
Abstract
Objective
Although tissue engineering is a promising option for articular cartilage repair, it has been challenging to generate functional cartilaginous tissue. While the synthetic response of chondrocytes can be influenced by various means, most approaches treat chondrocytes as a homogeneous population that would respond similarly. However, isolated cells heterogeneously progress through the cell cycle, which can affect macromolecular biosynthesis. As it is possible to synchronize cells within discrete cell cycle phases, the purpose of this study was to investigate the effects of cell cycle synchronization on the chondrogenic potential of primary articular chondrocytes.
Design
Different methods of cell synchronization (serum starvation, thymidine, nocodazole, aphidicolin, and RO-3306) were tested for their ability to synchronize primary articular chondrocytes during the process of cell isolation. Cells (unsynchronized and synchronized) were then encapsulated in alginate gels, cultured for 4 weeks, and analyzed for their structural and biochemical properties.
Results
The double-thymidine method yielded the highest level of cell purity, with cells synchronized in S phase. While the cells started to lose synchronization after 24 hours, tissue constructs developed from initially S phase synchronized cells had significantly higher glycosaminoglycan and collagen II amounts than those developed using unsynchronized cells.
Conclusions
Initial synchronization led to long-term changes in cartilaginous tissue formation. This effect was postulated to be due to the rapid auto-induction of TGF-βs by actively dividing S phase cells, thereby stimulating chondrogenesis. Cell synchronization methods may also be applied in conjunction with redifferentiation methods to improve the chondrogenic potential of dedifferentiated or diseased chondrocytes.
Introduction
Damage to the articular cartilage tissue is a common occurrence caused by a range of factors including acute injury 1 and deterioration by disease,2,3 which often leads to joint pain, loss of joint mobility, and the progressive destruction of tissue. Current strategies for articular cartilage repair, such as autologous osteochondral transfer and cell transplantation, aim to replace the damaged tissue with a repair tissue that has the same biochemical, mechanical, and functional properties as native articular cartilage. Autologous osteochondral transfer involves the transplantation of healthy cartilage plugs from non-load-bearing joint regions into the defect site. 4 Cell transplantation (e.g., autologous chondrocyte implantation and matrix-induced autologous chondrocyte implantation) involves isolating cells from non-load-bearing regions in the joint, in vitro expansion of chondrocytes, and implantation of the expanded cell population (with or without supporting matrices) at the defect site.5,6 While these approaches generally have good short-term clinical outcomes, these methods suffer from donor site morbidity4-6 and, in the case of cell transplantation, can be limited by the retention of cells within the treated defect.5,6 To overcome these limitations, cartilage tissue engineering has emerged as an alternative approach to provide replacement tissue suitable for articular cartilage repair.
While tissue engineering methods have been shown to generate cartilaginous tissues in vitro, the generation of engineered tissues of appropriate size, thickness, and functional properties is still a major challenge. Previous studies have extensively investigated the use of a variety of means to improve deposition of cartilaginous extracellular matrix and impart functional tissue properties (e.g., growth factors, 2 bioreactors,7,8 and/or mechanical stimuli9-16). While generally successful, these approaches tend to treat isolated chondrocytes as a homogeneous population, and hence would respond similarly to the imposed stimulus. However, potential variations exist within the isolated cell population, including their progression through the cell cycle, 17 which can affect macromolecular biosynthesis18-20 as well as their response to drugs and other treatments.21-23 As it is possible to synchronize cells within a discrete cell cycle phase (e.g., G0/G1, G2/M, S), we postulate that the growth of engineered cartilage can be improved by pre-synchronizing the isolated cell population. However, the difficulty with this approach is that commonly used chemical cell cycle arresting agents are applied during monolayer culture, which for chondrocytes can also elicit dedifferentiation. 24
Thus, the purpose of this study was to (1) develop methods to synchronize primary articular chondrocytes in situ and (2) to determine the effect of cell synchronization on cartilaginous tissue formation in vitro. We first evaluated the feasibility of adapting different chemical methods of cell synchronization of primary articular chondrocytes in situ. Then, using the method that yielded the highest degree of synchronization and viability, we tracked the progress of the synchronized cells through the cell cycle to determine the duration of cell synchrony. Finally, we compared the cellular content and extracellular matrix composition of engineered tissue constructs developed from populations of unsynchronized and synchronized cells.
Methods
Cell Isolation
Articular cartilage was harvested from the metacarpophalangeal joints of cows (<18 months old) obtained from a local slaughterhouse (Beverly Creek Farms, Milton, Ontario). The harvested cartilage tissues (<5 mm × 5 mm × 1 mm) were placed into a 100-mm petri dish containing complete media consisting of Ham’s F12 supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics/antimitotics. Cells were isolated by enzymatic digestion of the cartilage tissues with 0.5% w/v protease in Ham’s F12 for 1 hour followed by 0.15% w/v collagenase A in Ham’s F12 with 10% FBS for 12 hours at 37°C.
Cell Synchronization In Situ
Cell cycle synchronization of chondrocytes was achieved by serum starvation (G0/G1 phase) 25 or by applying commonly used cell cycle arresting agents, specifically, thymidine (S phase), 26 nocodazole (G2 phase), 27 aphidicolin (S phase), 28 or RO-3306 (G2 phase), 29 during the process of cell isolation.
Serum Starvation
The serum starvation method involved cell isolation in the absence of FBS by enzymatic digestion of the cartilage tissue with 0.5% w/v protease in Ham’s F12 for 1 hour followed by 0.15% w/v collagenase A in Ham’s F12 for 12 hours at 37°C.
Double Thymidine, Thymidine-Nocodazole, and Thymidine-RO-3306 Methods
The double thymidine, thymidine-nocodazole, and thymidine-RO-3306 methods involved first blocking the cells resident within the harvested tissues with 20 mL complete media containing 10 mM thymidine for 24 hours at 37°C, followed by a wash with 5 mL Ham’s F12 media. Cells were then released from the thymidine block in 20 mL Ham’s F12 media at 37°C for 8 hours (for double thymidine method), 3 hours (for thymidine-nocodazole method), or 5 hours (for thymidine-RO-3306 method). The media was then aspirated and replaced with 20 mL of 0.5% w/v protease in Ham’s F12 for 1 hour at 37°C. This was followed by a wash with 5 mL Ham’s F12 media and a second block in 20 mL Ham’s F12 media containing 10% FBS, 0.15% w/v collagenase A, and either 10 mM thymidine (double thymidine method), 100 ng/mL nocodazole (thymidine-nocodazole method), or 10 µM RO-3306 (thymidine-RO-3306 method) for 12 hours at 37°C.
Aphidicolin Method
The aphidicolin method involved serum starvation by incubating cartilage tissues with 20 mL Ham’s F12 media for 48 hours at 37°C, followed by incubation in 0.5% w/v protease in Ham’s F12 for 1 hour at 37°C. This was followed by a wash with Ham’s F12 media, and a subsequent 24-hour treatment with Ham’s F12 media containing 10% FBS, 0.15% w/v collagenase A, and 5 µg/mL aphidicolin.
Cell Encapsulation
To maintain the phenotype of chondrocytes in culture (synchronized or unsynchronized), isolated cells were encapsulated in alginate beads that were created by first mixing a high-density cell suspension of chondrocytes (20 × 106 cells/mL) in equal parts with a sterile 1.25% alginic acid solution (20 mM HEPES, 150 mM sodium chloride, pH 7.4) at room temperature. Cell-encapsulated alginate beads were then created by dropping 30 µL of the cell-alginate solution in a calcium chloride bath (102 mM CaCl2 containing 10 mM HEPES, pH 7.4). After 10 minutes, polymerized cell-encapsulated beads were extracted using forceps, placed into individual wells of a 24-well plate containing 1 mL Ham’s F12 media supplemented with 10% FBS and 100 µg/mL ascorbic acid, and cultivated at 37°C with 5% CO2 and 95% humidity. The culture media was changed every 48 hours and cell-encapsulated bead constructs were cultured for a maximum of 4 weeks.
Evaluation of Cell Viability and Cell Cycle Phase
Cell Recovery and Viability
Cells were recovered from encapsulation in order to assess cell viability and degree of cell cycle synchronization. Harvested cell-encapsulated alginate beads were dissociated at 37°C under light agitation (120 rpm) in a calcium chelating solution (55 mM EDTA in 1× PBS) for 15 minutes. Cell viability was then determined using the trypan blue exclusion method. 30
Assessment of Cell Cycle Phase Synchronization
Cell cycle phase synchrony was determined by flow cytometric analysis and gating of cells in the target cell cycle phase. Briefly, dissociated cells were pelleted by centrifugation (700 × g for 10 minutes), resuspended in Ham’s F12 media, and fixed in cold ethanol (80%). To prevent aggregation, cells were vortexed and left on ice for 30 minutes. Following fixation, the cells were pelleted by centrifugation (1000 × g for 5 minutes) and resuspended in PBS. Cells were then permeabilized using 10% Triton-x100, and then stained with propidium iodide (Abcam, ab139418). Flow cytometric analysis (BD Aria analytical cytometer) was performed by exciting the propidium iodide bound to DNA with a blue (488 nm) laser. The emission spectra of the stained cells were analyzed to determine DNA content. Unsynchronized control cell populations were used to establish cells in the G0/G1 phase. Cells that fluoresced with twice this intensity (i.e., twice the DNA content) were determined to be in the G2/M phase. Similarly, the population of cells distributed between the G0/G1 and G2/M peaks were determined to be within S phase. The DNA content of the cell population was then tracked over a 48-hour period following encapsulation (at 12-hour increments) to analyze the degree of synchrony of the encapsulated cell population in culture.
Analysis of Bead Constructs After Long-Term Culture
Biochemical Analysis
After 2 weeks and 4 weeks of cultivation, cell-encapsulated bead constructs were first lyophilized overnight and then digested by papain (40 µg/mL in 20 mM ammonium acetate, 1 mM EDTA, and 2 mM dithiothreitol) for 72 hours at 65°C. Biochemical assays were then performed on the papain digests to determine cellularity and extracellular matrix accumulation.15,31,32 DNA content was determined by the PicoGreen assay. 33 Proteoglycan content was determined by measuring the amount of sulfated GAG using the 1,9-dimethylmethylene blue (DMMB) dye binding assay.34,35 Collagen content was determined by hydrolyzing the papain digested samples in 6 N hydrochloric acid for 18 hours at 110°C and measuring the hydroxyproline content of the hydrolyzed samples using the chloramine-T/Ehrlich’s reagent assay. 36 Collagen content was estimated as 10 times the hydroxyproline content. 37
Histological and Immunohistochemical Evaluation
After 2 weeks and 4 weeks of cultivation, representative bead constructs were fixed in 4% paraformaldehyde for 24 hours. Fixed beads were then dehydrated in graded ethanol solutions and embedded in paraffin at 65°C. Sections were cut at 8 µm thickness and mounted on Superfrost slides. Sections were deparaffinized and rehydrated, and then stained with safranin-O with fast green counterstain or picrosirius red with hematoxylin counterstain for the visualization of GAG and collagen distribution, respectively. 15
For collagen I and collagen II immunostaining, rehydrated sections were treated with a solution of 3.9 kU/mL pepsin in 0.5% acetic acid for 30 minutes at 37°C, followed by 2 washes with PBS. The sections were then blocked with a solution of 1% bovine serum albumin (BSA) in PBS for 30 minutes at room temperature. After blocking, sections were incubated with an anti-collagen I (Abcam, ab90395) or anti-collagen II (Developmental Studies Hybridoma Bank, CIIC1) primary antibody (1:100 dilution in PBS containing 1% BSA) overnight at 4°C. Two washes with PBS were performed to remove the excess primary antibody, followed by staining with Texas red-labelled goat anti-mouse secondary antibody (Abcam, ab6787, 1:200 dilution in PBS containing 1% BSA) for 2 hours at room temperature. Three washes with PBS were performed, followed by mounting the slides with coverslips using Vectashield mounting medium containing DAPI.
Safranin O, picrosirius red, and collagen II staining were graded using the Bern Score, 38 according to 3 scoring categories: (1) stain uniformity and intensity, (2) cell distance, and (3) cell morphology (each scoring category with a score out of 3 for a total score of 9). The images were scored by 2 independent observers.
Statistical Analyses
All results were expressed as the mean with the standard error of the mean. Statistical differences were analyzed using one-way ANOVA and Tukey’s post hoc testing, and significance was associated with P values less than 0.05.
Results
Cell Cycle Synchronization of Primary Articular Chondrocytes
Primary articular chondrocytes were synchronized into different phases of the cell cycle using various chemical synchronizing agents during the process of cell isolation from harvested tissue. The best method was determined by analyzing the proportion of cell population synchronized within the phase of interest as well as viability of the synchronized cell population.
The double thymidine method yielded the highest percentage of cells synchronized within S phase, while maintaining a high cell viability ( Table 1 ). However, all other synchronization methods investigated (nocodazole, RO-3306, and aphidicolin) resulted in decreased cell viability (<90%) and/or a lack of highly synchronized cell populations (<90% within the cell cycle phase of interest) ( Table 1 ). For these reasons, the double thymidine method was chosen to synchronize primary articular chondrocytes for all subsequent studies, and the other synchronization methods were not investigated further.
Viability and Purity of Cell Populations Subjected to Different Synchronization Techniques a .
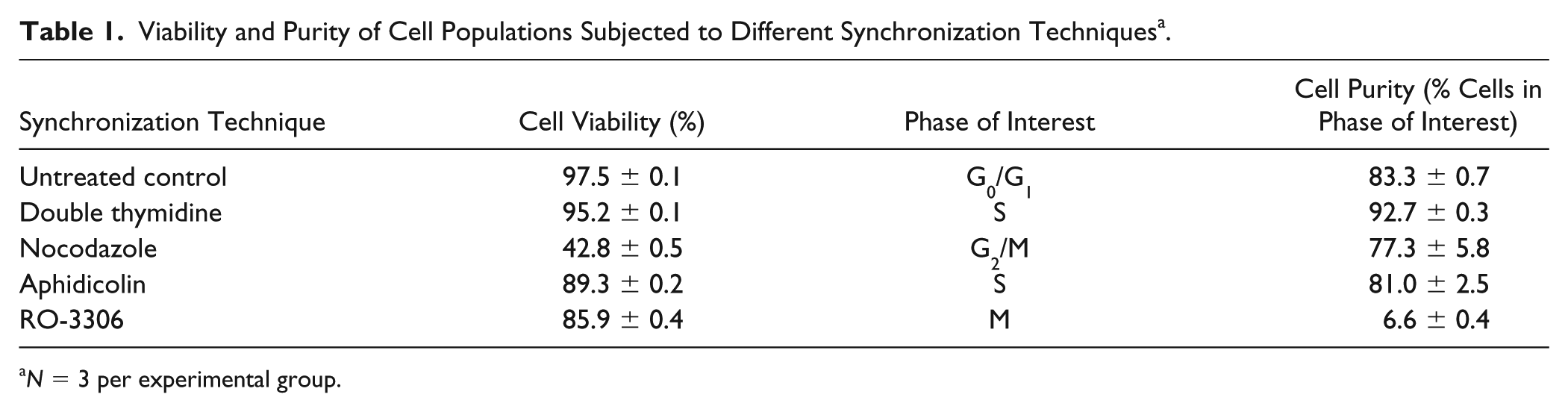
N = 3 per experimental group.
To assess the quality and longevity of the synchronization process using the double thymidine method, cell populations were recovered from encapsulation (via calcium chelation) and analyzed by flow cytometry at 12-hour intervals for up to 48 hours after synchronization ( Fig. 1A and B , Table 2 ). Initially, the synchronized cells were predominantly resident within the S phase (~93%). However, tracking of the cell cycle phase (at 12-hour increments post encapsulation) showed that while the cells proceeded to traverse through the cell cycle, synchronization of the population appeared to wane over time. A visible trailing peak of cells appeared to initiate at 24 hours post encapsulation, and with subsequent time in culture, this peak became visibly larger as evidenced by the broadening width of the histogram. At 48 hours post encapsulation, the percentage of cells resident within S phase declined to ~82%.
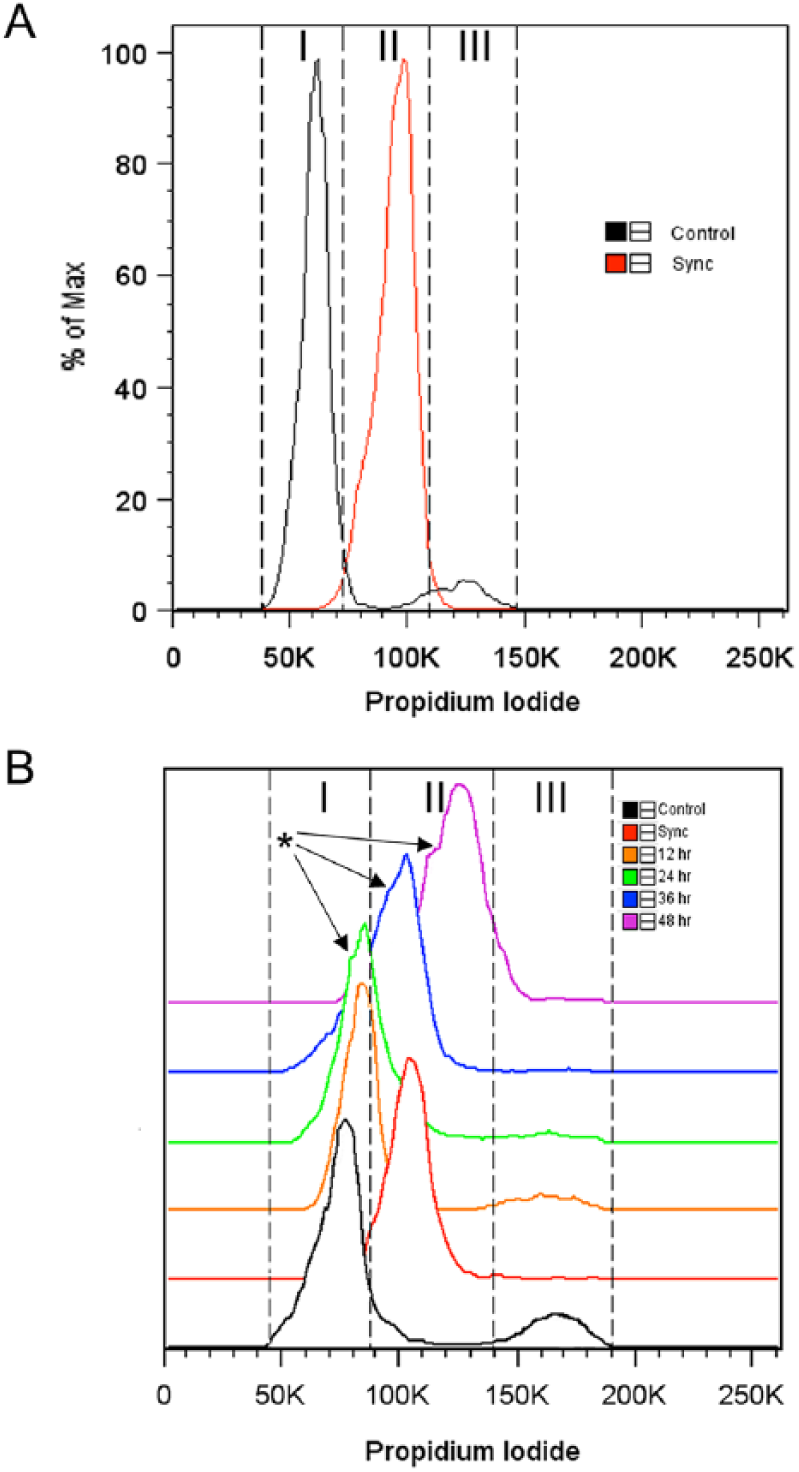
Cell cycle synchronization of primary articular chondrocytes. (
Percentage of Cells within Each Phase of the Cell Cycle at Different Time Points After Synchronization a .
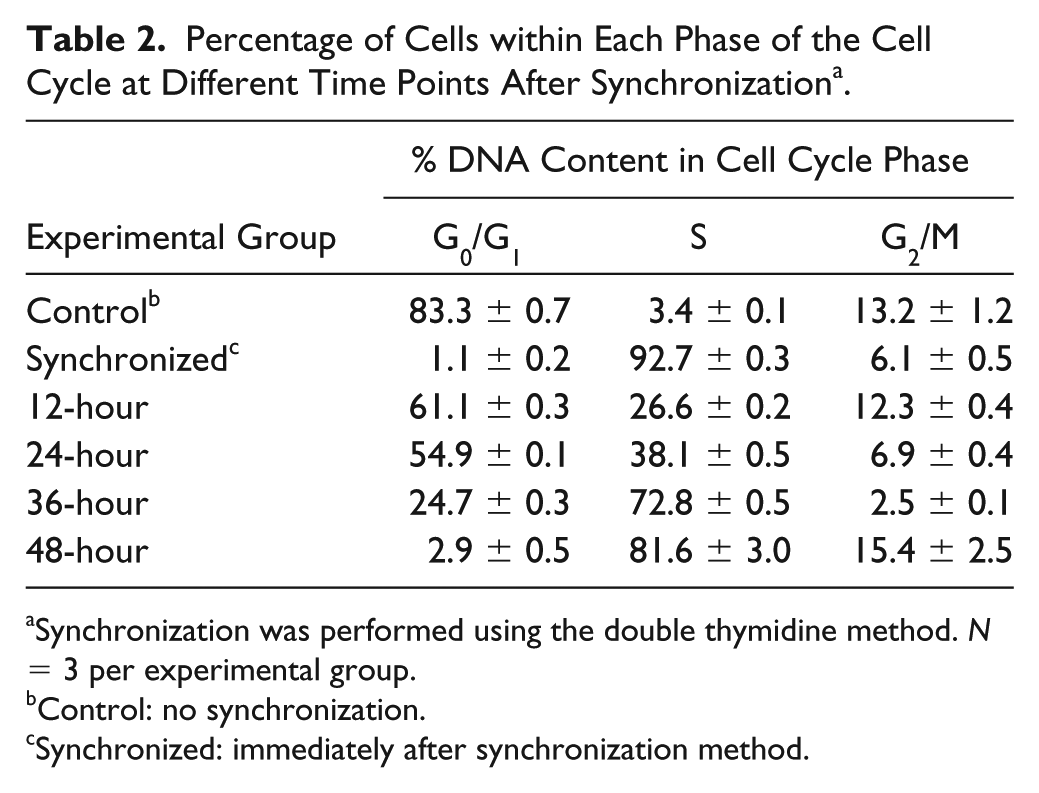
Synchronization was performed using the double thymidine method. N = 3 per experimental group.
Control: no synchronization.
Synchronized: immediately after synchronization method.
Effect of Cell Cycle Synchronization on Chondrogenesis
The biochemical composition and histological structure of the extracellular matrix accumulated within the alginate beads developed from unsynchronized and synchronized chondrocytes were analyzed and compared after 2 weeks and 4 weeks of culture. Amounts of DNA, sulfated GAG, and collagen (including comparison of type I and II collagen) were evaluated as primary markers of cartilaginous tissue synthesis.
While there were no significant differences in the biochemical composition between unsynchronized control and synchronized groups after 2 weeks of culture, the DNA, GAG, and collagen contents were significantly increased in the synchronized constructs compared with the unsynchronized controls after 4 weeks ( Fig. 2A - C , P < 0.05). Interestingly, the DNA content of the 4-week constructs were significantly lower than the 2-week constructs for both unsynchronized and synchronized groups ( Fig. 2A , P < 0.05). In contrast, GAG and collagen contents of the 4-week constructs were significantly higher than their respective 2-week constructs ( Fig. 2B and C , P < 0.05). When normalized to DNA content, the amount of GAG and collagen synthesis was similar in unsynchronized and synchronized groups at 2 weeks, but significantly increased with time in culture (4 weeks compared to 2 weeks) ( Fig. 2D and E , P < 0.05). The collagen-to-GAG ratio (which is often used as an index of chondrogenic growth) was significantly higher in the synchronized group compared with the unsynchronized control at 2 weeks, as well as in the 4-week constructs compared with their respective 2-week constructs ( Fig. 2F , P < 0.05).
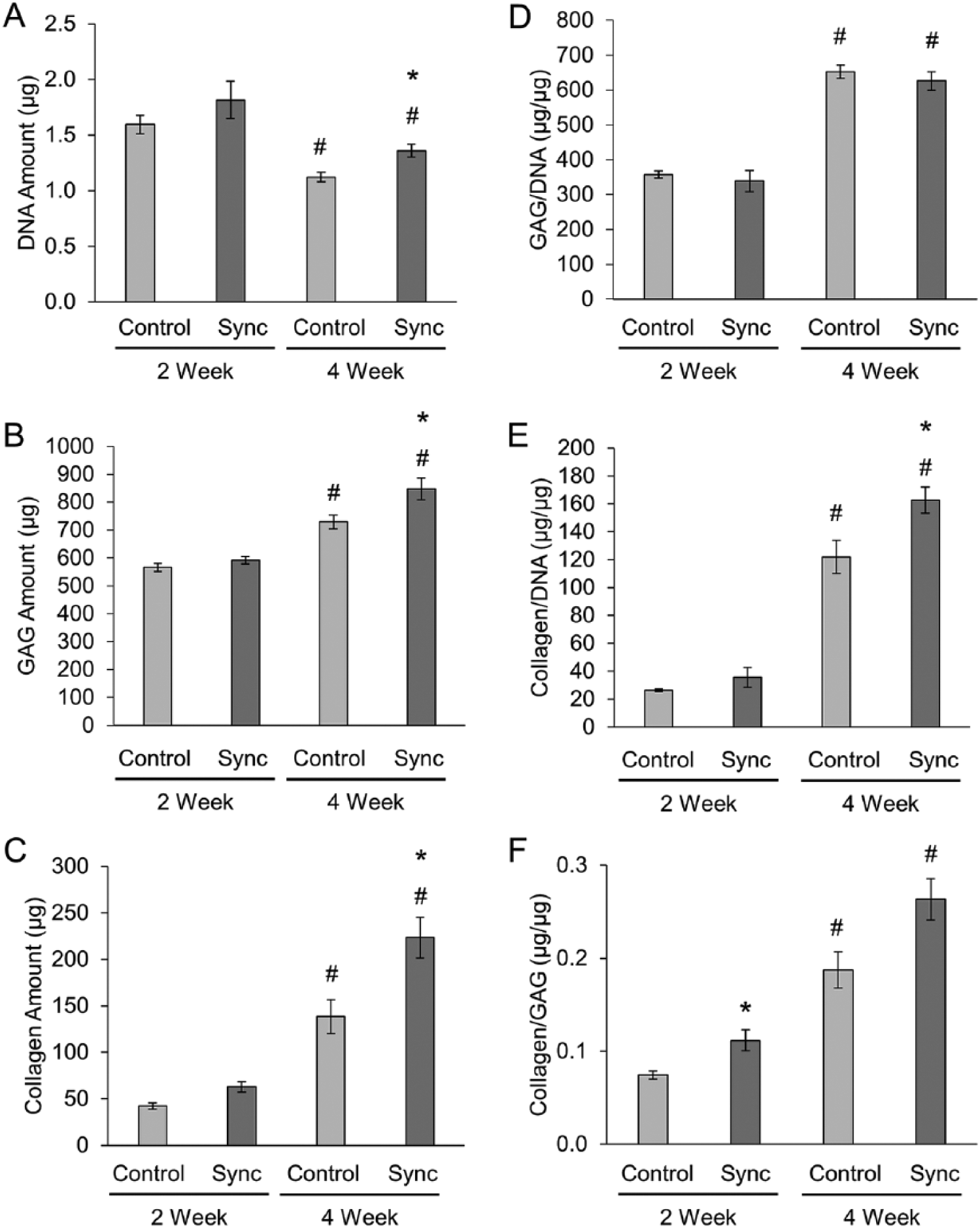
Biochemical properties of engineered cartilage constructs. (
Histological staining for sulfated proteoglycans and collagen displayed similar results. Safranin-O staining showed more accumulation of GAG in the extracellular matrix for the synchronized groups compared with the unsynchronized controls at both the 2-week and 4-week time points ( Fig. 3 , Table 3 ). Synchronized constructs generally appeared more cellular and showed increased presence of doublets, indicating active cellular division and increased formation of lacunae after longer times in culture (4 weeks compared with 2 weeks). Sirius red staining for collagen also showed similar results with increased collagen accumulation in the synchronized groups compared with the unsynchronized controls and increasing accumulation with time in culture ( Fig. 4 , Table 3 ). Immunohistochemical staining indicated that the collagen content within the accumulated extracellular matrix was primarily collagen II with negligible amounts of collagen I ( Fig. 5 , Table 3 ), thereby indicating a maintenance of chondrogenic phenotype irrespective of whether or not the cells were initially synchronized.
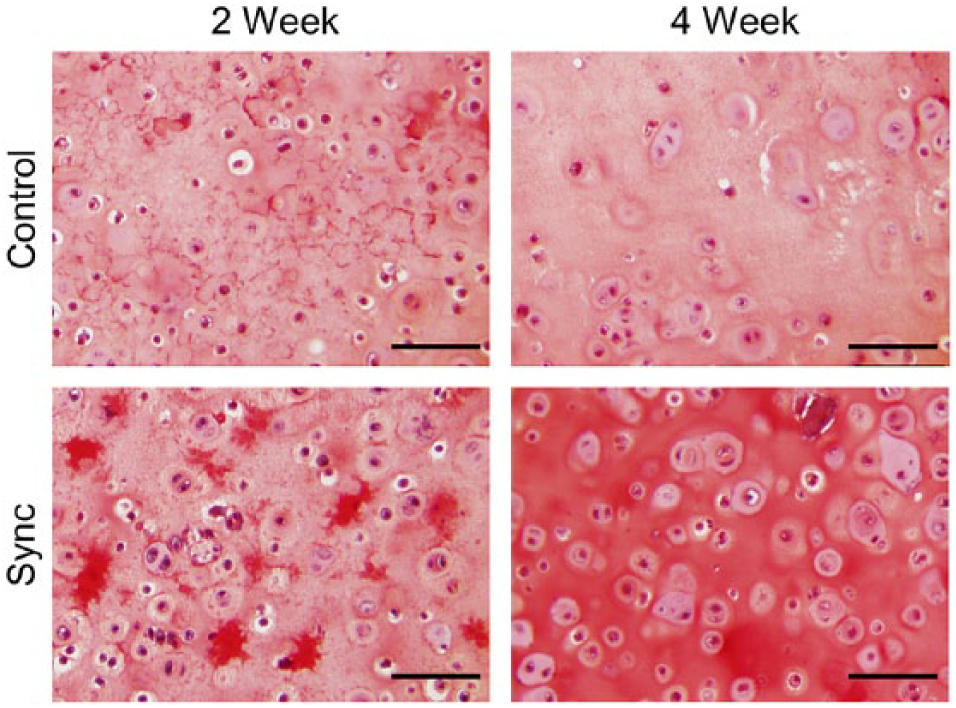
Representative microscopy images showing safranin-O staining of tissue constructs. Cartilage stained in red, nuclei stained in black, and background stained in bluish green. Scale bar = 100 µm. N = 6 per experimental group.
Grading Histological and Immunohistochemical Staining Using the Bern Score (Score Out of 9; n = 6 per Experimental Group).
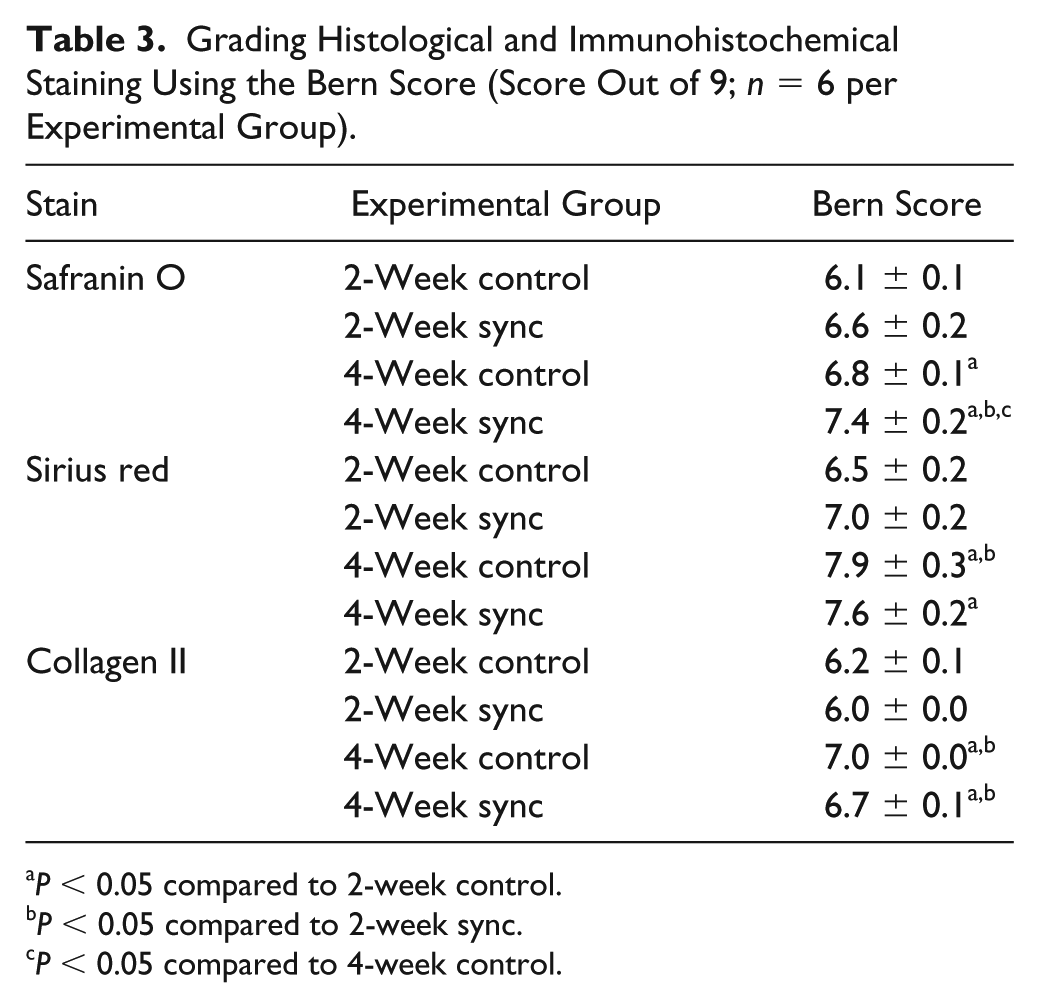
P < 0.05 compared to 2-week control.
P < 0.05 compared to 2-week sync.
P < 0.05 compared to 4-week control.
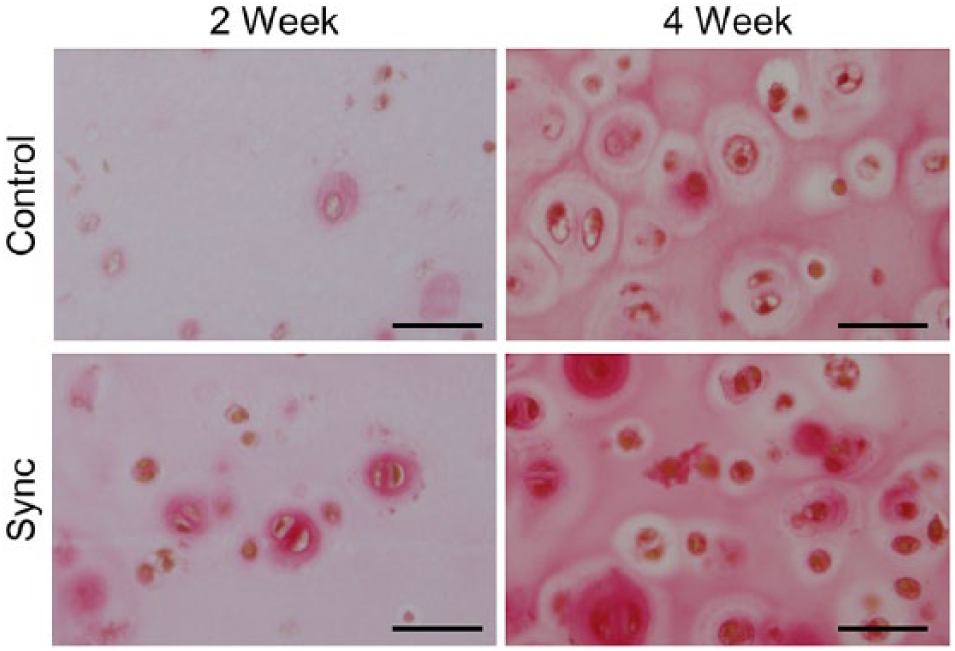
Representative microscopy images showing sirius red staining of tissue constructs. Collagen stained in red, nuclei stained in brown, and background stained in pale yellow. Scale bar = 100 µm. N = 6 per experimental group.
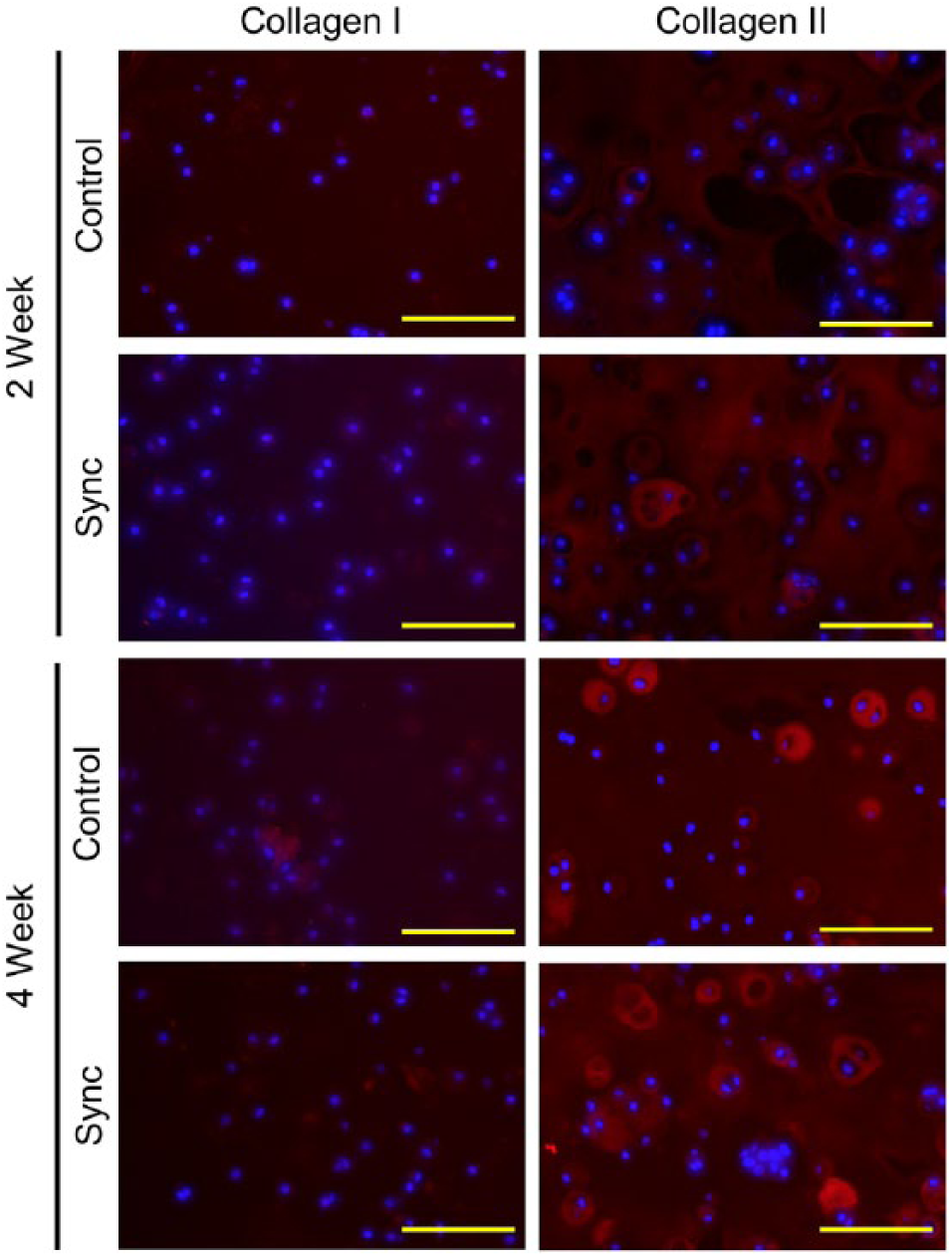
Immunohistochemical staining of collagen I and collagen II in tissue constructs. Collagen I and collagen II stained in red, and nuclei stained in blue. Scale bar = 100 µm. N = 6 per experimental group.
Discussion
As articular cartilage has a minimal capacity to repair itself, tissue engineering offers an alternative treatment option as tissue-engineered cartilage. However, it has been challenging to improve the deposition of cartilaginous extracellular matrix by the cells and, in turn, generate tissue with functional mechanical properties. Various studies have been performed to increase the deposition of cartilaginous extracellular matrix by articular chondrocytes, including growth factor stimulation,9,39 bioreactor cultivation,16,40 and/or application of mechanical stimuli.9-16 These approaches, however, tend to assume that the cultivated cells are a homogeneous population and hence would respond similarly to the imposed treatment(s). While there are several sources of inhomogeneity within the cell population, cells can also have varying properties due to their progression through the cell cycle. Similarly, it has been shown that cells synchronized within discrete phases of the cell cycle can respond more efficiently to biological treatments.23,25,41 As such, we hypothesized that pre-synchronization of isolated cells would facilitate improved tissue formation in vitro.
Cell cycle phase synchronization can be achieved by physical or chemical methods. 17 Physical methods (e.g., gradient centrifugation,42,43 membrane elution,44,45 fluorescence-activated cell sorting46,47) involve selection of cells based on their physical properties (size or density) and typically only result in the synchronization of a fraction of cells within the population. Alternatively, chemical synchronization methods rely on the reversible blocking of cells at a cell cycle phase by either deprivation or addition of chemical agents in the culture media during monolayer culture. Previous studies have achieved synchronization at different cell cycle phases by serum starvation (G1/G0) 25 or treatment with lovastatin (G1 phase), 48 aphidicolin or thymidine (S phase),26,28 and nocodazole or RO-3306 (G2/M phase).27,29 In this study, chemical synchronization methods were utilized due to their ability to yield higher levels of synchrony compared to physical methods. 48 However, traditional chemical synchronization methods cannot be directly translated to chondrocytes as these cells are known to dedifferentiate in monolayer culture. 24 For this reason, we adapted and evaluated different chemical synchronization techniques (serum starvation, thymidine, nocodazole, aphidicolin, and RO-3306) applied during the process of cell isolation from harvested cartilage. Among the evaluated techniques, the double thymidine method was found to yield the highest degree of cell synchrony without eliciting significant loss in chondrocyte viability. The other chemical agents investigated resulted in decreased cell viability and/or a lack of highly synchronized cell populations (within any phase) and were not investigated further.
Primary articular chondrocytes initially synchronized in S phase (by double thymidine method), and cultivated in alginate beads, led to the increased accumulation of cartilaginous extracellular matrix without observable changes in phenotype. Interestingly, this effect was sustained over long-term culture (4 weeks) even though the cells started to lose their synchrony at ~24 hours post encapsulation. The long-term effect on chondrogenesis, despite the cells losing their initial synchrony, may be attributed to the prolonged duration of the chondrocytes within S phase. Preliminary studies have shown that chondrocytes synchronized to S phase (in monolayer) are more responsive to growth factors compared with other phases of the cell cycle, resulting in enhanced chondrogenic tissue formation. 49 Chondrocytes express different transforming growth factor-β1 (TGF-β1) receptor systems depending on the cell cycle phase. 50 In particular, S phase synchronized cells express the lowest affinity TGF-β1 receptor (Kd = ~1100 pM).50,51 Similarly, transient exposure to TGF-β isoforms (~6 hours) can also enhance the endogenous expression of TGF-β peptides in S phase cells, thereby further magnifying the anabolic effects of endogenous released TGF-βs. 51 In general, transient exposure of chondrocytes to specific growth factors can have superior effects compared to sustained delivery.52,53 For example, while a sustained release of bFGF was shown to be more potent than bolus administration, the opposite has been observed for TGF-βs. 53 Last, TGF-βs have synergistic effects with other chondrogenic growth factors, such as bFGF 54 and IGF-1, 55 that also are endogenously expressed by chondrocytes. 56 Future work involving the quantification of growth factor expression at different time points will be performed to elucidate the mechanisms by which S phase synchronization increases extracellular matrix synthesis by primary articular chondrocytes.
The use of cell cycle synchronization to improve chondrogenic potential may also potentially be beneficial to improve the chondrogenic potential of passaged or osteoarthritic (OA) chondrocytes. In addition to the generally low biosynthetic rates of passaged and OA chondrocytes, these cells suffer from a loss of the characteristic chondrogenic phenotypic (e.g., dedifferentiation), thereby also requiring redifferentiation steps in order to produce cartilaginous tissue in vitro.39,57-59 As such, the cell synchronization approach developed in the present study may prove to enhance the responsiveness of these cells to redifferentiation methods and in turn enable the engineering of functional articular cartilage from these cells, which are either readily available (passaged chondrocytes) or found in abundance (OA chondrocytes).
Conclusions
In this study, we investigated the effects of cell cycle synchronization of primary articular chondrocytes on their potential to generate cartilaginous tissue in vitro. Different chemical blocking methods were evaluated to determine the most suitable cell cycle synchronization approach (thymidine, nocodazole, aphidicolin, and RO-3306). The double thymidine block method yielded the highest purity of cells synchronized in S phase. Although these cells became increasingly unsynchronized beyond 24 hours after synchronization, tissue constructs developed from these synchronized cells showed significantly higher amounts of cartilaginous extracellular matrix deposition (GAG and collagen II) as compared with constructs developed from unsynchronized cells after 4 weeks of cultivation. While the underlying mechanism(s) are unknown, we postulated that this effect may be due to the prolonged duration of the cells within S phase which influences the responsiveness to and release of specific chondrogenic growth factors.
Footnotes
Acknowledgments and Funding
The authors gratefully acknowledge Andjela Bajic for histological and immunohistochemical scoring. The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval was not sought for the present study because the tissue samples used in this study were purchased from a local abattoir.
Animal Welfare
Guidelines for humane animal treatment did not apply to the present study because the tissue samples used in this study were purchased from a local abattoir.