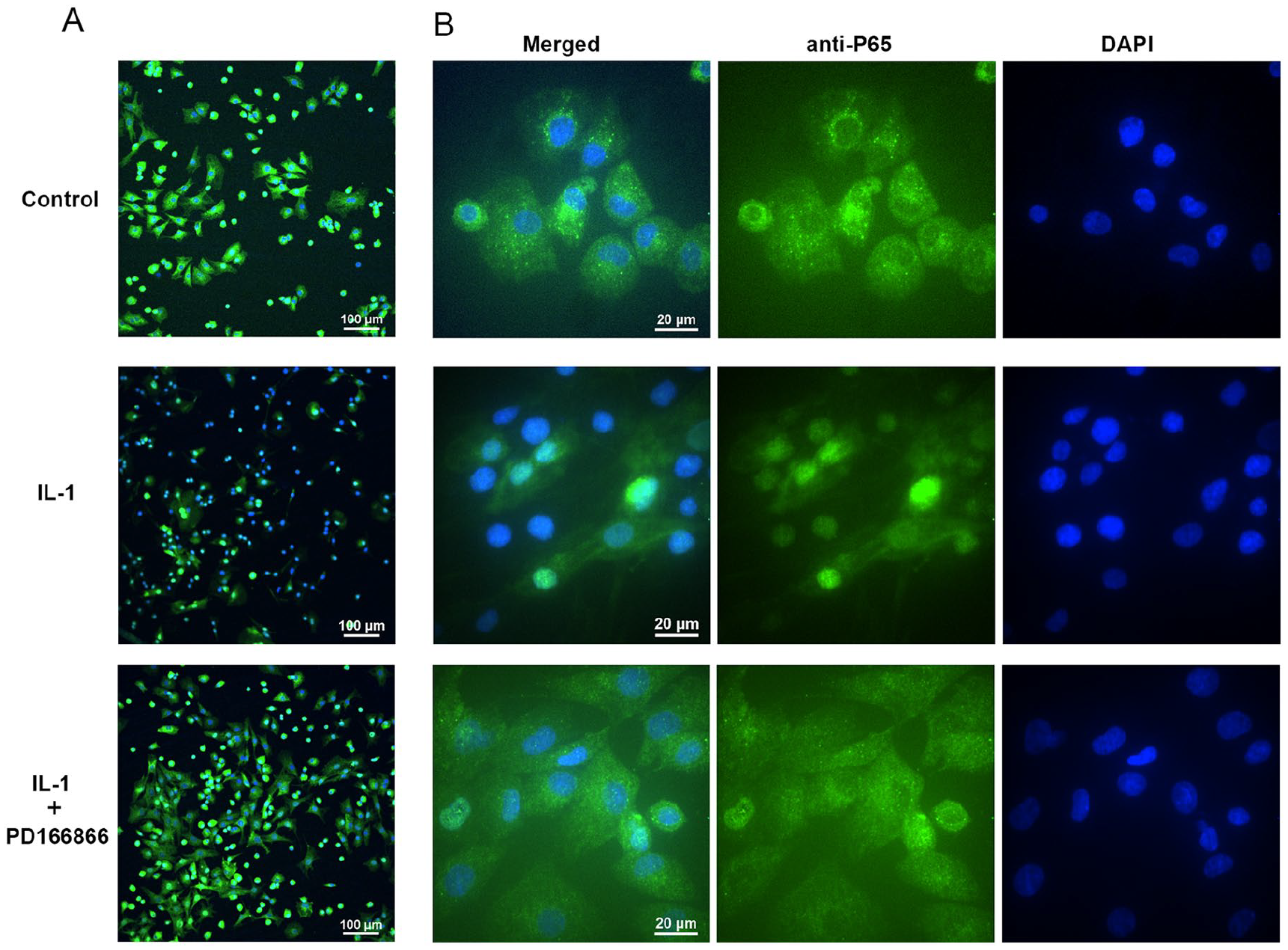
PD166866 treatment effectively suppressed the nuclear translocation of p65 in rat chondrocytes stimulated by interleukin-1β (IL-1β). (
Differential Expression Genes Caused by IL-1β and PD166866 in Rat Chondrocytes
The quality of total RNA of chondroytes was determined by Gel electrophoresis (data not shown). Then, whole genome-wide expression profiling was performed using oligonucleotide microarray. By using Affymetrix GeneChip Array analysis, we found that IL-1β treatment led to an up- or downregulation of the expression of 67 genes (P < 0.05, fold change >1.5) when compared with untreated controls. This IL-1β-induced gene expression pattern was used as a reference to explore the effects of Fgfr1 inhibitor on gene expression. In the presence of PD166866, 132 differentially expressed genes (DEGs) with more than 1.5-fold change were identified compared with IL-1β treatment alone. Of the 27,147 genes represented on the Affymetrix GeneChip rat gene 2.0 ST array, a total of 141 genes showed significant expression differences with more than 1.5-fold changes. The heat map of a total of 141 DEGs in three different groups is shown in Fig. 4A . However, only 19 thereof were coregulated by IL-1β and PD166866 simultaneously with more than 1.5-fold changes, and heat map was listed in Fig. 4B .
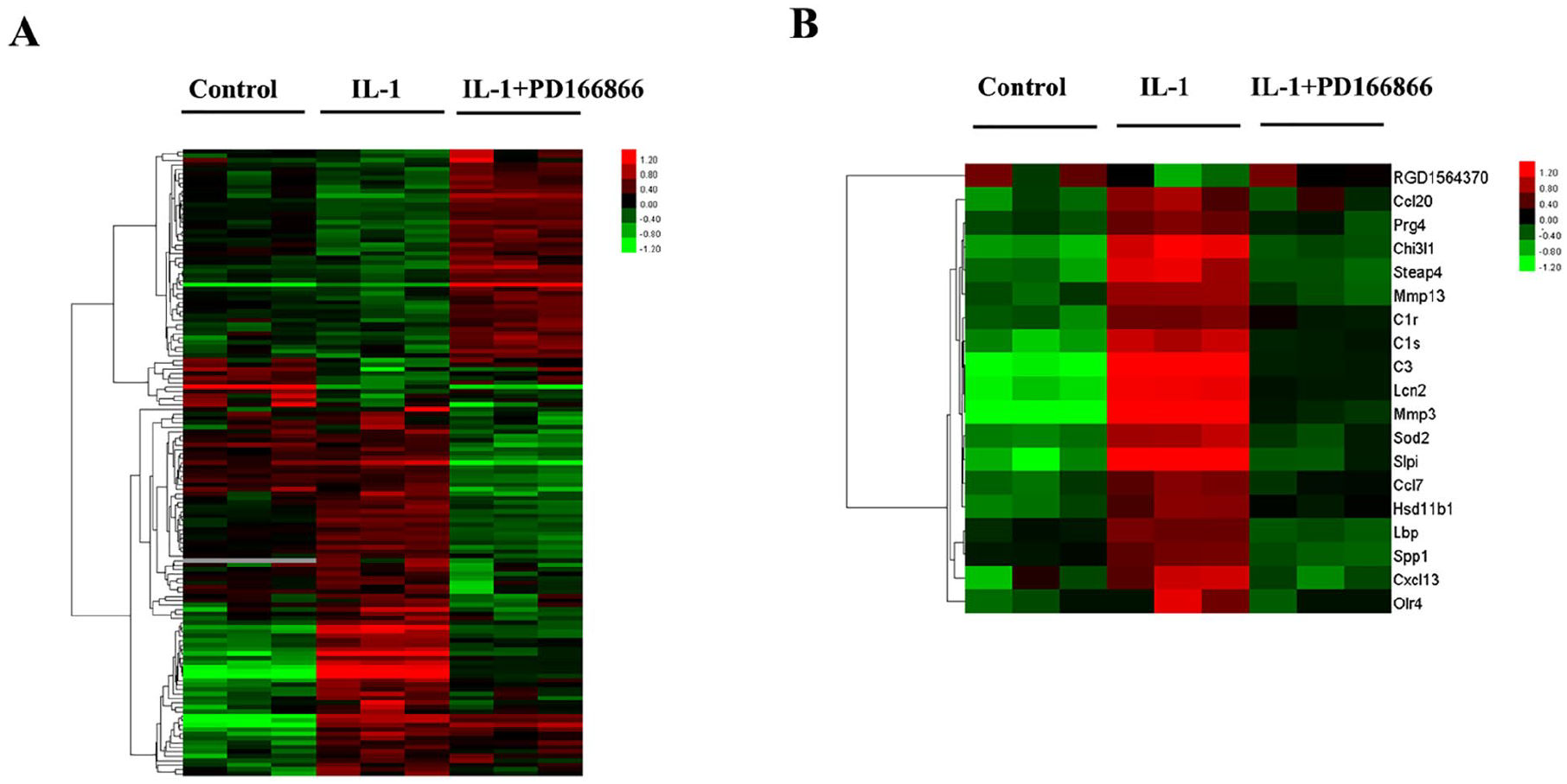
Global gene expression profile significantly affected by interleukin-1β (IL-1β) and/or PD166866. (A) Heat map of a set of union genes with more than 1.5-fold changes in the IL-1β-treated chondrocytes with or without PD166866. (
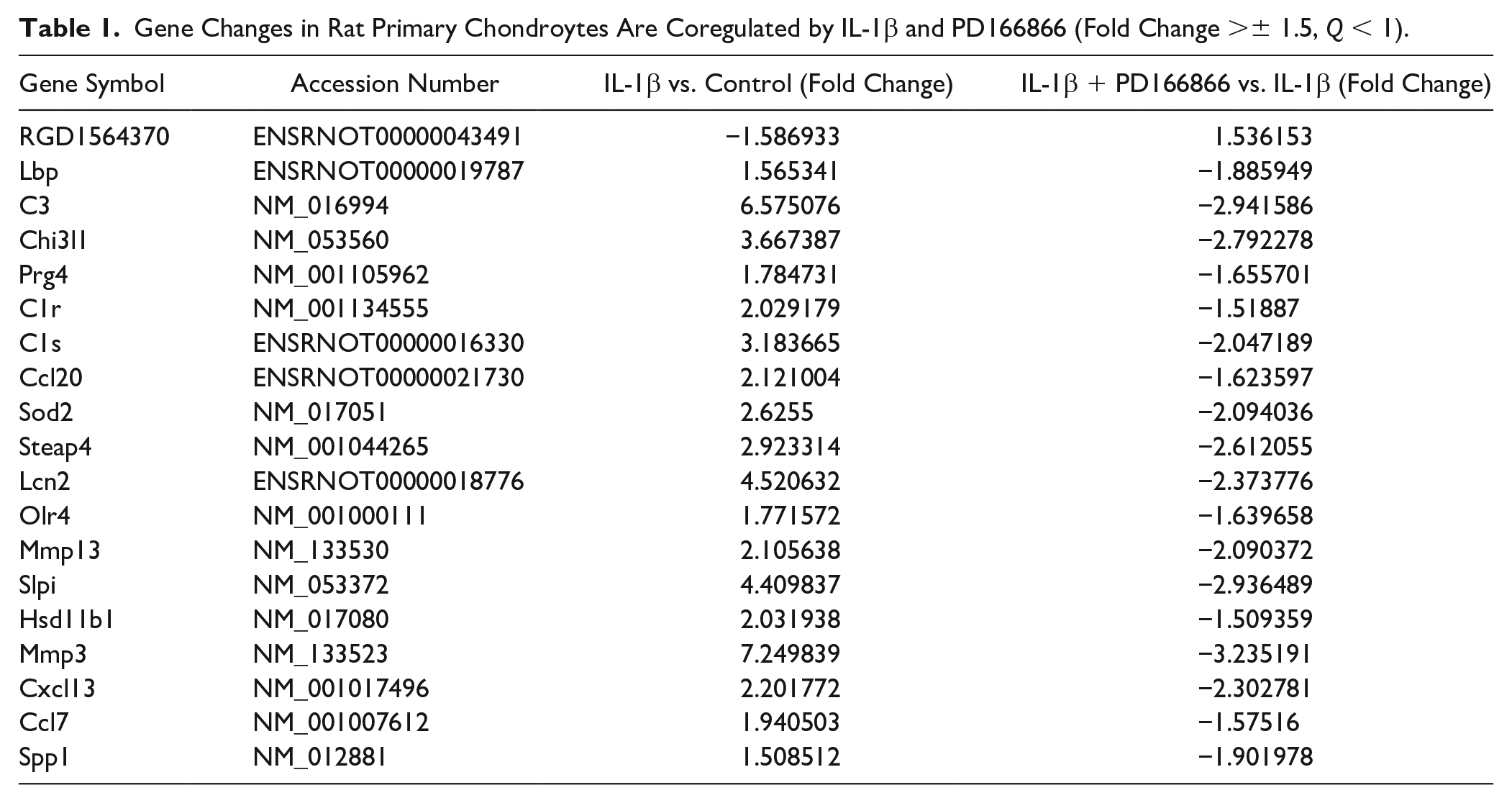
The list of known coregulated genes with their accession numbers and fold change for IL-1β and PD166866 regulation is shown in Table 1 . As expected, the mRNA for MMP-3 and MMP-13 significantly increased after IL-1β treatment and were suppressed by the cotreatment with PD166866. A subset of chemokines, including CCL1, CCL2, CCL7, CCL13, CCL20, CXCL1, and CXCL13 also significantly increased in the IL-1β-treated chondrocytes. Concurrent use of PD166866 and IL-1β suppressed the activation of CCL7, CCL20, and CXCL13, among those measured chemokines. Notably, some complement genes such as C3, C1r, and C1s were found to be coregulated by IL-1β and PD166866. To confirm the expression profiles suggested by microarray analysis, the expression of selected genes from among the co-regulated 19 genes was verified by real-time RT-PCR, including C1r, C1s, C3, Ccl20, Chi3l1, Hsd11b1, Lcn2, MMP-3, and MMP-13 ( Fig. 5 ). RT-PCR analysis confirmed that almost all these genes increased significantly in response to IL-1 and shifted the expression substantially under the cotreatment with PD166866.
Gene Changes in Rat Primary Chondroytes Are Coregulated by IL-1β and PD166866 (Fold Change >± 1.5, Q < 1).
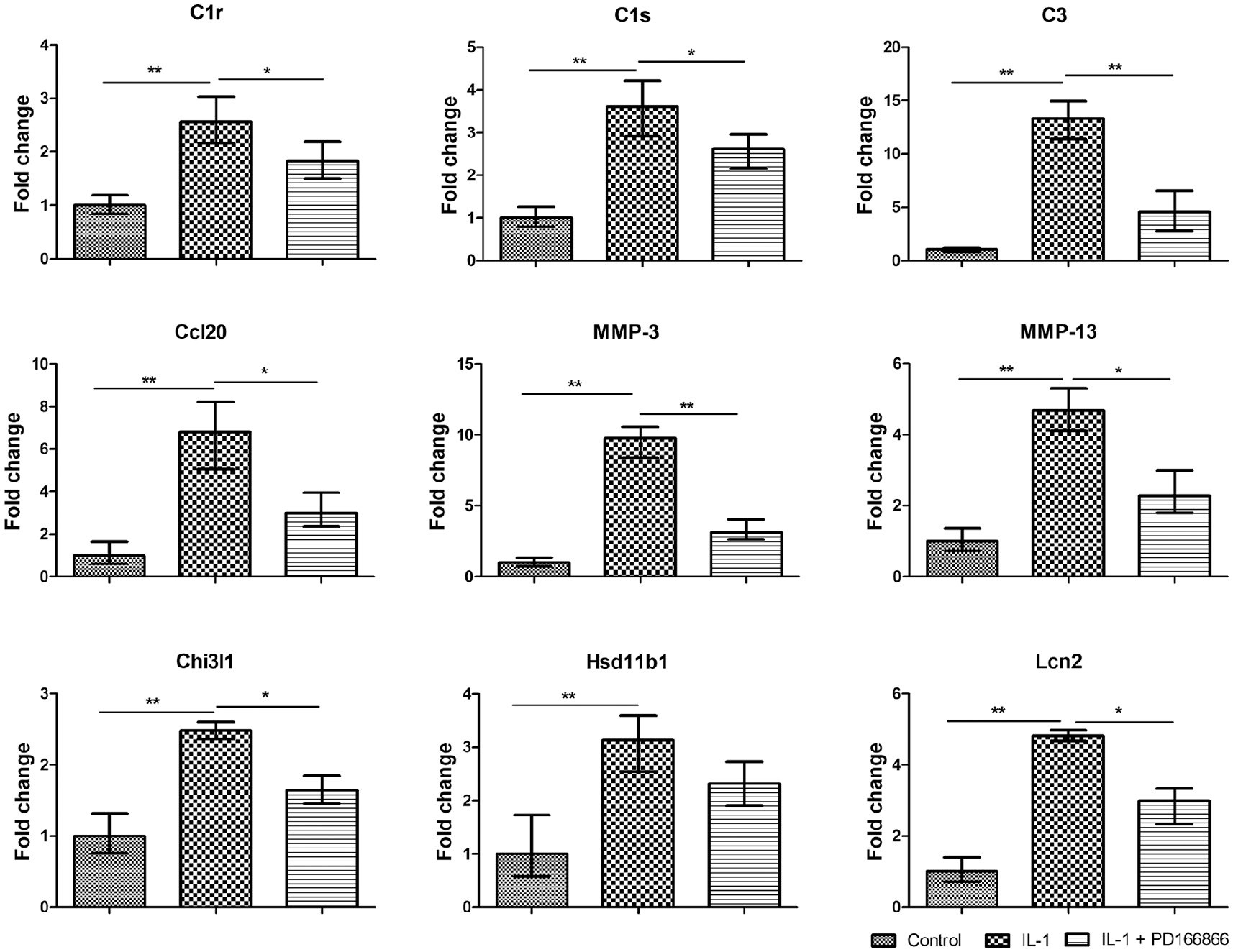
Validation of transcript levels by quantitative real-time polymerase chain reaction (PCR). Changes in mRNA levels in rat primary chondrocytes in response to interleukin-1β (IL-1β) and PD166866 were confirmed by quantitative real-time PCR. GAPDH was used as a control. The expressions of the following genes were determined: C1r, C1s, C3, Ccl20, MMP-3, MMP-13, Chi3l1, Hsd11b1, and Lcn2.
Bioinformatic Analysis of DEGs in Chondrocytes Affected by IL-1β and PD166866
Gene Ontology and Pathway Analysis
GO analysis was performed on 141 genes identified from 3 groups and the top 20 significant GO categories with P < 0.05 was presented ( Fig. 6A ). According to the threshold values, the highest GO terms were “inflammatory response,” “response to lipopolysaccharide,” “response to hypoxia,” and “positive response to angiogenesis.” Furthermore, we performed a pathway analysis to uncover significant pathways in which 141 DEGs participate. Pathway analysis was mainly based on the KEGG database, and the top 20 enriched pathways were presented ( Fig. 6B ). Based on KEGG pathway analysis, the most significantly changed pathways for P values from the KEGG pathway database, such as cytokine–cytokine receptor interaction, chemokine signaling pathway, and complement and coagulation cascades were observed ( Fig. 6B ).
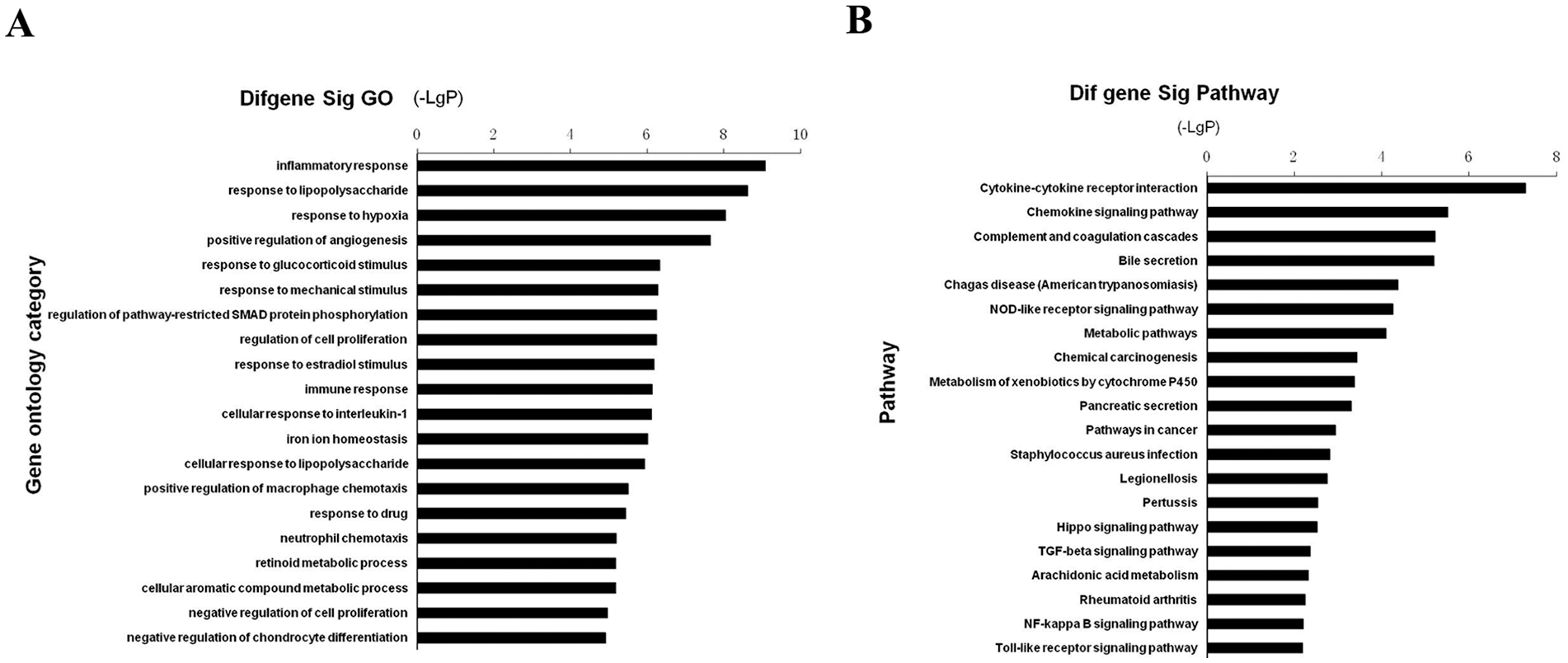
The gene ontology (GO) and pathway enrichment analysis of the differential expression genes in interleukin-1β (IL-1β)–treated chondrocytes with and without PD166866. (A) Significant GOs of differential expression genes. (
Serial Test of Cluster Analysis
Serial test of culture (STC) analysis showed 16 types of profiles for DEGs in the chondrocytes from 3 experimental groups. Among these profiles, 5 patterns (profiles 10, 15, 16, 14, and 5) were identified with significance and presented as red color (P < 0.05, Fig. 7A ). Of these, profiles 16 and 5 contained 26 and 21 genes, respectively, and showed the pattern of concurrent use of PD166866 and IL-1β significantly suppress high expression induced by IL-1β treatment. Detailed gene expression trends were shown in profiles 16 and 5 ( Fig. 7B ).
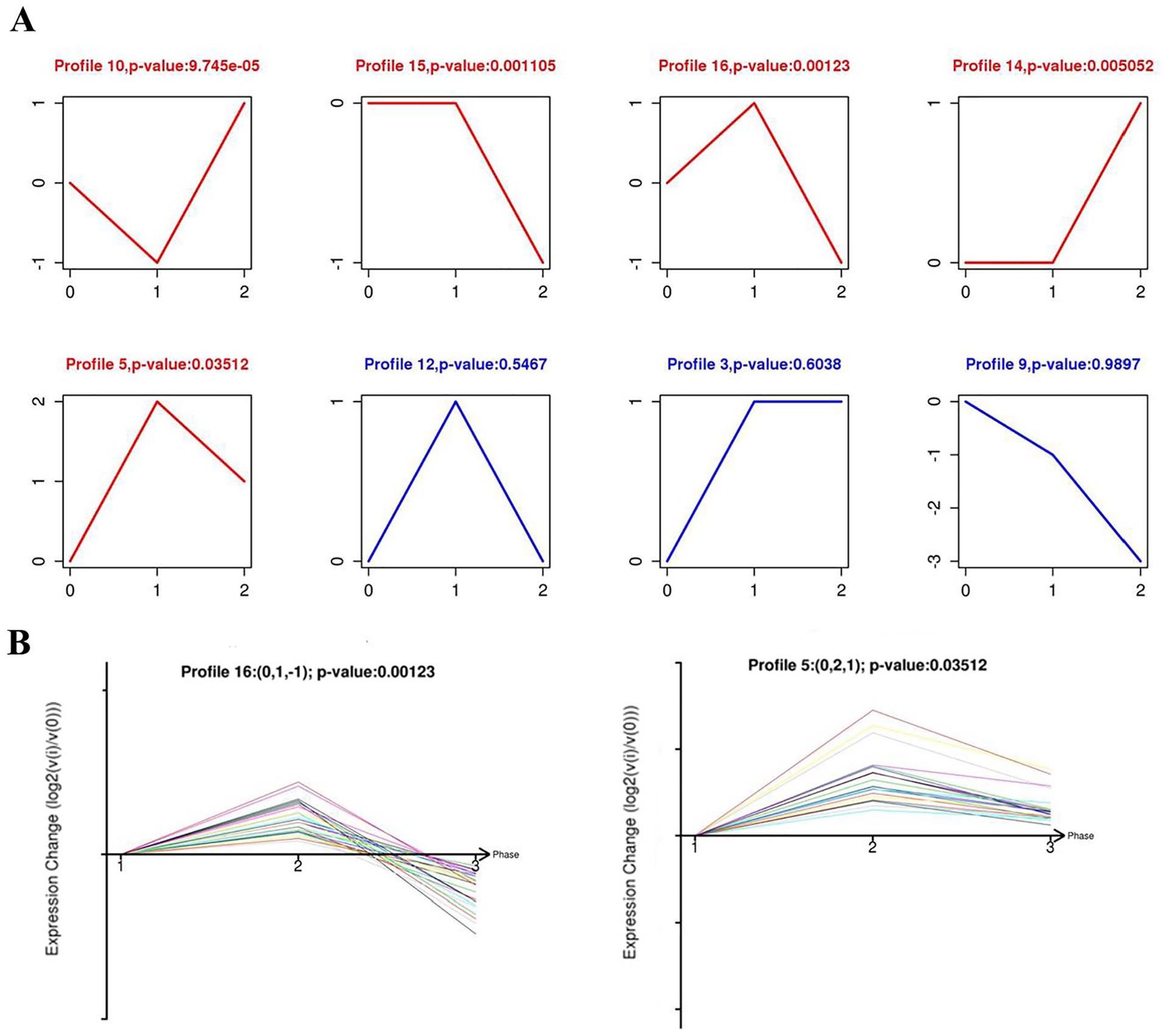
Serial test of culture (STC) analysis. (
Gene Coexpression Net
Gene coexpression net analysis was performed to screen the hub genes involved in PD166866-induced anticatabolic effects, and the co-expression net of 19 DEGs that screened from the intersection of 3 groups was showed ( Fig. 8A ). From the coexpression network, MMP-3, and C1s with the highest connectivity degree were significantly related to osteoarthritis development and had a great relationship with other genes, such as Lcn2, C3, Sod2, and Slpi ( Fig. 8A ). Meanwhile, the coexpression network of 141 DEGs, which screened by the union of 3 groups, suggested that Sema3d, Ifitm3, Cpz, and Slc40a1 were the hub genes with the highest connectivity degree ( Fig. 8B ).

The network of differentially expressed genes (DEGs) in chondrocytes among 3 groups. (
Discussion
In this study, we demonstrated the protective effects of Fgfr1 inhibition with PD166866 against IL-1β-induced catabolic effects on normal porcine explants cartilage and primary rat chondrocytes. The evaluation of 2 kinds of cartilage and chondrocytes made the results more solid and is helpful for us to understand the Fgfr1 target and its inhibitor PD166866 in OA therapy.
Our results and previous studies suggested that blockade of Fgfr1 is a potential strategy for the treatment of OA, which encouraged us to explore more thoroughly its physiologic effect on cellular and molecular levels. It has been reported that the changes of cartilage gene expression induced by IL-1β are time dependent and it is very important to consider the time point when analyzing the IL-1β effects. 15 The main purpose of IL-1β stimulation of chondrocytes for 2 hours is to study the early genetic changes and acute inflammatory gene changes. 16 Considering that chondrocytes were in a low level of inflammatory microenvironment for an extended period of time during OA, the dysregulated transcriptional program was a chronic and indirect effect. Therefore, we selected the samples treated with IL-1 and/or PD166866 for 24 hours for genome-wide microarray analysis. To our knowledge, this is the first time to screen downstream genomic targets of Fgfr1 inhibition possibly relevant for therapeutic application in an established IL-1β-stimulated cell culture model.
Many typical molecular changes of IL-1β stimulation, including the upregulation of a subset of chemokines and MMPs, were confirmed in our microarray study. Our results showed that IL-1β-induced upregulation of a subset of chemokines, including CCL1, CCL2, CCL7, CCL13, CCL20, CXCL1, and CXCL13, similar to the previous reports.15,16 We also found that the IL-1β-induced upregulation of MMP-3 and MMP-13 were greatly suppressed by Fgfr1 inhibitor in this OA in vitro model. Our findings here are in a good agreement with previous single-gene analyses, which showed that genetic inhibition of Fgfr1 protects mouse from OA progression by suppression MMP-13 expression. 9 Based on microarray analysis, a total of 132 DEGs with more than 1.5-fold change were identified in the PD166866 cotreatment compared with IL-1β treatment alone. Among these genes, a key set of 19 genes were identified and regulated by IL-1β and PD166866 simultaneously. These genes may largely contribute to the therapeutic effect of Fgfr1 inhibitor against IL-1β-stimulated catabolic effects in cartilage.
It has been reported that chemokines and chemokine receptors are expressed by cells within the joint, exerting catabolic effects on articular cartilage either through paracrine and/or autocrine mechanisms. 17 CCL2/CCR2 signal increased MMP-3 expression in human articular chondrocytes and enhanced proteoglycan loss from human cartilage in vitro. 18 CCL20/CCR6 might play a critical role in the pathogenesis of OA by inducing changes in phenotype and catabolic gene expression in chondrocytes. 19 Many studies have suggested that chemokines are relevant to OA pathogenesis, but their regulation and downstream of chemokine/receptor in chondrocytes needs further elucidation. In this study, we found that the IL-1β-stimulated upregulation of mRNA for CCL-7, CCL-13, as well as CXCL-13 was markedly suppressed by cotreatment with PD166866 in chondrocytes. These results indicated that Fgfr1 inhibition prevented the transcriptional activation of chemokines responding to inflammation in chondrocytes. Our results suggest that the expression of several chemokines can be regulated by pharmacologically manipulation of Fgfr1 signals, thus delayed the progression of OA relevant phenotypic changes.
Another important finding of our microarray analysis is that the Fgfr1 inhibitor can suppress the induction of complement molecules in this OA in vitro model. We identified that C3, C1r, and C1s were coregulated by IL-1β and PD166866. Previously, microarray analysis suggested aberrant expression and activation of complement components, as well as decreased complement inhibitors in human OA joints compared with controls. 20 Complement activation in synovial joints has been reported to be critical for the development of OA. 20 Recently, the involvement of complement and complement activation products in the pathogenesis of OA has been accepted and intensively reviewed. 21 However, precise regulation of complement expression and cross-talk with other signaling molecular in joint related tissues are still not fully understood. Inflammatory cytokine was reported to be able to regulate complement synthesis in cultured chondrocytes.22,23 Our results demonstrated that there was a direct relationship between Fgfr1 blockade and suppression of complement system activation, and the specific mechanism of FGF signal regulating complement expression is being further studied.
In the current study, we also showed that blockade of Fgfr1 with PD166866 appears to completely abolish the translocation of the p65 NF-κB subunit from the cytoplasm to the nucleus in chondrocytes on IL-1β stimulation. NF-κB pathway is a central player in inflammatory signal transduction and activation of which greatly contribute to the cartilage destruction on injury. 24 Combined with the inhibition of NF-κB activation, complement molecule and chemokine secretion by Fgfr1 inhibitors, these results provide a novel insight that Fgfr1 inhibitor served as a potential therapeutics for the development of anti-inflammatory drugs in the synovial joint. PD166866 may be an innovative molecule to counteract the inflammation process during OA.
In order to clarify the overall effect of PD166866, GO analysis and KEGG pathway analysis were performed to reveal the involvement of a variety of biological processes. Predominantly affected GO terms were “inflammatory response,” “response to lipopolysaccharide,” and “response to hypoxia.” The most significantly changed pathways were “cytokine–cytokine receptor interaction,” “chemokine signaling pathway,” and “complement and coagulation cascades.” In addition, the protein-protein interaction network was constructed, and hub genes were also revealed. Overall, the detailed interpretation of IL-1β- and PD166866-induced regulations of genes showed a strong anti-inflammatory effect of Fgfr1 inhibition in versatile fields of cellular response.
Using a gene chip containing more than 27,000 genes, we screened 141 differential genes more than 1.5-fold changes in 3 groups, of which 19 genes were coregulated by IL-1β and PD166866. Among these 19 genes, in addition to the chemokines, complements, and MMPs mentioned above, we conducted a more in-depth analysis of the remaining genes. Glycoprotein chitinase 3-like-1 (CHI3L1) and lubricin (Prg4) are 2 glycoproteins functionally related to the development of OA. The expression of CHI3L1 was considered as a potential marker for the progression of OA, 25 whereas Prg4 appears to be chondroprotective and could retard the progression of cartilage degeneration.26,27 Lipocalin-2 (LCN2) is a catabolic adipokine contributing to the obesity and OA pathologies, which was induced by inflammation and this induction can be reversed by TGF-β1 and IGF-1. 28 Secretory leucocyte proteinase inhibitor (Slpi) was found upregulation in OA superficial chondrocytes 29 , and secreted phosphoprotein 1 (Spp1) was also highly expressed in OA chondrocytes and regulated by PI3K-AKT pathway. 30 In addition, we found that some genes, such as Hsd11b1, 31 lipopolysaccharide binding protein (Lbp), 32 and STEAP4 33 are associated with rheumatoid arthritis, but their roles in OA are unknown. The relationship of these genes and Fgfr1 inhibition as well as OA development remains further investigation.
In summary, we showed that PD166866 might act as an anti-inflammatory agent in porcine cartilage and primary rat chondrocytes, and present initial data of a genome-wide analysis of Fgfr1 inhibition in an inflammation OA in vitro model. Besides well-described effects, we found new aspects of cellular reactions in response to Fgfr1 inhibition such as alterations in the chemokine signaling pathway and complement and coagulation cascades. It is anticipated that pathways and the novel DEGs identified in this study may shed light on the underlying molecular interaction between chondrocyte-intrinsic Fgfr1 signal and proinflammatory cytokine.
Footnotes
Authors’ Note
This work was performed at the Department of Orthopaedics, the Fourth Medical Centre, Chinese PLA General Hospital.
Acknowledgments and Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval for this study was obtained from Animal Care Committee of the Fourth Medical Centre of Chinese PLA General Hospital.
Animal Welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.