
Abstract
Oncostatin M (OSM) is an interleukin-6 (IL-6) family cytokine that has been implicated in a number of biological processes including inflammation, hematopoiesis, immune responses, development, and bone homeostasis. Recent evidence suggests that OSM may promote breast tumor invasion and metastasis. We investigated the role of OSM in the formation of bone metastases in vivo using the 4T1.2 mouse mammary tumor model in which OSM expression was knocked down using shRNA (4T1.2-OSM). 4T1.2-OSM cells were injected orthotopically into Balb/c mice, resulting in a greater than 97% decrease in spontaneous metastasis to bone compared to control cells. Intratibial injection of these same 4T1.2-OSM cells also dramatically reduced the osteolytic destruction of trabecular bone volume compared to control cells. Furthermore, in a tumor resection model, mice bearing 4T1.2-OSM tumors showed an increase in survival by a median of 10 days. To investigate the specific cellular mechanisms important for OSM-induced osteolytic metastasis to bone, an in vitro model was developed using the RAW 264.7 preosteoclast cell line co-cultured with 4T1.2 mouse mammary tumor cells. Treatment of co-cultures with OSM resulted in a 3-fold induction of osteoclastogenesis using the TRAP assay. We identified several tumor cell–induced factors including vascular endothelial growth factor, IL-6, and a previously uncharacterized OSM-regulated bone metastasis factor, amphiregulin (AREG), which increased osteoclast differentiation by 4.5-fold. In addition, pretreatment of co-cultures with an anti-AREG neutralizing antibody completely reversed OSM-induced osteoclastogenesis. Our results suggest that one mechanism for OSM-induced osteoclast differentiation is via an AREG autocrine loop, resulting in decreased osteoprotegerin secretion by the 4T1.2 cells. These data provide evidence that OSM might be an important therapeutic target for the prevention of breast cancer metastasis to bone.
Introduction
Breast cancer is the most commonly diagnosed cancer in women worldwide, with approximately 200,000 new cases being reported each year in the United States alone. 1 Seventy percent of breast cancer patients with advanced disease have bone metastasis. Although not a direct cause of death, these metastases often lead to weakening of weightbearing bones due to osteolytic lesions, resulting in severe pain, pathological fractures, and hypercalcemia. 2 While mechanisms of breast cancer progression have been researched extensively, metastasis specifically to osseous tissue is understudied, and therefore, the role of inflammatory factors in this process is not well understood. Recent data from human serum samples indicate that the promotion of metastasis to bone may be caused by inflammatory cytokines in the interleukin-6 (IL-6) family. 3
Oncostatin M (OSM) is a pleiotropic IL-6 family cytokine important in inflammation and other cellular processes such as development, hematopoiesis, liver function, neurogenesis, and bone homeostasis. 4 OSM activates the JAK/STAT, MAPK, and PI3K/AKT pathways via binding its receptors, OSM receptor β (OSMRβ) or leukemia inhibitory factor receptor β (LIFRβ) dimerized with a common gp130 subunit.5-8 During oncogenesis, OSM often acts as an antiproliferative factor for multiple types of cancers including multiple myeloma, lung cancer, and breast cancer.9,10 Paradoxically, OSM has also been shown to increase the proliferation of prostate and ovarian cancer cells, although this discrepancy is not well defined.11,12 Although OSM may promote or inhibit tumor cell proliferation, recent research suggests a contributing role for this cytokine in metastasis.13-16
In breast cancer, OSM has been shown by our group and others to 1) increase the transition of various cancer cell types from an epithelial to mesenchymal phenotype (EMT), 15 2) promote breast tumor cell-substrate detachment and invasion in vitro,13,14 3) upregulate proteases such as matrix metalloproteinases (MMPs) and cathepsins that degrade the local extracellular matrix (unpublished results),14,16 and 4) induce proangiogenic factors such as vascular endothelial growth factor (VEGF).17,18 In human tissue, OSM is expressed at low levels in normal mammary epithelial cells and at higher levels in ductal carcinoma in situ and invasive breast carcinoma (unpublished results). 19 High levels of OSM are also found in the breast tumor microenvironment in both tumor-associated macrophages and neutrophils that express and release OSM in response to breast carcinoma cells in vitro.20-22 These studies suggest a role for both autocrine and paracrine signaling by OSM in tumor cell detachment and invasion during early stages of the metastatic cascade. The role of OSM in bone colonization and osteolysis has not been addressed previously, but OSM, IL-6, and other gp130-related factors have been shown to regulate normal bone homeostasis.23-26
Bone homeostasis is maintained via a constant balance between mineralized bone formation and bone resorption. These processes are mediated primarily by the osteoblasts that synthesize mineralized bone and the osteoclasts that resorb bone. The role of OSM in maintaining normal bone homeostasis has been documented in several in vitro and in vivo models including OSM and OSMRβ knockout mice that show reduced osteoclast differentiation and activity.27-30 There is also evidence that OSM induces mineralization in mouse osteoblast cells 31 ; however, during disease states such as osteoarthritis and osteolytic bone metastasis, the balance of OSM and other IL-6/gp130 cytokines has been shown to be severely disrupted, often leading to bone degradation. 25
Breast cancer–derived bone metastasis leads to the dysregulation of IL-6, IL-11, and various growth factors such as the receptor activator of NFκB (RANKL), macrophage colony stimulating factor (MCSF), and VEGF by different cell types including osteoblasts, stromal cells, and immune cells.4,23,32,33 These factors promote osteoclast differentiation and activity, leading to increased bone resorption and compromised structural bone integrity. Currently, there are no published investigations addressing the in vivo role of OSM in breast cancer metastasis to bone.
To investigate the effect of OSM on bone metastasis, we utilized the 4T1.2 syngeneic mouse model of breast cancer. The 4T1.2 cell line 34 is a subclone of 4T1 cells that were originally derived from a spontaneous mammary tumor in a Balb/c/C3H mouse. 35 When injected orthotopically, 4T1.2 cells metastasize to bone as well as to the lung, adrenal glands, and lymph nodes, 34 a metastatic pattern similar to that seen in breast cancer patients. In this study, we generated modified cell lines by knocking down the expression of OSM (4T1.2-OSM) and show that a lack of OSM expression is sufficient to inhibit metastasis to bone from the primary mammary tumor as well as increase survival. Additionally, osteolysis observed after intratibial injection of 4T1.2-OSM cells is significantly reduced in mice compared to control cells. By treating co-cultures of 4T1.2 and RAW 264.7 cells (peripheral blood mononuclear cells [PBMCs], a cellular model of preosteoclasts) with OSM, we show that OSM-induced secretion of IL-6, VEGF, and amphiregulin (AREG) promotes osteoclastogenesis. Altogether, these findings indicate that targeting OSM expression and signaling provides a novel therapeutic approach for the treatment of metastatic breast cancer.
Results
OSM increases the invasive potential of mammary tumor cells in vitro
Previous results from our group indicate that OSM may play an important role in early stages of metastasis.13,14,20 Mouse mammary tumor 4T1.2 cells were characterized in vitro to establish them as a credible model for studying OSM signaling in vivo. First, we evaluated OSM and OSMRβ expression levels by comparing the highly metastatic 4T1.2 mammary tumor cells to a nonbone metastatic cell line, 66c14, also derived from the same Balb/c/C3H mammary tumor. 4T1.2 cells secreted 2-fold more OSM and expressed 2.5-fold more OSM receptor (OSMRβ) as compared to 66c14 cells (Fig. 1A and 1B). The treatment of 4T1.2 cells with OSM (25 ng/mL) resulted in a 20% reduction in adherent cells by day 10 (Fig. 1C, left) and a 60-fold induction of viable tumor cell detachment by day 8 (Fig. 1C, right). The decrease in adherent cell number could be explained by the increase in cell detachment or effects on proliferation.
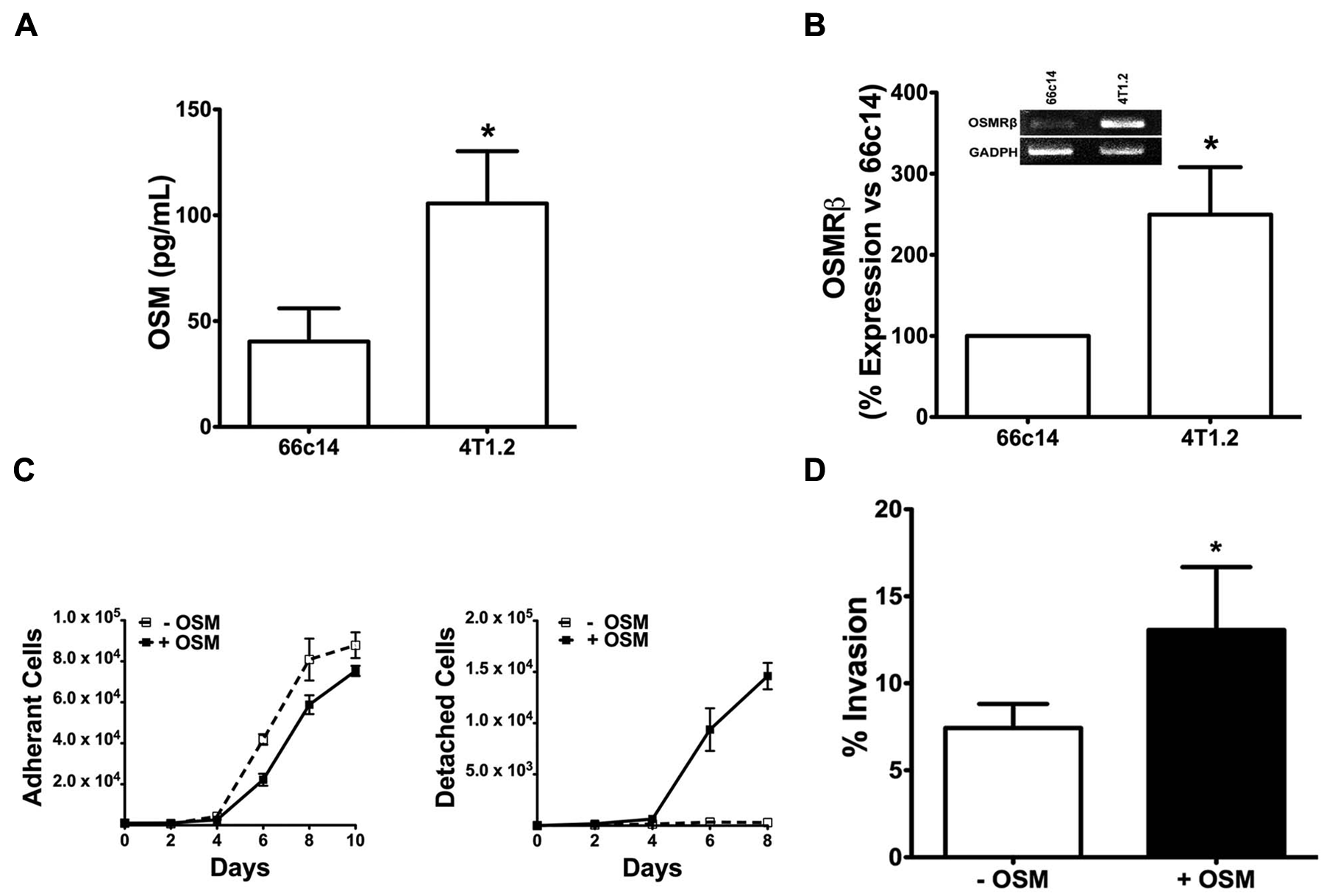
OSM-induced invasive potential of mouse mammary tumor cells in vitro. (
In addition, we observed an approximate 50% increase in 4T1.2 cell invasive potential after a 24-hour exposure to OSM using a Matrigel invasion assay (BD Biosciences, Bedford, MA) (Fig. 1D). The above experiments confirmed that OSM signaling is intact in 4T1.2 cells and that these cells can be used to study the contribution of OSM to metastasis to bone in vivo.
OSM is necessary for mammary tumor metastasis to bone
To examine whether OSM is required for mammary tumor metastasis to bone, we undertook stable knockdown of OSM expression (Fig. 2A, left). Three independent OSM shRNA sequences were cloned into the pSilencer4.1 vector and transfected into 4T1.2 mouse mammary tumor cells, and 2 clonal cell populations showed reduced OSM expression. Of the 2 clones, 4T1.2-OSM2 cells secreted the lowest levels of OSM (72% less OSM; 14.9 ± 2.3 pg/mL) by ELISA compared to 4T1.2-LacZ control cells (53.1 ± 8.8 pg/mL) (Fig. 2A), and 4T1.2-OSM1 cells secreted 30% less OSM (37.0 ± 5.9 pg/mL) (Fig. 2A). OSM mRNA expression levels were confirmed by semiquantitative RT-PCR (Fig. 2A, inset). To test the effects of OSM on mammary tumor metastasis in vivo, control 4T1.2-LacZ, 4T1.2-OSM1, and 4T1.2-OSM2 cells were injected orthotopically into the mammary fat pads of Balb/c mice. As OSM has been shown to have antiproliferative effects on breast cancer cells in vitro,9,10 we hypothesized that low levels of OSM should result in increased tumor growth in vivo. Injection of 4T1.2-OSM2 cells, which displayed the largest decrease in tumor cell–secreted OSM, led to a significant increase in average tumor growth (1,628 mm3 for 4T1.2-OSM2 cells at day 30) (Fig. 2B) compared to control 4T1.2-LacZ cells (860 mm3 at day 30) (Fig. 2B) and parental 4T1.2 cells (870 mm3 at day 30) (data not shown). Injection of 4T1.2-OSM1 cells, which displayed a modest decrease in tumor cell–secreted OSM, resulted in tumor growth similar to control cells (861 mm3 at day 30) (Fig. 2B).
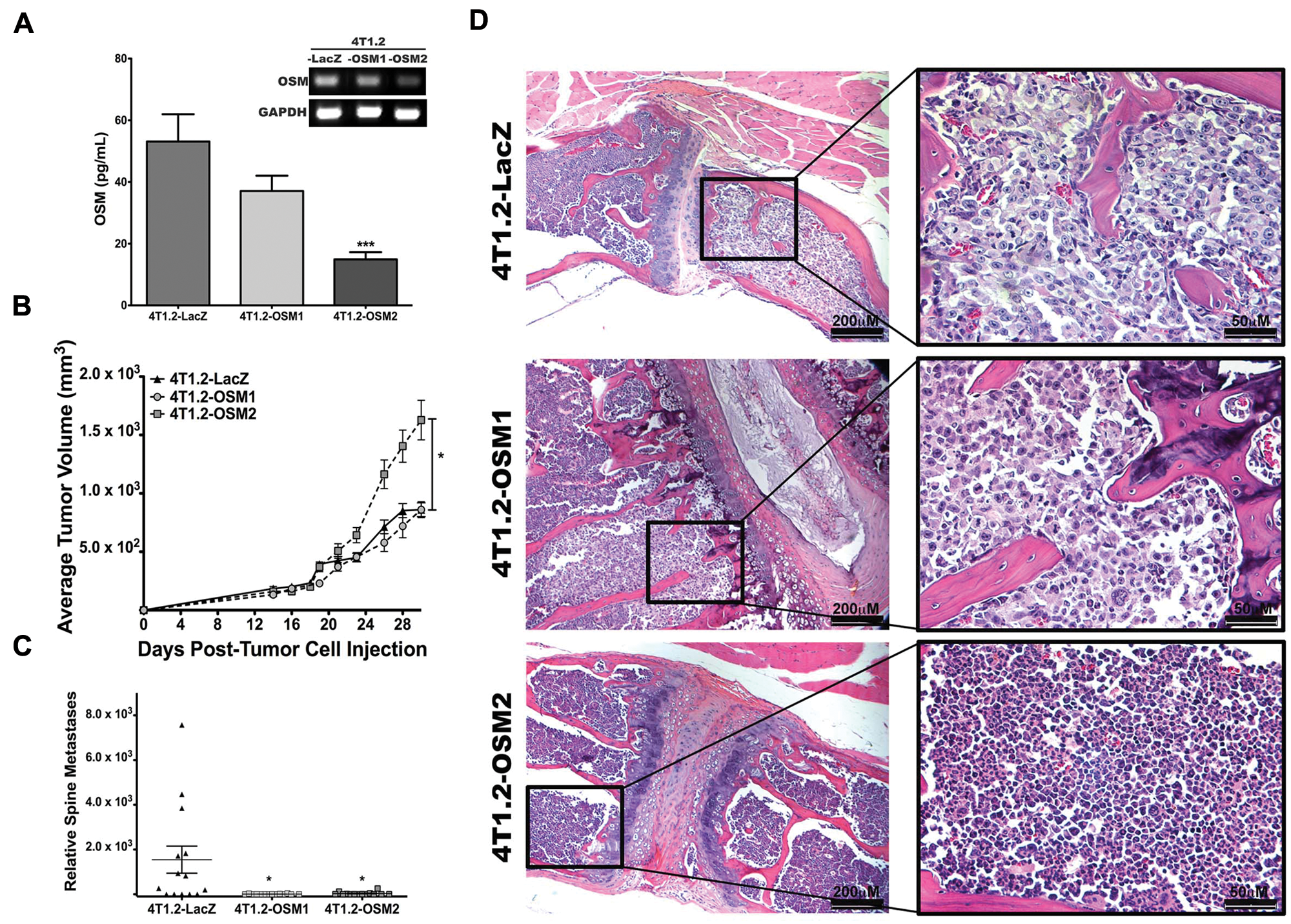
4T1.2 cells with reduced OSM expression are less metastatic to bone. (
Metastasis to bone in mice injected with control 4T1.2-LacZ, 4T1.2-OSM1, and 4T1.2-OSM2 cells was quantified by qPCR, which measures the amount of vector DNA contained in all 3 cell types. Mean bone metastatic burden in the spine was significantly lower in mice that received 4T1.2-OSM1 or 4T1.2-OSM2 cells compared to 4T1.2-LacZ control cells (Fig. 2C). Histological evaluation of vertebrae by H&E staining revealed the presence of metastatic tumor cells in the bone marrow of mice injected with 4T1.2-LacZ control cells but not in the bone marrow of mice injected with 4T1.2-OSM2 cells (Fig. 2D). After careful inspection, a small intraosseous metastasis was identified in vertebrae from a mouse injected with 4T1.2-OSM1 cells, explainable by the fact that these cells did not demonstrate as robust an OSM expression knockdown as the 4T1.2-OSM2 cells. Overall, these results suggest that tumor cell–expressed OSM is necessary for mammary tumor metastasis to bone.
In addition to the increased tumor growth and decreased metastasis to bone observed in mice injected with 4T1.2-OSM2 cells in vivo, these cells also demonstrate decreased invasive potential in vitro. Specifically, 4T1.2-OSM2 cells displayed an increase in adherent cell number over time (data not shown), a decrease in viable cell detachment (Suppl. Fig. S1A), and a decrease in invasive potential (Suppl. Fig. S1B) compared to 4T1.2-LacZ control cells. These results demonstrate that 4T1.2-OSM2 cells display a reduced invasiveness in vitro and provide an appropriate model for subsequent in vivo experiments.
OSM promotes mammary tumor cell–mediated osteolysis in vivo
4T1.2-OSM2 and control cells were injected into the tibia of Balb/c mice. Legs excised 4 weeks after the injection of control 4T1.2-LacZ cells showed significant osteolysis in the proximal end of the tibia, as analyzed by micro–computed tomography (micro-CT). The same region in mice injected with 4T1.2-OSM2 cells showed less osteolysis, more closely resembling PBS-injected animals (Fig. 3A); however, a mixed osteolytic/osteoblastic phenotype was seen. Analysis of trabecular bone revealed extensive osteolysis due to 4T1.2-LacZ cells, prohibiting 3-dimensional reconstruction of the region of interest (ROI) in all but 1 animal (Fig. 3B, top, and Suppl. Fig. S2A). Mice injected with 4T1.2-OSM2 cells displayed a trabecular bone morphology more similar to that of PBS-injected mice (Fig. 3C, top, and Suppl. Fig. S2A). A similar pattern was observed in the cortical bone (Fig. 3B, bottom, and Suppl. Fig. S2B). Quantitative analysis of trabecular and cortical bone was performed using ROIs (50 × 10 µm slices). Mice injected with 4T1.2-OSM2 cells displayed a dramatically greater total trabecular bone volume and trabecular thickness when compared to control 4T1.2-LacZ cells (Fig. 3C). Both cortical bone volume/total volume and cortical bone mineral density were significantly higher in 4T1.2-OSM2 cell–injected mice as compared to mice injected with control cells (Fig. 3C). Overall, these quantitative results indicate that OSM expression is important for osteolytic activity of 4T1.2 cells in the bone microenvironment.
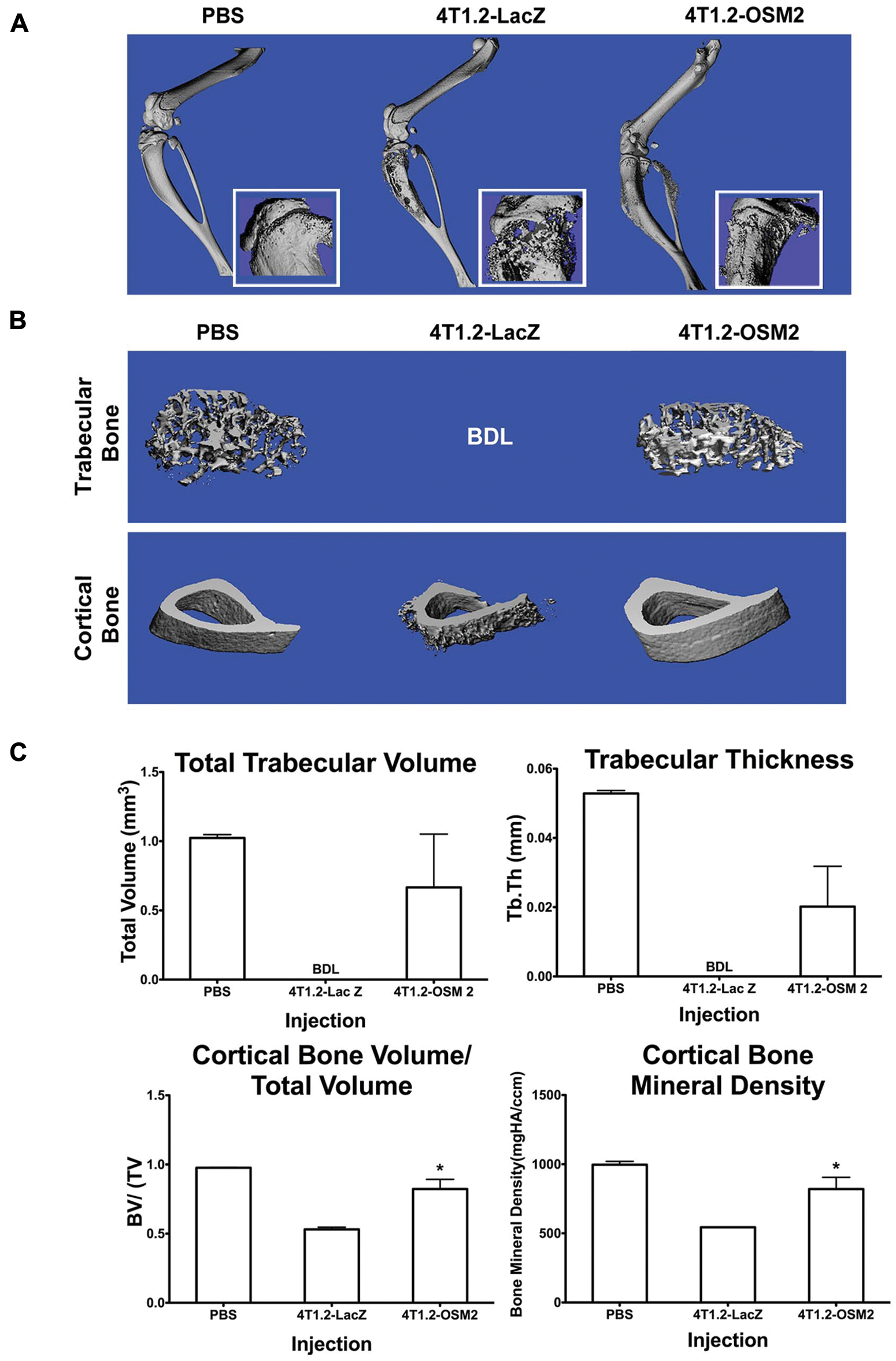
Reduced OSM expression by 4T1.2 cells protects against bone osteolysis. (
Lack of mammary tumor cell–derived OSM increases survival in vivo
To determine the influence of decreased OSM expression on bone metastasis independent of its effects on primary tumor growth, we utilized a tumor resection model. Orthotopic mammary fat pad injections were performed using control 4T1.2-LacZ and 4T1.2-OSM2 cells, primary tumors were resected at day 14 (Fig. 4A), and mice were monitored until end-point criteria were met (see Materials and Methods). The survival time for mice that received 4T1.2-OSM2 cell injections significantly increased by a mean of 10 days and a maximum of 42 days compared to control 4T1.2-LacZ cells (Fig. 4B). In addition, mice injected with 4T1.2-OSM2 cells had no detectable metastases in the spines, as analyzed by qPCR, whereas all of the mice injected with 4T1.2-LacZ cells demonstrated high levels of metastases in the spine (Fig. 4C). Lung metastases were detectable in both the 4T1.2-LacZ and 4T1.2-OSM2 groups of mice at the time of sacrifice; however, the time for maximum metastasis to the lungs was significantly delayed in the 4T1.2-OSM2–injected group (data not shown). These results indicate that following primary tumor resection, decreased OSM expression in mammary tumor cells delayed bone metastasis and increased the animal survival rate.
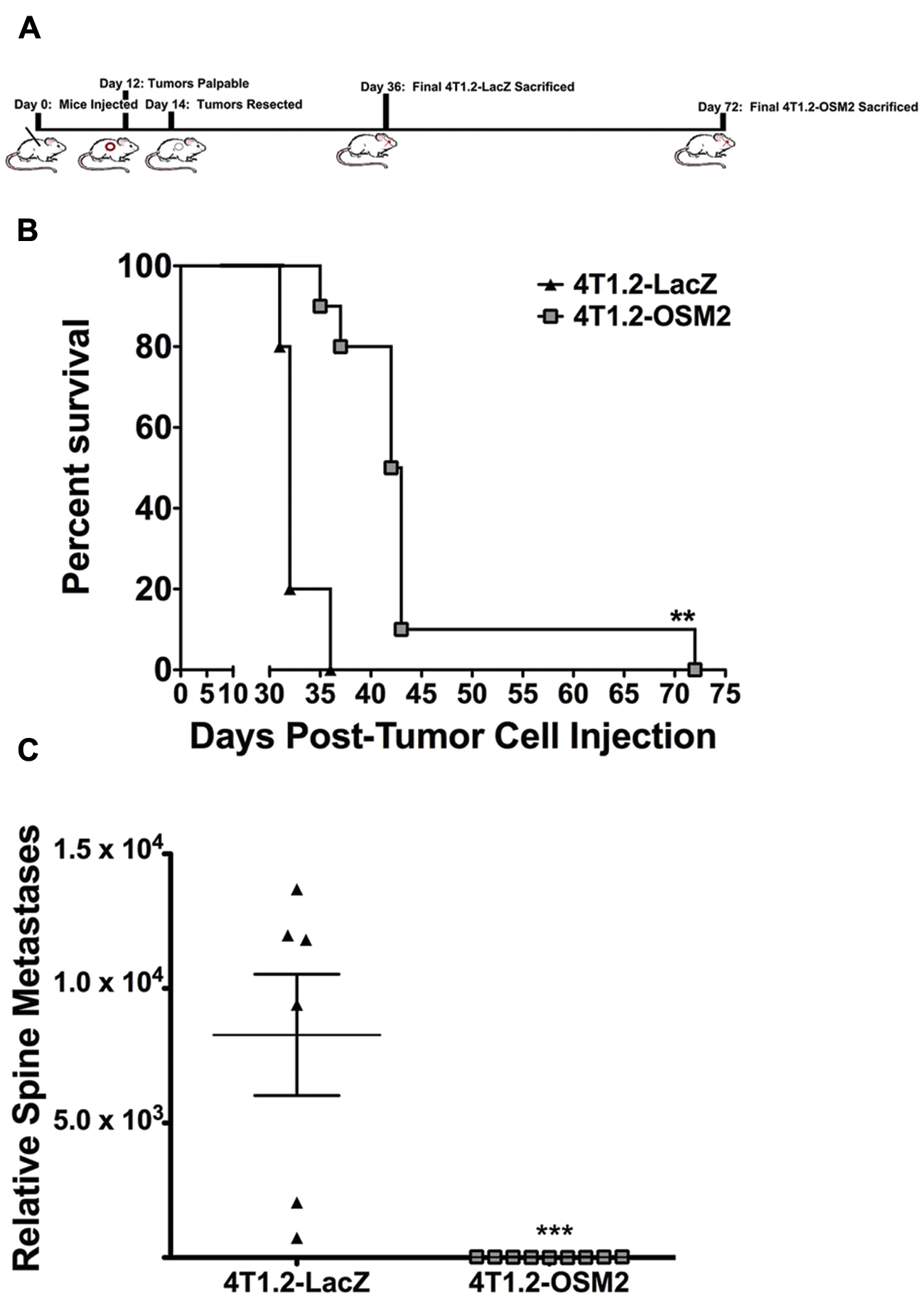
Reduced OSM expression by 4T1.2 tumors increases survival. (
OSM induces tumor cell–mediated osteoclastogenesis
To identify the mechanism by which mammary tumor cell–derived OSM causes osteolysis, an in vitro model of osteoclastogenesis mimicking PBMC-derived osteoclasts was employed using RAW 264.7 mouse monocytic cells co-cultured with 4T1.2 cells. The capacity of each cell type to respond to OSM and other OSM-related factors was analyzed by semiquantitative RT-PCR. Both the RAW 264.7 cells, which are able to undergo osteoclast differentiation, and the 4T1.2 mouse mammary tumor cells expressed the gp130 subunit necessary for all IL-6 family receptors (Fig. 5A). 4T1.2 cells expressed OSMRβ, LIFRβ, and IL-6R, while RAW 264.7 cells expressed IL-6R and barely detectable levels of LIFRβ (Fig. 5A). Therefore, in our in vitro model of osteoclastogenesis, we predicted that the 4T1.2 cells should respond to OSM, LIF, and IL-6 and that the RAW 264.7 cells should not respond to OSM.

OSM induces 4T1.2 tumor cell–mediated osteoclastogenesis in vitro. (
The ability of 4T1.2 cells to induce osteoclastogenesis in response to OSM was assessed. A 7.5-fold increase in TRAP-positive (TRAP+) cells, a marker of osteoclast differentiation, was observed in OSM-treated RAW 264.7 and 4T1.2 co-cultures compared to untreated co-cultures in the presence of 5 ng/mL MCSF and 10 ng RANKL (Fig. 5B). To demonstrate the importance of OSM-mediated secreted factors from 4T1.2 cells, conditioned medium was collected from 4T1.2 cells treated with or without OSM and applied to RAW 264.7 cells. An approximate 3-fold increase in TRAP+ osteolcasts was observed in RAW 264.7 cells cultured with conditioned media from OSM treated 4T1.2 cells compared to conditioned medium from untreated 4T1.2 cells. (Fig. 5B). No significant difference in the level of osteoclastogenesis was observed when RAW 264.7 cells alone were treated with OSM (Fig. 5B).
Mammary tumor cells secrete pro-osteoclastogenic factors in response to OSM
To identify which 4T1.2 cell–secreted pro-osteoclastogenic factors were increased in response to OSM, ELISA and Western blot analyses were performed. No difference in the levels of 4T1.2 cell–derived RANKL, MCSF, transforming growth factor β (TGFβ), or tumor necrosis factor α (TNFα) was observed after treatment with OSM (Suppl. Table S2). However, levels of IL-6 and VEGF, whose receptors are both present on RAW 264.7 cells (Fig. 5A), were significantly increased in response to OSM (Fig. 5C and 5D). 4T1.2 cells alone, as well as RAW 264.7 and 4T1.2 cell co-cultures, showed a 3- to 4-fold increase in secreted IL-6 levels when treated with OSM (Fig. 5C). RAW 264.7 cells alone did not show a significant increase in IL-6 in response to OSM (Fig. 5C). VEGF secretion by 4T1.2 cells was increased 2-fold following treatment with OSM versus untreated cells (Fig. 5D). Secreted VEGF levels increased 7-fold in treated co-cultures compared to untreated 4T1.2 cells, suggesting that in addition to OSM treatment, a direct interaction between the 2 cell types may cause an increase in VEGF.
OSM-induced AREG in mammary tumor cells increases osteoclastogenesis
AREG, a member of the epidermal growth factor (EGF) family and previously unknown to be induced by OSM, was identified by proteome array analysis (R&D Systems, Minneapolis, MN) of conditioned medium from 4T1.2 cells treated with OSM (see Supplementary Materials and Methods). A greater than 2-fold increase in secreted AREG was verified by ELISA in conditioned medium collected from OSM-treated 4T1.2 cells as compared to untreated cells (Fig. 6A, left). Co-culturing RAW 264.7 cells with 4T1.2 cells had no effect on OSM-induced AREG secretion levels, and RAW 264.7 cells alone did not increase AREG secretion in response to OSM (Fig. 6A, left). To determine whether AREG plays a direct role in osteoclastogenesis in our in vitro model, recombinant AREG (50 ng/mL) was added to co-cultures of RAW 264.7 and 4T1.2 cells in the presence of 5 ng/mL MCSF and 10 ng RANKL. AREG supplementation increased the number of TRAP+ osteoclasts by 2-fold in these co-cultures, and the addition of OSM with AREG further increased the number of TRAP+ osteoclasts by nearly 9-fold compared to untreated controls (Fig. 6A, right). AREG showed a minimal effect on osteoclastogenesis in cultures of RAW 264.7 cells alone (Fig. 6A, right). Inhibition of AREG signaling using a neutralizing antibody in RAW 264.7 and 4T1.2 cell co-cultures resulted in a near complete suppression of OSM-mediated osteoclastogenesis (Fig. 6B, left). No significant effect was seen in RAW 264.7 cultures (Fig. 6B, left). A combination of anti–IL-6, anti-VEGF, and anti-AREG antibodies did not significantly reduce osteoclastogenesis beyond co-cultures treated with anti-AREG only (Fig. 6B, right). Anti–IL-6 or anti-VEGF antibodies alone had less effect on the inhibition of OSM-induced osteoclastogenesis in co-cultures than anti-AREG (Fig. 6B, right). Thus, OSM-induced AREG appears to be a key player in mammary tumor cell–induced osteoclastogenesis in vitro.
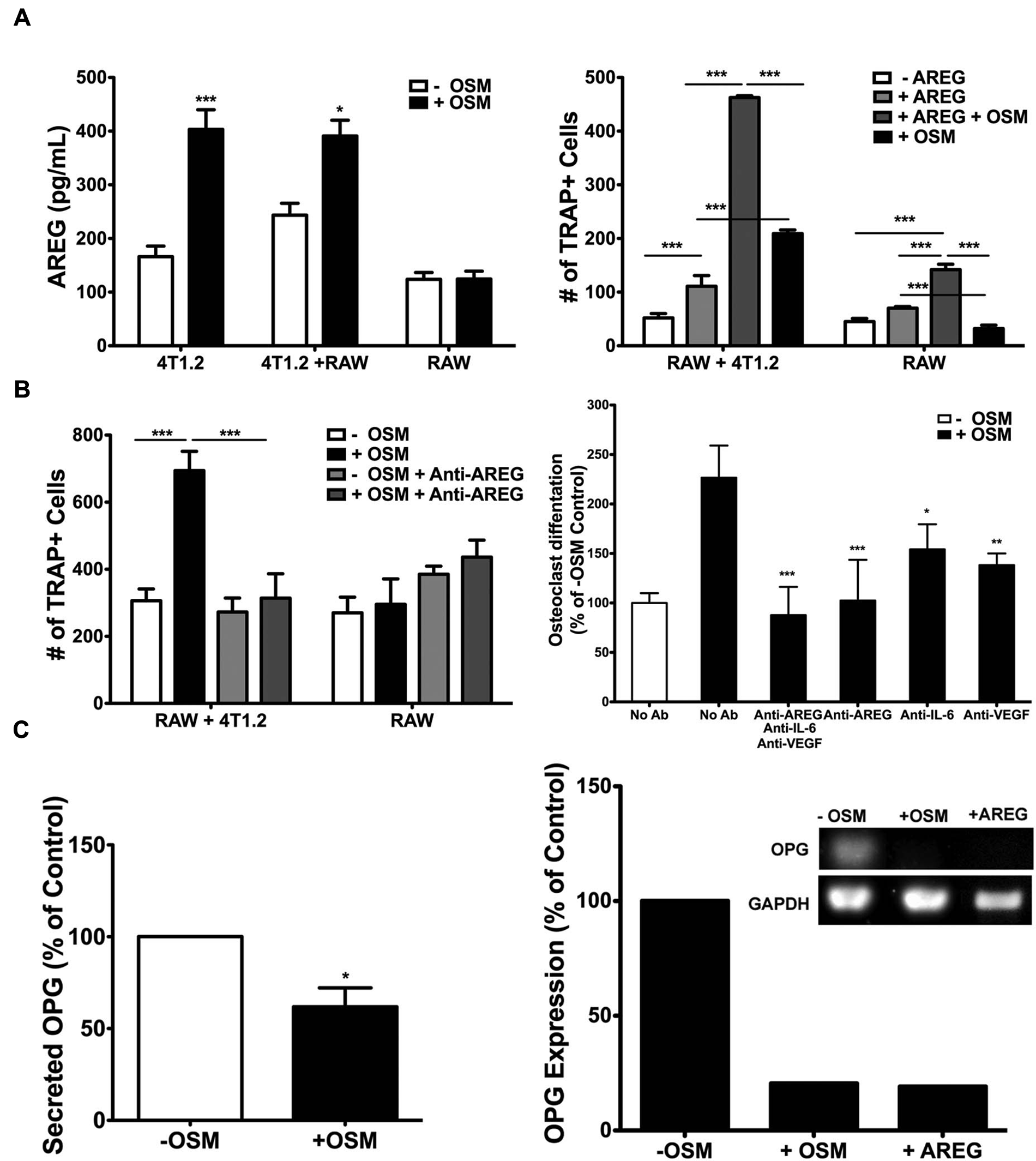
OSM-induced AREG secretion drives 4T1.2 cell–mediated osteoclastogenesis in vitro. (
OSM inhibits osteoprotegerin expression in tumor cells
Osteoprotegerin (OPG), the decoy receptor for RANKL, is an important endogenous inhibitor of osteoclastogenesis. Preliminary data from our group have shown that expression of OPG is decreased by RT-PCR in a panel of human breast cancer cells treated with OSM (data not shown). Therefore, we investigated the effect of OSM on OPG expression by 4T1.2 mouse mammary tumor cells by both RT-PCR and ELISA. OSM treatment of 4T1.2 cells resulted in a 40% reduction in secreted OPG levels by ELISA (Fig. 6C, left) and an 80% reduction in mRNA expression by RT-PCR (Fig. 6C, right). Interestingly, the addition of recombinant AREG (25 ng/mL) also resulted in an 80% decrease in OPG expression (Fig. 6C, right), suggesting that OSM-mediated osteoclastogenesis may act through AREG suppression of OPG.
Discussion
This study provides evidence that tumor cell–secreted OSM promotes osteolytic breast cancer metastasis to bone. Our data indicate that OSM is important in late stages of the metastatic process but may also be necessary for earlier stages. We show specifically that 1) OSM induces 4T1.2 mouse mammary tumor cell detachment and invasion in vitro, 2) mice orthotopically injected with 4T1.2 tumor cells with reduced OSM expression exhibit decreased bone metastases and increased survival, 3) intratibial injections of the above 4T1.2 cells in mice result in decreased osteolysis, and 4) a novel mechanism for OSM promoting tumor cell–secreted AREG during osteoclast differentiation was identified.
The syngeneic 4T1.2 mouse model is the only reliable model for spontaneous mammary tumor metastasis to bone currently available. Intracardiac or intratibial injections of osteolytic human breast cancer cells into immunocompromised athymic mice to study the role of OSM in bone metastasis are problematic due to a lack of homology between human and mouse OSM signaling 36 as well as the need for intact immune function to adequately study the effect of an inflammatory cytokine on metastasis. Using our 4T1.2-OSM2 orthotopic mouse model, we observed a dramatic decrease in bone metastasis despite increased tumor growth. A dual role as an antiproliferative and yet prometastatic factor in breast cancer metastasis has been shown with other breast cancer cell–secreted factors such as transforming growth factor β (TGFβ). TGFβ is not regulated by OSM in 4T1.2 cells (Suppl. Table S2) but is known to increase invasion and metastasis of breast tumors along with its tumor cell growth inhibitory effects. 37 To date, this is the first study demonstrating an independent and yet similar role for OSM driving breast cancer metastasis to bone, despite inhibitory effects on primary tumor growth.
Bone is the most common site of metastatic disease in breast cancer patients, 1 although metastasis also occurs to the lung, liver, adrenal glands, brain, kidney, spleen, heart, and lymph nodes. Specific quantification of metastatic burden by qPCR was conducted for the spine and lung only (data not shown) (Fig. 2C), and both were significantly reduced in mice orthotopically injected with 4T1.2-OSM1 and 4T1.2-OSM2 cells. By utilizing a tumor resection model, we were able to increase the time it took to reach overall maximum metastatic burden and identify other important metastatic sites modulated by OSM. Upon dissection of these mice at maximum metastatic end point, sites with visual metastases, in addition to the spine, were the lung, liver, and lymph nodes. A reduction in the number of lung metastases or prolongation of the time to lung metastasis development may explain the significant increase in survival seen in mice that had resected primary tumors with reduced OSM expression. Therefore, the lung was identified as another major metastatic site affected by OSM and is being investigated further. Overall, OSM is likely critical for both late as well as early stages of metastasis, resulting in an increase in total metastatic burden. However, our data suggest a more significant role for OSM during later steps of the metastatic process, specifically in the bone microenvironment.
4T1.2-OSM2 tumor cells injected directly into the bone microenvironment of the tibia revealed that tumoral OSM expression is critical for induction of osteolysis in both trabecular and cortical bone. In general, 4T1.2-OSM2 cell–injected mice showed a mixed osteolytic/osteoblastic phenotype, similar to what is seen in human metastatic breast cancer patients. 38 Previous studies analyzing intrafemoral injection of 4T1 cells (the parental cells for the 4T1.2 subclone used in this article) show only osteolytic activity by micro-CT. 39 This article is the first study presenting micro-CT data from intratibial injections of the 4T1.2 subclone. 4T1.2 cells injected into the mouse tibia resulted in some osteoblast activity, although overall, the osteolytic phenotype was predominant (Suppl. Fig. S2). OSM has been shown previously to play a dual role in the normal bone microenvironment, stimulating both bone resorption as well as bone formation. Specifically, OSM has been shown to promote osteoclastogenesis and osteoclast activity by STAT3-induced RANKL expression in osteoblast/stromal cells 40 yet will stimulate osteogenesis in mesenchymal stem cells and mineralization in murine osteoblast cells.31,40 The role of OSM in osteoblastogenesis and mineralization in our breast cancer metastasis model is currently being conducted. However, we have measured osteoclast activity in vitro using a synthetic bone resorption assay, and increased bone resorption was detected using a mixed population of mouse primary preosteoclasts and preosteoblasts cultured in conditioned medium from 4T1.2 cells treated with OSM (data not shown). Increased osteolysis in the animals injected with 4T1.2-LacZ cells compared to parental 4T1.2 cells was also noted (Suppl. Fig. S2). siRNA/shRNA technology has been shown to cause generally unavoidable off-target effects related to immune system activation, as the siRNA machinery is physiologically associated with viral infections41,42 that can potentially lead to aberrant osteoclastogenesis and osteolysis. Overall, our studies suggest that tumor cell–derived OSM leads to an increase in both osteoclast differentiation and activity that result in osteolytic bone lesions.
Using an in vitro co-culture system of osteoclast differentiation, this work presents evidence for several OSM-induced tumor-secreted factors that could mediate the observed in vivo osteolysis. The RAW 264.7 mouse monocytic cell line was chosen as a source of preosteoclast cells in our in vitro model of tumor cell/bone microenvironment interaction instead of bone marrow cells derived from Balb/c mice. This decision was based on recent studies suggesting that PBMCs that include monocytes are a major source of osteoclasts in pathological osteolysis. PBMCs isolated from patients with bone metastases have been shown to undergo spontaneous osteoclastogenesis,43-45 while similar effects with bone marrow cells isolated from cancer patients have not been reported. Furthermore, we were able to maximize OSM-induced effects on osteoclastogenesis in our co-culture system by the addition of very low levels of RANKL and MCSF to all the assays. The low level of these key osteoclast-promoting factors resulted in the formation of predominantly mononuclear TRAP+ cells, which nonetheless had direct osteolytic activity (data not shown). 46 The significant increase in RAW 264.7 TRAP+ cells in OSM-treated co-cultures with 4T1.2 cells or conditioned media from 4T1.2 cells treated with OSM was mediated in part by IL-6 and VEGF. Both IL-6 and VEGF have been shown by our group and others to promote osteoclast differentiation and activity,47,48 and VEGF expression has been detected in osteolytic bone metastases. 49 To identify novel OSM-induced factors, a proteome array analysis of conditioned medium from 4T1.2 cells treated with OSM was performed, and AREG was identified. AREG is membrane bound until cleaved by the protease ADAMTS1 (a disintegrin and metalloproteinase with thrombospondin motifs 1), the expression of which was also induced by OSM in 4T1.2 cells (Suppl. Table S2). AREG has been associated with several mechanisms of breast cancer progression including tumor cell proliferation and the expression of proteases such as MMP2 and MMP9 50 and is expressed at moderate to high levels in human infiltrating breast carcinoma.51,52
The role of AREG in breast cancer metastasis to bone has not been studied extensively. AREG has been shown to increase the expression of parathyroid hormone–related protein (PTHrP) from breast cancer cells in vitro; however, in studies of human breast cancer bone metastases, it is unclear as to whether PTHrP expression drives the metastasis to bone or the bone microenvironment induces PTHrP expression. 53 In our 4T1.2 co-culture model, OSM-induced AREG did not promote the expression or secretion of PTHrP by RT-PCR and ELISA (Suppl. Table S2) but instead resulted in the decreased secretion of the decoy receptor for RANKL, OPG. This is the first time that AREG and OSM have been shown to decrease OPG secretion from tumor cells, although a previous study demonstrated that AREG inhibits OPG secretion by osteoblasts, the primary source of this factor. 54 AREG is known to bind to the epidermal growth factor receptor (EGFR or ErbB1) for downstream signaling; however, a recent publication and our Western blot analysis indicate that 4T1.2 cells do not express this receptor. 55 With 4T1.2 cells, it is possible that AREG binds an alternative receptor such as ErbB3/HER3 56 to inhibit OPG expression. OPG is associated with decreased osteoclastogenesis, and increased OPG expression in breast cancer cells has been shown to protect against osteolytic tumor growth in bone. 57 A proposed model summarizing our in vitro results is illustrated in Figure 7 in which OSM induces VEGF, IL-6, and AREG secretion from breast tumor cells. Secreted VEGF and IL-6 bind to preosteoclasts to promote osteoclast differentiation, while AREG signals tumor cells to inhibit the production of the antiosteoclastic factor OPG.
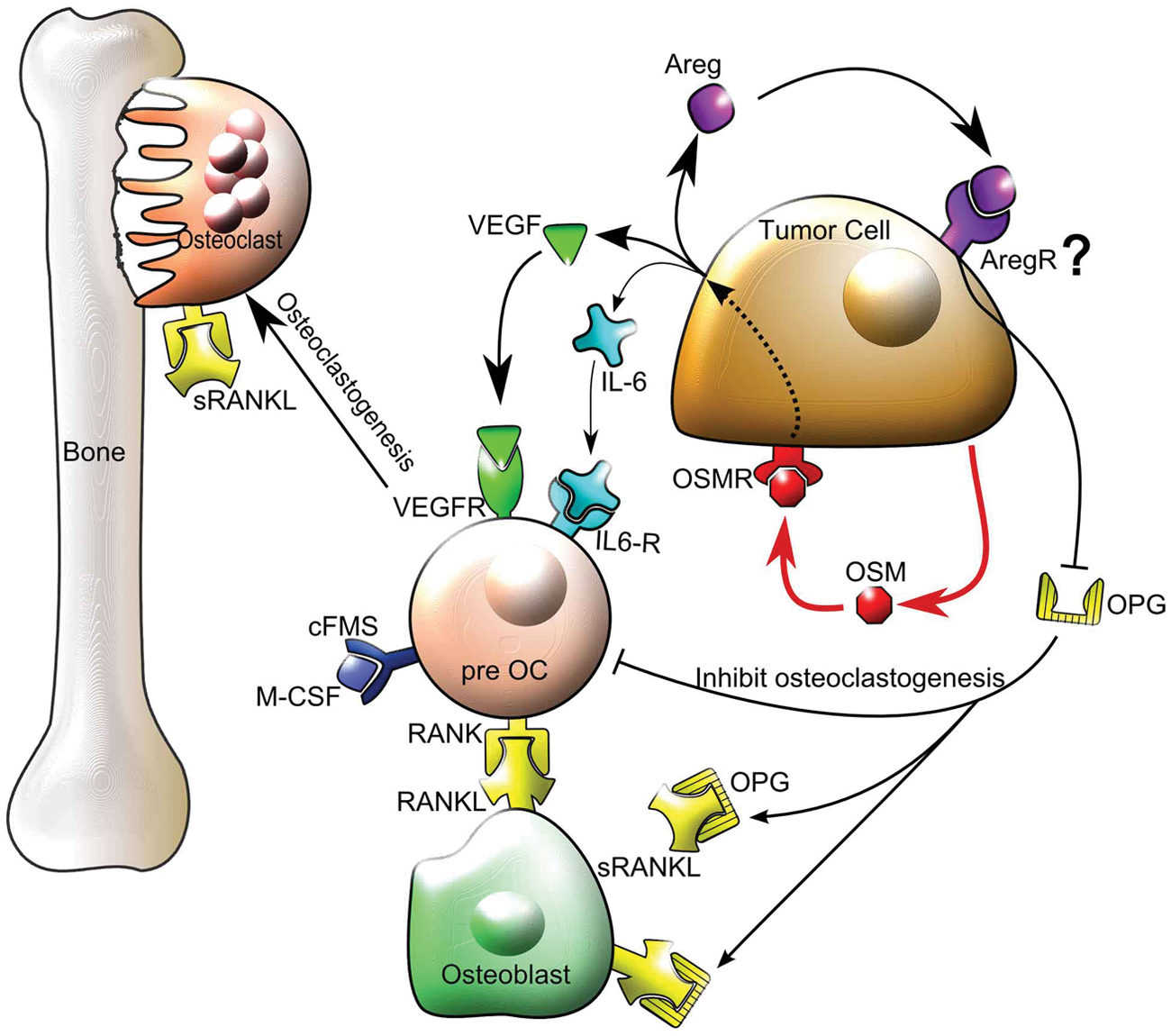
Proposed model for the mechanism of OSM in tumor cell–mediated osteoclastogenesis. OSM increases VEGF and IL-6 secretion from tumor cells that then bind to preosteoclasts and mature osteoclasts on their respective VEGF and IL-6 receptors (VEGFR and IL-6R) to promote osteoclastogenesis and osteoclast activity in the presence of RANKL and MCSF, resulting in bone resorption. OSM also induces tumor cell–secreted AREG that signals via an unknown receptor to suppress OPG production and reduce its ability to bind RANKL and inhibit osteoclastogenesis. Thus, membrane-bound RANKL on osteoblasts or soluble RANKL (sRANKL) is more available to bind to RANK on preosteoclasts, thereby promoting osteoclastogenesis. cFMS = receptor for MCSF.
In conclusion, this study demonstrates that tumor cell–derived OSM promotes osteolytic bone metastasis. We propose this may be through a novel OSM-induced tumor-secreted factor AREG that leads to an increase in osteoclast activation by suppressing OPG. Current therapies for osteolytic bone metastases, such as bisphosphonates, alleviate patient symptoms but have a limited effect on inhibiting tumor cell metastasis to bone and relevant specific breast tumor–derived factors responsible for osteolysis. Based on this study, we suggest that OSM could be a viable therapeutic target for patients with surgically removed primary tumors to reduce bone metastasis and prevent bone loss.
Materials and Methods
Cell lines and culture conditions
66c14 and 4T1.2 cells were maintained in DMEM media supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, and 100 U/mL each of penicillin and streptomycin. RAW 264.7 cells were obtained directly from the American Type Culture Collection (Rockville, MD), cultured in MEMα media with the same supplements, and passaged for no more than 6 months. Cells were maintained at 37°C, 5% carbon dioxide, and 95% humidity. All media and supplements were obtained from Hyclone (Logan, UT). 66c14 and 4T1.2 mouse mammary cell lines were generated by the authors; therefore, there are no external lines against which they can be authenticated.
Adherent cell count/proliferation, detachment, and invasion assays
To assess the number of viable adherent cells correlating with proliferation, 1.0 × 103 cells were seeded in 24-well plates with 1 mL of complete medium and treated with or without 25 ng/mL recombinant mouse OSM (R&D Systems). Cells were detached using 0.25% trypsin (Hyclone) and counted using a hemocytometer every 2 days. OSM-stimulated tumor cell detachment and invasion were conducted as described previously 20 with the following modifications to the invasion assay. A total of 2.5 × 104 cells were plated with or without 25 ng/mL OSM in the upper chamber of the Matrigel-coated invasion chambers (BD Biosciences) in normal growth medium without serum, and normal growth medium containing 10% FBS was added to the bottom chamber as a chemoattractant. Staining of the invasion chambers was achieved with 0.05% crystal violet. Each Matrigel-coated (BD Biosciences) or uncoated control membrane was counted in its entirety for each experiment. Data for invasion assays are presented as percentage of invasion (number of cells that migrated through Matrigel-coated membranes/number of cells that migrated through control membranes), according to the manufacturer’s instructions.
ELISA
To measure OSM, AREG, and OPG in conditioned medium, a direct or sandwich ELISA was developed (see Supplementary Materials and Methods). For the measurement of mouse VEGFA and mouse IL-6 in conditioned medium, ELISA kits (R&D Systems) were utilized according to the manufacturer’s instructions. For all ELISAs, 1 × 105 cells were plated per well in a 24-well plate in serum-free media, and conditioned medium was collected 48 hours later.
RT-PCR
To determine the relative expression of various receptors in 4T1.2 and RAW 264.7 cells, RNA was extracted from these cells using RNA-STAT60 (Teltest, Friendswood, TX) according to the manufacturer’s instructions, and RNA integrity was assessed using the bleach gel method as recently described. 58 cDNA was synthesized using the GeneAmp RNA PCR kit (Applied Biosystems, Carlsbad, CA) and amplified using the GoTaq PCR kit (Promega, Madison, WI). Primer sequences and annealing temperatures (TA) are listed in Supplementary Table S1. The thermocycler conditions for all PCR reactions were as follows: 95°C for 2 minutes, 29 cycles at 95°C for 1 minute, TA for 30 seconds, and 72°C for 1 minute and 72°C for 1 minute after the 29 cycles. Relative amount of RNA was quantified by densitometry of PCR products run on 0.5% agarose gels containing ethidium bromide.
Plasmid construct design and cell transfections
To create OSM knockdown vectors, OSM shRNA sequences were cloned into the pSilencer 4.1 plasmid (Ambion, Austin, TX). A total of 3 shRNA constructs (1-3) were tested for OSM knockdown as well as a LacZ control using the same loop sequence of TTCAAGAGA and the following sense (s) and antisense (a) sequences: OSM1s: GGACAGAGTCTT GTACCAACT, OSM1a: AGTTGGTACAAGACTCTGT CC; OSM2s: GTACCAACTGGATGCTTTA, OSM2a: TAAAGCATCCAGTTGGTACAA; OSM3s: GCACAATA TCCTCGGCATAAG, OSM3a: CTTATGCCGAGGATATTGTGC; LacZs: GCTCCAAAGAAGAAGCGTT, LacZa: AACGCTTCTTCTTTGGAGC. 4T1.2 cells were transfected with the pSilencer 4.1 constructs (Ambion) containing the various mOSM shRNAs using Lipofectamine LTX (Invitrogen, Carlsbad, CA) reagent, as per the manufacturer’s instructions. Stably transfected cell lines were grown in the presence of 0.3 mg/mL of the neomycin analog G418 (Sigma-Aldrich, St. Louis, MO). All established cell lines were checked for OSM expression by ELISA.
Animals and tumor cell injections
Six-week-old female Balb/c mice were obtained from the National Cancer Institute’s Animal Production Facility (Frederick, MD). For orthotopic injections, each mouse was anesthetized by intraperitoneal injection of 6.25 mg/kg sodium pentobarbital or with 2.5% isoflurane, and 1.0 × 105 cells diluted in 10 µL of PBS containing 10% medium were injected into the fourth mammary fat pad. For intratibial injections, 1 × 104 cells in 30 µL PBS containing 10% medium were injected, and needle placement was verified by X-ray as previously described. 59 For tumor resection, mammary tumors were surgically excised 14 days after orthotopic injections. All animal studies were conducted in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Boise Veterans Affairs Medical Center (Boise, ID), Washington University School of Medicine (St. Louis, MO), or the Peter MacCallum Cancer Centre (Melbourne, VIC, Australia). Starting at 2 weeks after injection, tumor length and width were measured by mechanical calipers 3 times a week, and tumor volume was extrapolated using the following equation: tumor volume = (length × width2)/2. “Survival end point” was defined by the IACUC as a tumor size greater than 20 mm in diameter, 10% or more weight loss, and/or the appearance of cachexia.
qPCR
Spines dissected from mice bearing mammary tumors were stripped of any soft tissue, snap-frozen in liquid nitrogen, and pulverized into a fine powder. DNA was extracted using an STE buffer containing 20 µL/mL of proteinase K and purified by 2 phenol/chloroform (1:1 v/v) extractions followed by ethanol precipitation. The ratio of cancer cells to normal cells was quantified by measuring genomic DNA levels for the neomycin resistance gene (neor) found integrated in tumor cells versus the DNA levels for a control gene vimentin present in all cells, as described previously. 60 Taqman PCR was performed on an Applied Biosystems 7500 real-time thermocycler. Probe and primer sequences are listed in Supplementary Table S1. The cycling conditions were run as follows: 50°C for 5 minutes, 95°C for 2 minutes, and then 40 cycles at 95°C for 1 minute and 60°C for 45 seconds. Fluorescence was measured every cycle after the annealing step, and threshold cycle number (Ct) values were calculated. The data were analyzed using the comparative ΔCt method. 61
Histology
To verify the location of spine metastasis, spines from each experimental group (4T1.2-LacZ: n = 2; 4T1.2-OSM2: n = 2; and 4T1.2-OSM1: n = 1) were placed in ultralight fixative (Ultralight Histology, Nampa, ID), paraffin-embedded, and sectioned (Bi-Biomics, Nampa, ID). For each spine, four 1-µm sections were collected 10 µm apart in the lumbar region and H&E stained.
Micro-CT analysis of in vivo osteolysis
For determining the 3-dimensional architecture of the trabecular and cortical bone, mice were euthanized, and the hindlimb was removed and placed in 10% formalin overnight before being transferred to 70% ethanol for analysis using a vivaCT40 scanner (Scanco Medical AG, Wangen-Brüttisellen, Switzerland). Tube settings were at 55 kVp and 145 µA with a tube diameter of 21.5 mm. The entire hindlimb (femur, tibia, and fibula) was scanned at a 10-µm isometric resolution with a 250-ms integration time. For trabecular bone analysis, the end of the growth plate was used as the landmark, and a 0.525-mm (50 slices) region below the landmark (metaphysis) was considered as the ROI. Contours were drawn on 50 slices distal to the landmark around the trabecular bone only (endocortical boundary). An additional analysis of the cortical bone below the growth plate was performed from the trabecular bone analysis scans. The mid-diaphyseal region (approximate middle of the proximal-end and tibia-fibula junction) of the tibia was considered as the ROI for cortical bone analysis. Two contours were drawn on 50 slices of the ROI: one at the periosteal surface (inclusive) including the cortical bone, and the other at the endosteal surface of the bone (exclusive) to exclude the medullary region. A Gaussian filter (with Gaussian sigma of 0.4, and support of 1) was applied on all slices before image segmentation. All the contoured slices were segmented and analyzed with the global threshold value of 250 “per-mille” (linear attenuation coefficient (µ) of 2.00 [1/cm]).
Osteoclastogenesis assay
There were 1.0 × 104 RAW 264.7 cells that were added to each well of a 24-well plate in duplicate in complete MEMα medium containing 50 mg/L L-ascorbic acid. There were 1.0 × 103 4T1.2 cells that were plated in co-culture with the RAW 264.7 cells or alone, and 25 ng/mL OSM and/or 50 ng/mL AREG were added. All osteoclastogenesis assays included very low levels of RANKL and MCSF (10 ng/mL RANKL and 5 ng/mL MCSF). Two hours before the addition of OSM, AREG, VEGF, and IL-6 neutralizing antibodies were added at a final concentration of 1 µg/mL where indicated. All mouse factors and antibodies were obtained from R&D Systems. The co-cultures were maintained for 9 to 10 days without any changes of the medium, and osteoclasts were stained using a tartrate-resistant acid phosphate (TRAP) stain kit (Sigma-Aldrich) according to the manufacturer’s instructions. TRAP-positive cells that stained purple were counted and quantified per well.
Statistical analysis
Data are displayed as mean ± standard error of the mean (SEM). Data were analyzed using an unpaired Student t test or analysis of variance (ANOVA) with Tukey multiple comparison post hoc test where appropriate, using Prism GraphPad 5.0b software (GraphPad Software Inc., San Diego, CA). Survival data were analyzed using the log-rank (Mantel-Cox) test. Asterisks denote the following: *P < 0.05, **P < 0.01, or ***P < 0.001.
Footnotes
Acknowledgements
Special thanks is given to John Englebach, Joel Garbow, and Tarpit Patel for assistance with micro-CT experiments, Dr. Dondelinger and Tyson Nielson (Bi-Biomics Inc., Nampa, ID) for assistance with H&E and pathology, and Christina Restall (Peter MacCallum Cancer Centre, Melbourne, VIC, Australia) for assistance with the orthotopic mouse model and qPCR.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the American Cancer Society (ACS RSG-09-276-01-CSM), Susan G. Komen for the Cure (KG100513), National Institutes of Health (NIH/NCRR P20RR016454), National Aeronautics and Space Administration (NASA NNX10AN29A), and National Breast Cancer Foundation (NBC, Australia [to R.A.]). Mice were maintained at the Veterans Affairs Medical Center (Boise, ID), which is supported in part by the National Institutes of Health (NIH/NIAMS P30AR057235).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.