
Abstract
Not all leukemia T cells are susceptible to high levels of phorbol myristate acetate (PMA)–mediated apoptosis. At micromolar levels, PMA induces apoptosis of Jurkat T cells by causing mitochondrial polarization/de-polarization, release of cytosolic granules, and DNA fragmentation. Chemical inhibitors U0126 and PD98059 block mitogen-activated protein kinase kinase 1 (MEK1)–mediated phosphorylation of extracellular signal–regulated kinase (ERK) and prevent apoptosis. Mechanistically, proapoptotic tumor suppressor WOX1 (also named WWOX or FOR) physically interacts with MEK1, in part, in the lysosomes in Jurkat cells. PMA induces the dissociation, which leads to relocation of MEK1 to lipid rafts and WOX1 to the mitochondria for causing apoptosis. U0126 inhibits PMA-induced dissociation of WOX1/MEK1 complex and supports survival of Jurkat cells. In contrast, less differentiated Molt-4 T cells are resistant to PMA-induced dissociation of the WOX1/MEK1 complex and thereby are refractory to apoptosis. U0126 overturns the resistance for enhancing apoptosis in Molt-4 cells. Together, the in vivo MEK1/WOX1 complex is a master on/off switch for apoptosis in leukemia T cells.
Introduction
T lymphoid cells possess a plethora of multiple functions in cell-mediated immunity for host defense. During thymus development, progenitor T cells are selectively committed to naive CD4+ or CD8+ T cells.1,2 Once naive T cells encounter antigens, they become activated through T cell receptor (TCR)–CD3 signaling, which leads to the release of interleukin (IL)–2 and other cytokines. IL-2 activates T cells for clonal expansion. Later, these activated T cells are eliminated via apoptosis, so that the ongoing immune response is turned off. 3 Nonetheless, the mechanisms underlying T cell death are largely unknown. Deciphering the molecular events for T cell activation-induced apoptosis is crucial in drug interventions targeting leukemia T cells.
Phorbol 12-myristate 13-acetate (PMA) is a potent inducer of T cell differentiation and death and has diverse biological effects, such as tumor promotion, platelet aggregation, cell differentiation, metabolic alterations, and apoptosis induction. 4 In T cells, PMA-mediated protein kinase C activation is an early event mimicking the TCR signaling. At nanomolar concentrations, PMA triggers differentiation in various types of cells.5,6 PMA causes T cell differentiation via alteration of cellular enzymatic activities, expression of maturation-linked antigens, and induction of suppressor/cytotoxic T cell phenotypes.7-11 PMA induces extracellular signal–regulated kinase (ERK) phosphorylation and nuclear translocation for upregulating p21, a cell cycle inhibitor. 12 However, at micromolar levels, PMA induces apoptosis of T cells.
Here, we show that tumor suppressor WW domain– containing oxidoreductase, known as WWOX, FOR, or WOX1,13-15 is involved in PMA-induced T cell death. Human WWOX gene is located on a common fragile site FRA16D on chromosome 16q23.2.16-20 WWOX gene possesses approximately 1 million bases with 9 exons and codes for a 46-kDa protein containing 414 amino acids.13-15 WWOX/WOX1 has 2 N-terminal WW-domains, a C-terminal short-chain alcohol dehydrogenase/reductase (SDR) domain and a nuclear localization signal (NLS) in between the WW domains. Loss or alteration of WWOX gene has been shown in a variety of human malignancies.16-20 Targeted deletion of murine Wwox gene at exons 2 to 4 results in spontaneous tumor formation in mice. 21 Wwox gene knockout mice have a shortened life span and defects in bone metabolism, splenic atrophy, and other deficiencies.22,23
WWOX/WOX1 is involved in multiple signal networks, particularly in stress signaling, growth and apoptosis regulations, and control of the activation of transcription factors, including p53, p73, AP2γ, and c-Jun.16,18-20,24-26 Tyr33-phosphorylated WOX1 (p-WOX1) is essential for binding and stabilizing Ser46-phosphorylated p53. 24 The protein complex is critical for apoptotic response.15,25,26 Tyrosine kinase Src phosphorylates Tyr33 in WOX1.24,25,27-32 Also, Tyr33 becomes phosphorylated when cells are exposed to sex steroid hormones, 27 transforming growth factor, 28 complement C1q, 29 UV light,24,25 and anisomycin. 25 During neuronal injury, WOX1 undergoes Tyr33 phosphorylation and accumulation in the mitochondria and nuclei.30-32 Activated tyrosine kinase 1 (Ack1) phosphorylates WOX1 on Tyr287 for polyubiquitination and protein degradation in prostate cancer cells. 26 Interestingly, WOX1 enhances the NF-κB-regulated promoter activation. 32
Both Jurkat and Molt-4 leukemia T cells were used in this study.33,34 PMA mimics the function of diacylglycerol in activating PKC and regulating the Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) pathway, which affects cell growth, differentiation, and death. 35 At nanomolar concentrations, PMA triggers differentiation of human lymphoid leukemia cell lines8,9 and protects Jurkat T cells from Fas- and death receptor–mediated apoptosis, which depends on the activity of ERK and NF-κB.36-38 Nonetheless, PMA, at micromolar levels, exerts cytotoxicity in many cancer cell lines.39,40 We determined that inhibition of MEK1 (mitogen-activated protein kinase kinase) by U0126 protected Jurkat from PMA-induced apoptosis but sensitized Molt-4 for apoptosis. In light of these findings, we explored the role of WOX1 and MEK1 in inducing apoptosis and found the WOX1/MEK1 complex as a switch in controlling leukemia T cell death.
Results
Jurkat is sensitive to PMA-induced apoptosis, but less differentiated Molt-4 is refractory
To better understand the molecular mechanisms underlying T cell activation and death, we used Jurkat and Molt-4 T cell lines and exposed them to various amounts of PMA (nM-µM). Jurkat cells (11.33 ± 1.34 µm in diameter; n = 35) are significantly smaller than Molt-4 cells (13.60 ± 1.37 µm in diameter; n = 54) (Fig. 1A,B). Jurkat cells possess numerous surface microvilli or protrusions, whereas Molt-4 cells appear to be relatively smooth. Compared to Jurkat, Molt-4 cells have a lower expression of a differentiation marker CD3 (Fig. 1B). The observation is in agreement with a previous report. 34
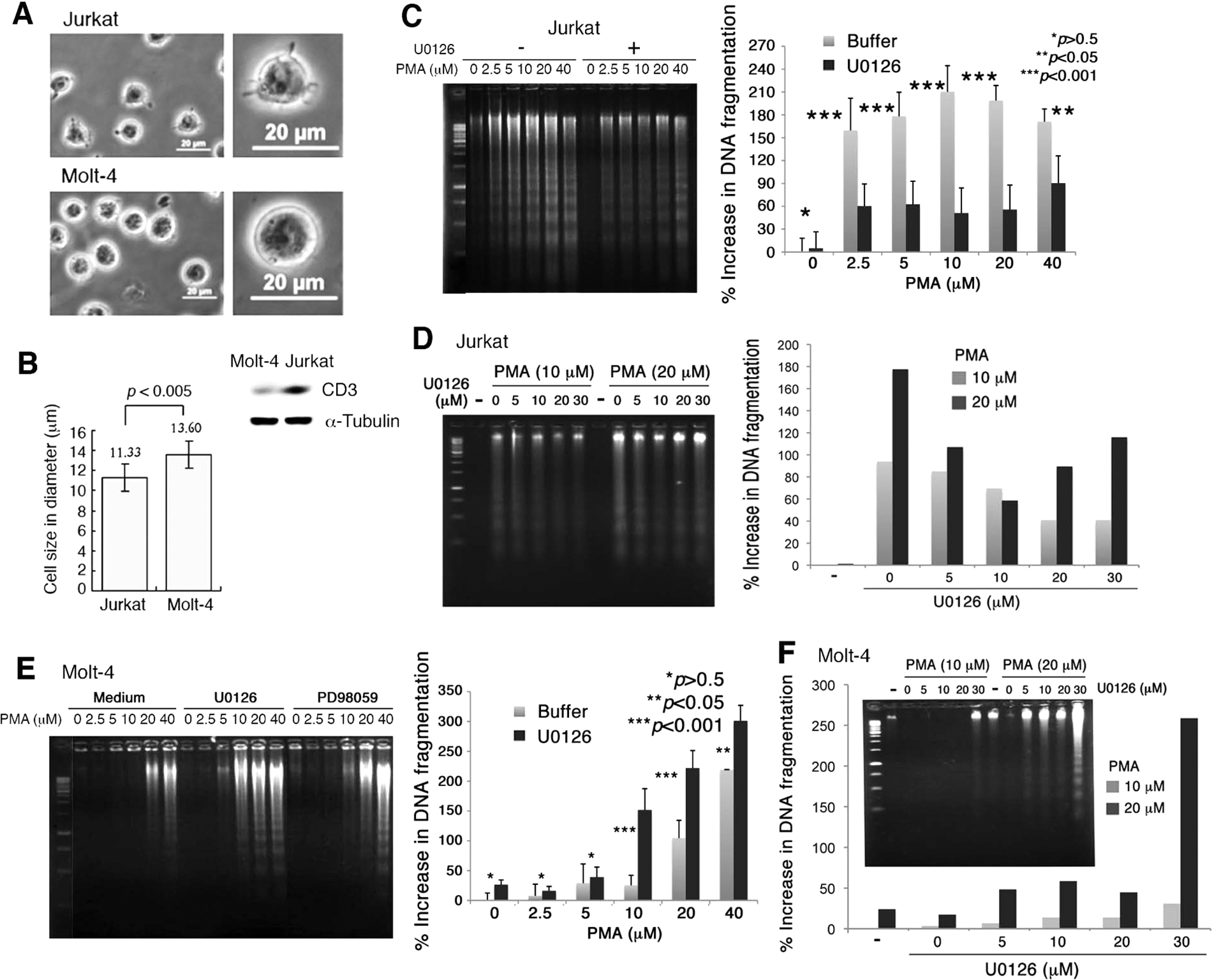
Jurkat T cells are more sensitive to phorbol myristate acetate (PMA)–induced apoptosis than Molt-4 T cells. (
Both cells were exposed to PMA (2.5-40 µM) for 24 hours, followed by determining the extent of cell death by DNA fragmentation assays and cell cycle analyses. PMA induced internucleosomal DNA fragmentation in Jurkat cells in a dose-dependent manner (Fig. 1C). In contrast, Molt-4 cells were refractory to PMA-induced apoptosis. DNA fragmentation occurred only when high concentrations of PMA (≥20 µM) were used in treating Molt-4 cells (Fig. 1E).
Inhibition of MEK by U0126 blocks PMA-induced apoptosis in Jurkat but enhances apoptosis in Molt-4
Next, we used specific MEK inhibitors U0126 and PD98059 to block the Ras/Raf/MEK/ERK signaling. Both chemicals are highly selective inhibitors of MEK1 and MEK2.41,42 U0126 may block T cell proliferation via inhibition of the Ras/Raf/MEK/ERK pathway. 42 Jurkat T cells were pretreated with U0126 for 1 hour, followed by exposure to PMA for 24 hours. U0126 suppressed PMA-induced DNA fragmentation and death in Jurkat T cells (Fig. 1C,D). In stark contrast, Molt-4 T cells were refractory to PMA-induced apoptosis, and interestingly, U0126 enhanced the cellular sensitivity to death by PMA (Fig. 1E,F).
Cell cycle analyses were carried out to further verify the above observations. Suppression of MEK by U0126 significantly reduced PMA-mediated apoptosis of Jurkat cells, as evidenced by decreased populations of cells in the subG1 phase of the cell cycle (Fig. 2A,B). Also, compared to control cells, U0126 increased G1 growth arrest of Jurkat cells by 25% to 50%, treated with or without PMA (Fig. 2A). By Western blotting, PMA induced ERK phosphorylation, and that U0126 blocked the phosphorylation (Fig. 2C).
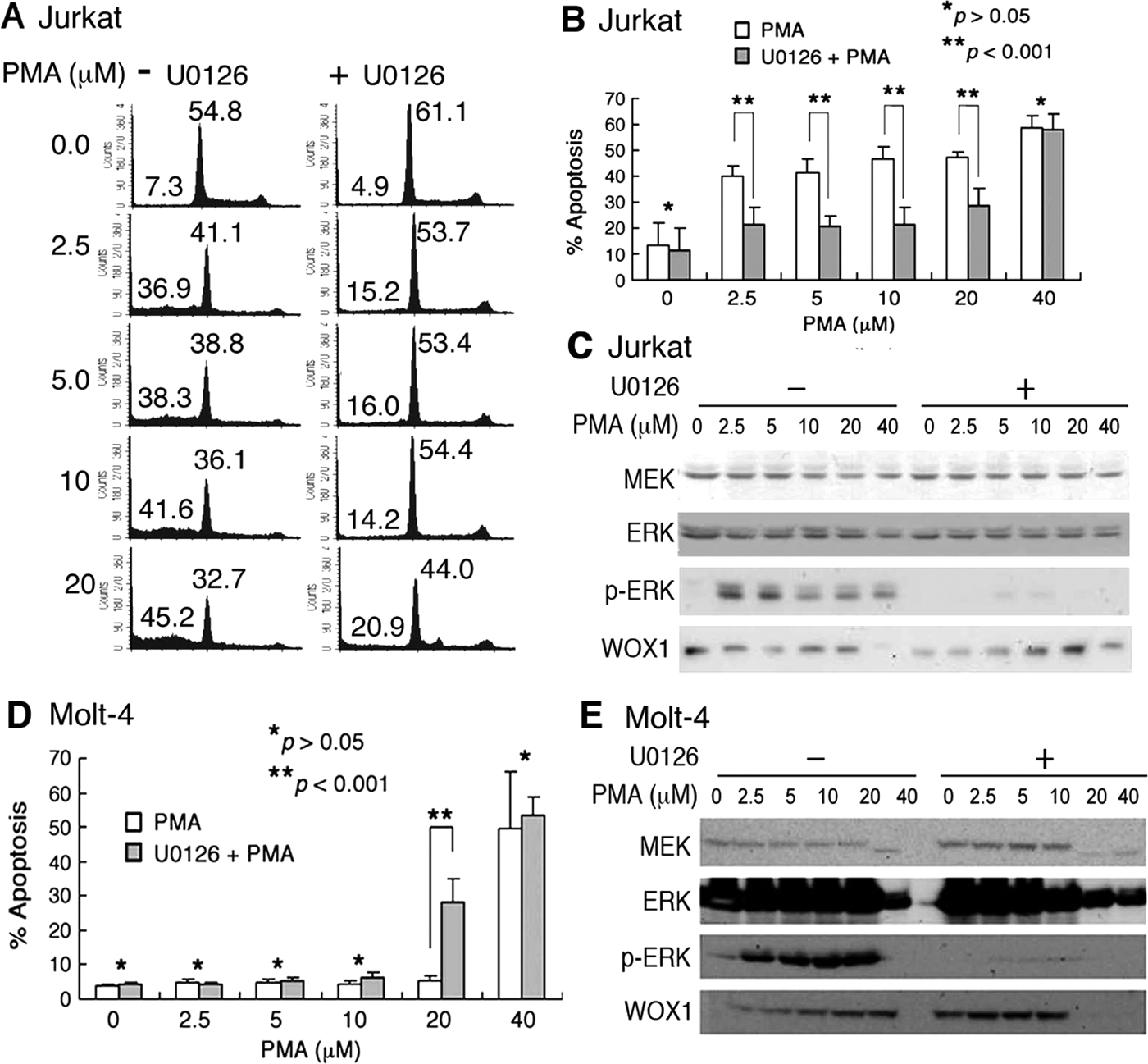
Inhibition of MEK by U0126 protects Jurkat cells from phorbol myristate acetate (PMA)–induced apoptosis. (
In comparison, when Molt-4 cells were sensitized with U0126 for 1 hour and then co-treated with 20 µM PMA for 24 hours, apoptosis was enhanced (see subG1 phase in Fig. 2D and Suppl. Fig. S1). U0126 did not appear to cause G1 growth arrest of Molt-4 cells (Suppl. Fig. S1). Also, U0126 suppressed PMA-induced ERK phosphorylation (Fig. 2E). WOX1 protein expression levels were affected by both PMA and/or U1026 (Fig. 2E).
Induction of Molt-4 differentiation restores their sensitivity to PMA-induced apoptosis
The status of T cell differentiation probably plays a critical role in determining their sensitivity to PMA-induced cell death. Molt-4 cells were exposed to a low dose of PMA (10 nM) for inducing differentiation for 9 days and then challenged with micromolar levels of PMA (Suppl. Fig. S2A). Upon induction of differentiation, Molt-4 cells underwent morphological changes, including reduction in cell sizes, increase in cell adherence, and increased cell surface microvilli (Suppl. Figs. S2B and S2C). The cell sizes were significantly reduced from 13.06 ± 1.39 (n = 33) to 11.85 ± 1.18 µm (n = 27) in diameter in 9 days (Suppl. Fig. S2D). When treated with PMA at 5 µM, these cells underwent apoptosis (Suppl. Fig. S2E). Pretreatment of these cells with U0126 protected them from PMA-induced apoptosis (Suppl. Fig. S2E). Similar results were observed by treating Molt-4 T cells with 50 or 100 nM PMA for 9 days (data not shown). Together, these observations support the notion that the differentiation status of T cells is critical in determining their sensitivity to PMA-induced apoptosis.
In comparison, monocytic THP-1 cells were exposed to U0126 for 1 hour and then treated with PMA for 24 hours. THP-1 cells were refractory to PMA-induced apoptosis, whereas U0126 sensitized these cells to apoptosis by PMA (Suppl. Fig. S3A). Similarly, naive THP-1 cells were stimulated with 10 nM PMA for 9 days (see the treatment scheme in the Suppl. Fig. S2A), followed by treating with or without U0126 for 1 hour and then exposure to PMA for 24 hours. These stimulated THP-1 cells did not acquire protection from U0126 treatment (Suppl. Fig. S3B).
In addition, we examined a panel of 11 cell lines and determined which cell line becomes protected upon pretreatment with U0126 and then challenge with PMA. These cells included adherent human breast cancer MCF7, MDA-MB-231, and MDA-MB-435S cells; human colon cancer HCT116 cells; human lung cancer NCI-H1299 cells; human neuroblastoma SK-N-SH cells; human skin squamous cell carcinoma SCC4, SCC9, and SCC15 cells; and mouse L929 fibrosarcoma and melanoma B16F10 cells (Suppl. Fig. S4). Most of these cells are resistant to apoptosis caused by PMA and/or U0126 (e.g., SCC cells). Like Molt-4, HCT116 and THP-1 cells were sensitized to apoptosis by pretreatment with U0126 and then co-exposure to PMA for 24 hours (Suppl. Figs. S3 and S4). U0126 blocked ERK phosphorylation induced by PMA in the aforementioned cells (data not shown). Together, none of the tested cells act similarly as that in Jurkat T cells in response to U0126 and PMA.
PMA induces mitochondrial polarization and release of cytosolic granules in Jurkat T cells, and U0126 blocks the events
Jurkat T cells were loaded with JC-1 for staining mitochondria. Upon exposure to PMA, mitochondria rapidly underwent polarization, as revealed by significant accumulation of JC-1 aggregates in red-orange color in time-lapse microscopy (Fig. 3A,B and Suppl. Fig. S5). Mitochondria were then depolarized, as the aggregates disappeared and the green monomer was accumulated with time. The time-dependent JC-1 aggregation and monomer formation were quantified (Fig. 3A,B and Suppl. Fig. S5). Instead of shrinking, the cells gradually swelled up and then popped out a “big balloon” or underwent blebbing for rupture (Fig. 3A,B and Suppl. Fig. S5). Pretreatment of cells with U0126 to inhibit MEK resulted in suppression of PMA-induced mitochondrial polarization-depolarization (Fig. 3B).
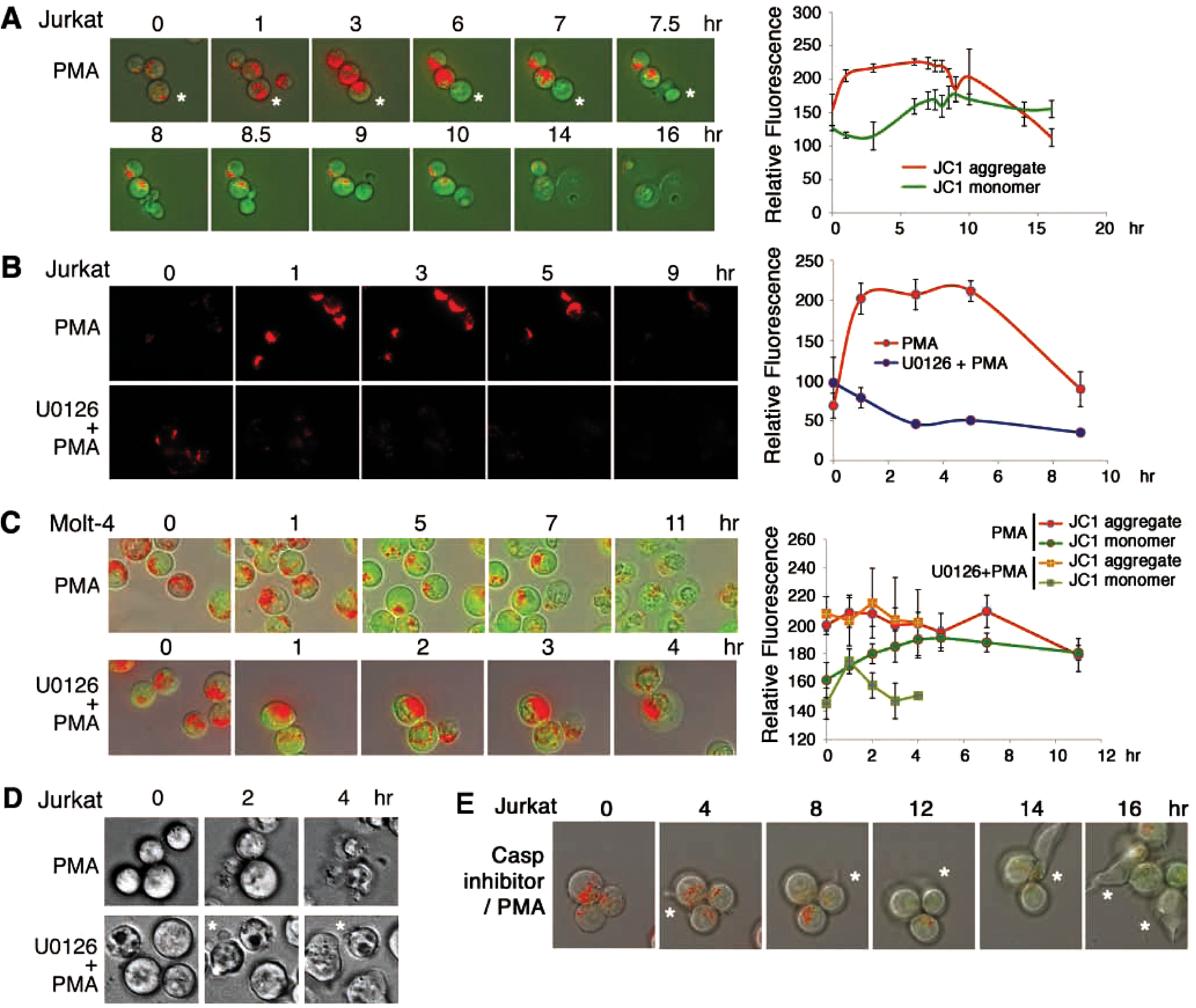
U0126 blocks phorbol myristate acetate (PMA)–induced mitochondrial polarization in Jurkat but accelerates the polarization in Molt-4. (
In Molt-4 T cells, PMA also induced mitochondrial polarization within 1 hour (Fig. 3C). The depolarization event lasted a prolonged period for 3 to 11 hours (Fig. 3C), which correlates with the resistance of Molt-4 to PMA-induced apoptosis (Fig. 3C). U0126 enhanced the mitochondrial polarization, as shown by significantly increased aggregation of JC-1. The U0126/PMA-treated Molt-4 cells directly underwent rupture without mitochondrial depolarization in 4 hours (Fig. 3C).
Cytosolic granules are frequently present in Jurkat and Molt-4 cells. PMA induced release of the cytosolic granules from Jurkat cells with time (Suppl. Fig. S6A), and U0126 blocked the release (Suppl. Fig. S6B). Similarly, Molt-4 cells were pretreated with U0126 for 1 hour to become sensitized to PMA-mediated cell death. PMA rapidly induced the release of cytosolic granules from cells (Suppl. Fig. S7). Without prior treatment with U0126, no release of cytosolic granules was shown in Molt-4 cells.
Suppression of MEK and caspases restores PMA-induced membrane blebbing: wrestling with death
PMA-induced apoptosis was not involved in cell shrinkage and membrane blebbing (top panel of Fig. 3D and Suppl. Fig. S8A). Pretreatment of Jurkat cells with U1026 for 1 hour, followed by exposure to PMA, resulted in restoration of membrane blebbing under the PMA stress (bottom panel of Fig. 3D and Suppl. Fig. S8B). The induced membrane blebbing could be regarded as a manner by which the cell struggles with the PMA-induced death stress. Alternatively, suppression of caspase 8 by IETD-fmk resulted in PMA-induced membrane blebbing in Jurkat T cells (Fig. 3E). Mitochondrial polarization was suppressed and no cell death observed (Fig. 3E). Similar results were observed by treating cells with a pan-caspase inhibitor zVAD-fmk (data not shown).
WOX1 physically interacts with MEK1, and PMA rapidly dissociates the WOX1/MEK1 complex in Jurkat but not in Molt-4 cells
WOX1 is a tumor suppressor and proapoptotic protein.16-20 Activated WOX1 with phosphorylation at Tyr33 may translocate to mitochondria and nuclei to induce apoptosis both in vitro and in vivo.15,24-25,27-31 Whether WOX1 participates in T lymphoid cell death is largely unknown. From yeast 2-hybrid cDNA library screens, we determined that MEK1 is one of the proteins that physically interacts with WOX1 (data not shown). Jurkat T cells were exposed to PMA (10 µM) for indicated durations, followed by processing immunoprecipitation. In resting Jurkat cells, endogenous WOX1 physically interacted with MEK1, and that PMA rapidly dissociated the binding in 10 minutes (Fig. 4A). Pretreatment of the cells with U0126 resulted in the inhibition of PMA-mediated dissociation of the WOX1/MEK1 complex (Fig. 4B). In comparison, exposure of Molt-4 T cells to PMA did not effectively cause dissociation of the WOX1/MEK1 complex (Fig. 4C), which correlates with their resistance to PMA-induced apoptosis. The antibody recognizes MEK1 and MEK2, whereas no apparent MEK2 was shown in both cells. The validity of anti-MEK1 IgG was further verified (Fig. 4D). Nonspecific protein bands were not observed using nonimmune sera for immunoprecipitation (Fig. 4D).
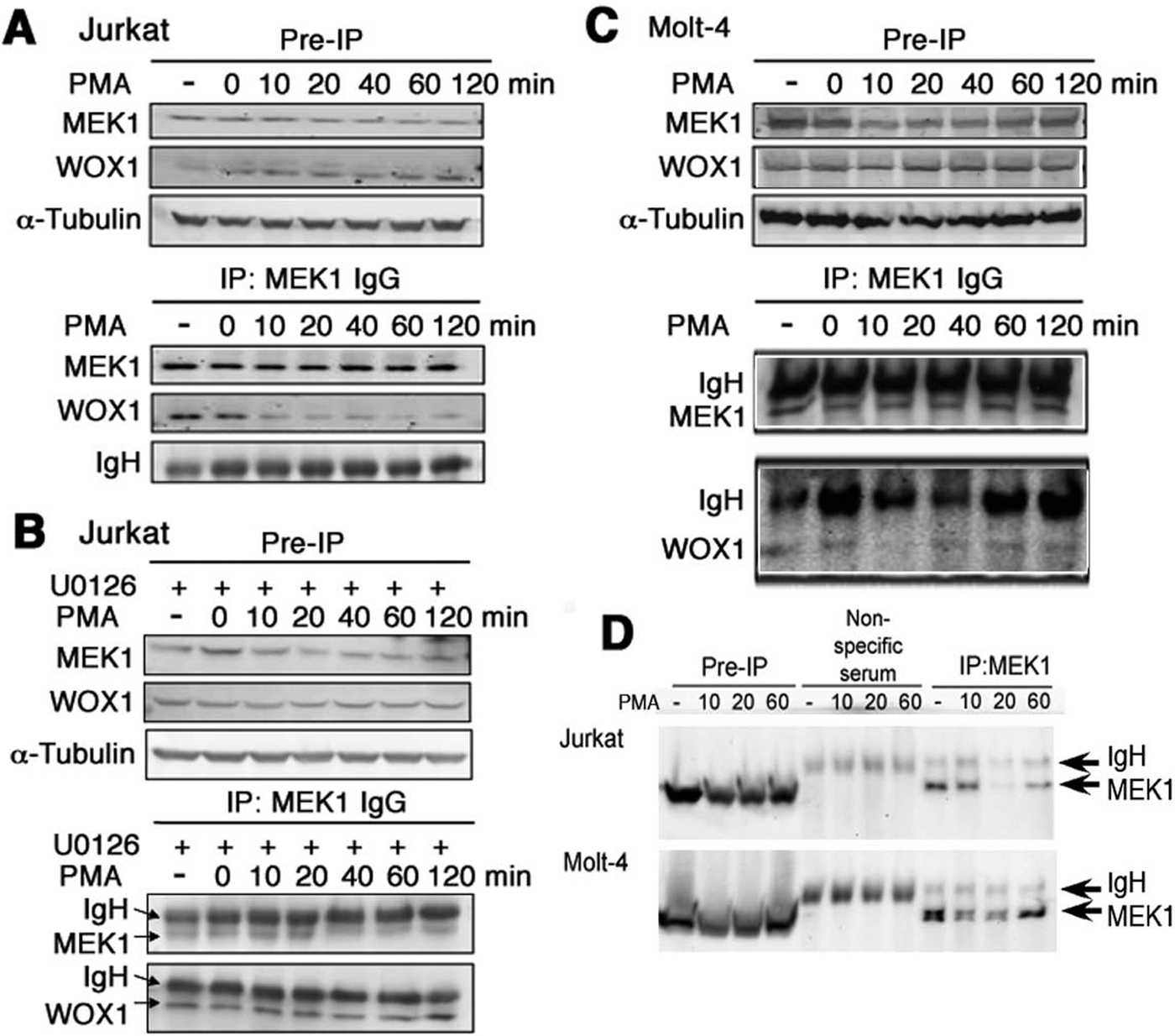
Phorbol myristate acetate (PMA)–induced dissociation of an in vivo WOX1/MEK1 complex in Jurkat T cells. Jurkat cells were exposed to PMA (10 µM) for indicated durations, followed by preparing whole-cell lysates and processing Western blotting and immunoprecipitation using specific anti-MEK1 antibody and protein A agarose beads. (
Dominant negative WOX1 suppresses PMA-induced cell death
Transiently overexpressed WOX1 or its N-terminal WW domain (WOX1ww) enhanced PMA-induced apoptosis of Jurkat cells, as determined by DNA fragmentation analysis (Fig. 5A). Control cells were overexpressed with enhanced green fluorescent protein (EGFP) only (Fig. 5A). We have generated dominant negative WOX1 (DN-WOX1) constructs, which block WOX1 activation by inhibiting Tyr33 phosphorylation and preventing WOX1- and p53-mediated apoptosis.15,24,25,31 Jurkat cells were electroporated with the wild-type WOX1, DN-WOX1 constructs (EGFP-tagged DN-WOX1 and WW domain DN-WOX1ww), and a control EGFP vector, respectively. Compared with EGFP controls, transiently overexpressed DN-WOX1 inhibited PMA-induced apoptosis (Fig. 5B,C) and ERK phosphorylation in Jurkat T cells (Fig. 5D). In parallel, knockdown of WOX1 expression by small interfering RNA (WOX1si) abolished U0126-mediated protection of Jurkat cells from PMA-induced apoptosis (Fig. 5E).
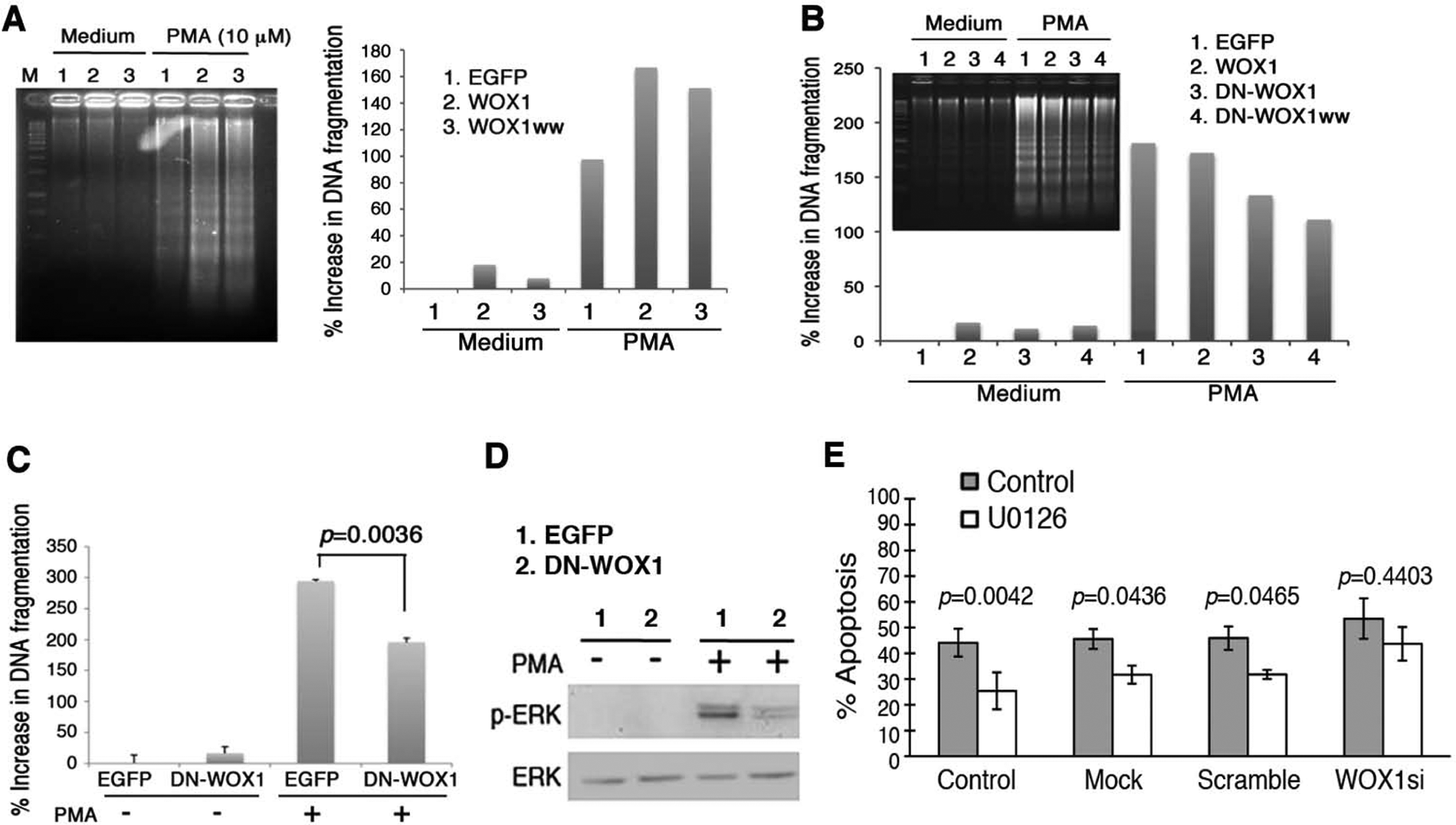
Inhibition of phorbol myristate acetate (PMA)–induced apoptosis by dominant-negative WOX1 and knockdown of WOX1 in Jurkat cells. (
Also, we verified the results by time-lapse microscopy and determined the inhibitory effect of DN-WOX1ww on PMA-mediated cell death (Suppl. Fig. S9). Control cells, expressing EGFP only, underwent whole-cell rupture at 120 to 160 minutes posttreatment with PMA, and no membrane blebbing was observed (data not shown). Nontransfected control cells underwent rupture at 120 to 160 minutes without membrane blebbing (Suppl. Fig. S9). For comparison, in DN-WOX1ww-expressing cells, membrane blebbing was observed at 100 to 140 minutes, followed by rupture at 200 to 220 minutes posttreatment (Suppl. Fig. S9). That is, DN-WOX1ww restricted cell death and restored membrane blebbing.
MEK1 and WOX1 co-localize in part in the lysosome, and PMA induces relocation of MEK1 to the lipid rafts and WOX1 to the mitochondria. We examined the localization of MEK1 in Jurkat T cells. By epifluorescence and confocal microscopy, a major portion of MEK1 is localized in the lysosome, which appears as a large “aggregate” (yellow punctate) (Fig. 6A,B and Suppl. Fig. S10). Phosphorylated MEK1 is also present, in part, in the lysosome (data not shown).
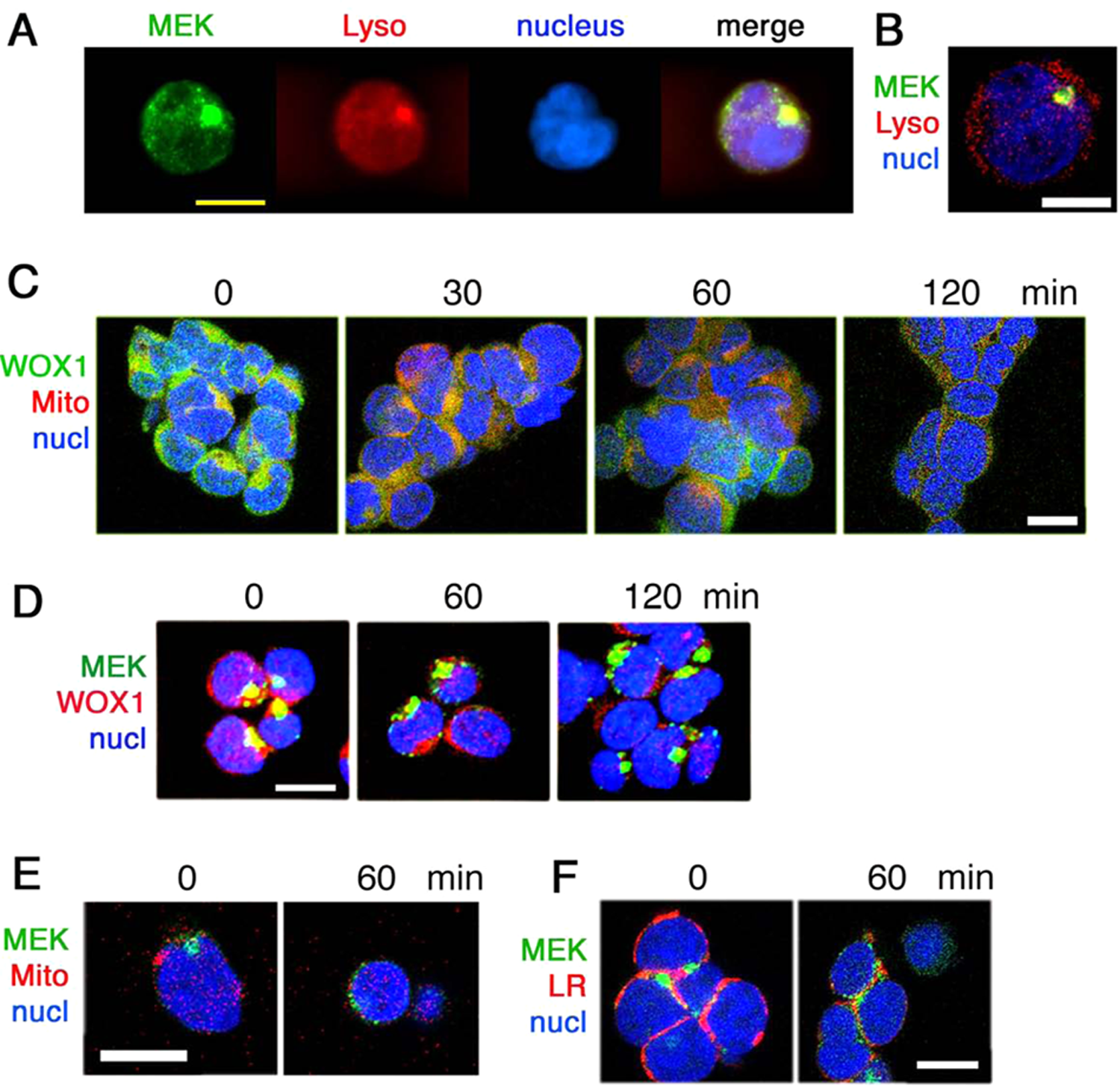
Presence of MEK1/WOX1 complex in the lysosome of Jurkat cells, and phorbol myristate acetate (PMA) induces relocation of WOX1 to the mitochondria and MEK1 to the lipid raft. (
In response to stress stimuli, WOX1 may relocate to the mitochondria both in vivo and in vitro.15,16,30,31 Confocal microscopy analysis revealed that PMA rapidly induced relocation of WOX1 to the mitochondria approximately within 30 minutes (see the yellow punctate; Fig. 6C). After 1 hour, WOX1 was released from the mitochondria to the cytoplasm (Fig. 6C). Indeed, transiently overexpressed EGFP-WOX1 was localized, in part, in the mitochondria, and PMA caused release of WOX1 from the mitochondria to the cytoplasm, as determined in time-lapse microscopy (Suppl. Fig. S11).
We determined whether WOX1 and MEK1 are co-localized in Jurkat cells. Confocal imaging analysis showed the co-localization of endogenous WOX1 with MEK1 and that PMA reduced the co-localization in a time-related manner (see disappearance of yellow punctate with time; Fig. 6D and Suppl. Fig. S12). MEK1 is not localized in the mitochondria and did not relocate to the mitochondria of Jurkat cells in response to PMA (see the green punctate; Fig. 6E). However, PMA stimulated relocation of MEK1 to the lipid rafts (see the yellow punctate; Fig. 6F).
PMA increases the binding of MEK1 with the C-terminal SDR domain of WOX1
Finally, we mapped the domain/domain binding of MEK1 with WOX1. COS7 fibroblasts were co-transfected with expression constructs for both destabilized red fluorescence protein (DsRed)–MEK1 and EGFP-WOX1 (or other WOX1 constructs) and treated with PMA for time-lapse microscopy. PMA increased the binding of MEK1 with WOX1, in which MEK1 interacted most strongly with the SDR domain of WOX1 with time (Fig. 7A,B). In the presence of a C-terminal D3 region, PMA did not increase the binding of MEK1 to SDR/D3 (Fig. 7A,B). In negative controls, no binding interactions were observed between EGFP and DsRed (Fig. 7A,B). In parallel experiments, PMA did not effectively induce dissociation of the endogenous MEK and WOX1 complex in COS7 cells, as determined by co-immunoprecipitation (data not shown). COS7 cells are resistant to apoptosis by PMA.
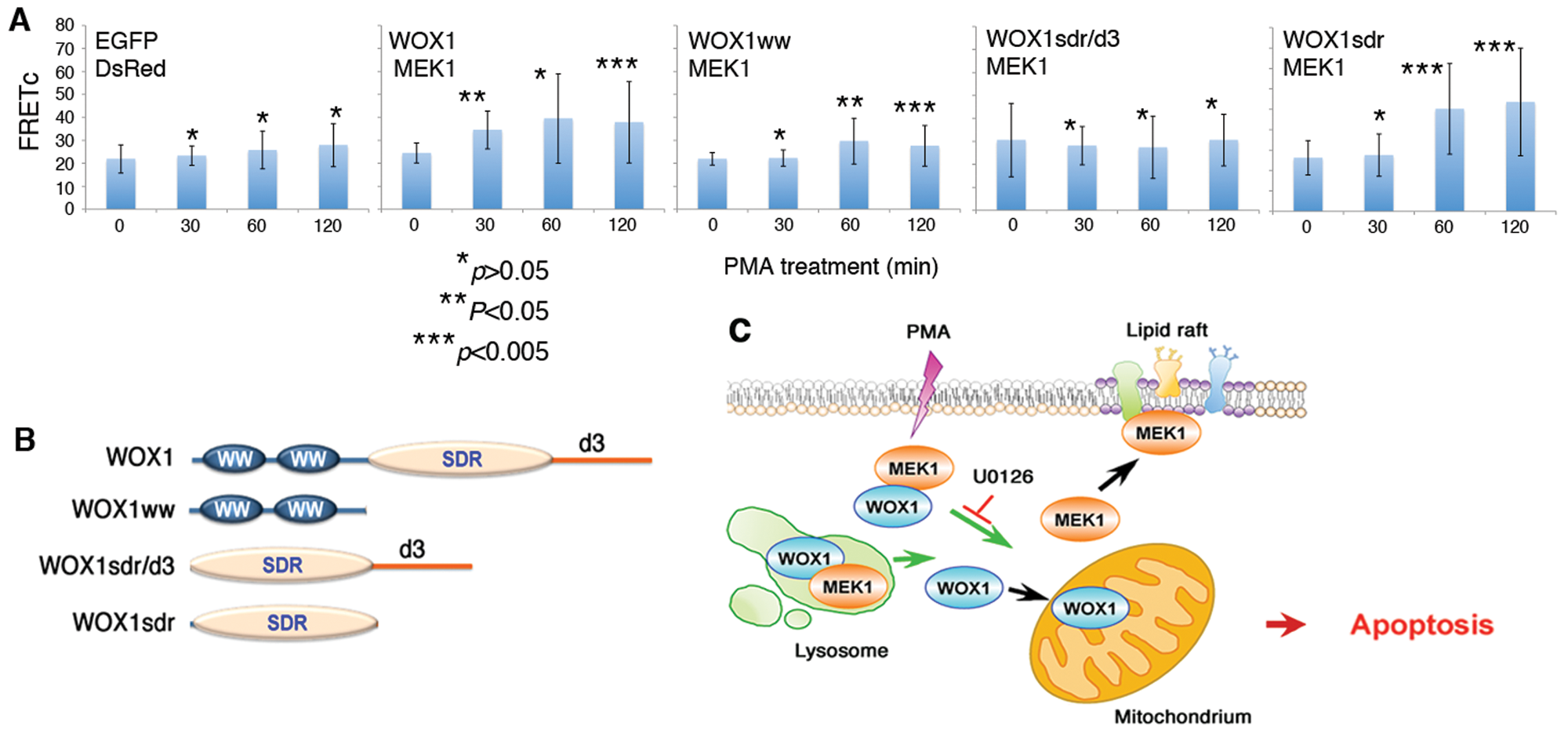
PMA increases the binding of MEK1 with WOX1: critical role in death signaling pathways. (
Discussion
In this study, we have discovered for the first time the in vivo complex of MEK1 and WOX1, which is associated with the death event of activated T cells. 35 PMA, at µM levels, induces dissociation of the complex and ultimately leads to cell death. Jurkat T cells are sensitive to PMA-induced dissociation of the MEK1/WOX1 complex for causing apoptosis. Inhibition of MEK1 in the Ras/Raf/MEK/ERK pathway by U0126 and PD98059 prevents the complex from dissociation and subsequent apoptosis. The less differentiated Molt-4 T cells are refractory to PMA-mediated dissociation of the MEK1/WOX1 complex for leading to apoptosis, suggesting that T cells respond differently to PMA depending on their status of differentiation. To further substantiate this postulation, preinduction of cell differentiation by nanomolar PMA sensitizes Molt-4 T cells to PMA-induced apoptosis at µM levels. Also, these treated Molt-4 cells become protected by U0126 upon challenge with PMA.
In addition, we have tested 13 cell lines regarding their sensitivity to PMA-induced apoptosis and the protective effect of U0126. Most of these cells are resistant to apoptosis caused by PMA and/or U0126. Like Molt-4, HCT116 cells become sensitized to apoptosis by pretreatment with U0126 and then co-exposure to PMA for 24 hours. Unlike Molt-4, monocytic THP-1 cells could not be induced to become a Jurkat-like phenotype. Yet, U0126 significantly increased the extent of PMA-induced apoptosis. PMA could not induce dissociation of the MEK1/WOX1 complex in COS7 cells and did not effectively cause apoptosis (data not shown). Thus far, we have determined that the Jurkat cell is the only cell type that becomes protected from PMA-mediated apoptosis upon pretreatment with U0126.
WOX1 physically interacts with MEK1, and both proteins co-localize, in part, in the lysosomes. Indeed, the presence of MEK/ERK in the endosomal/lysosomal compartment has been shown. 43 Confocal imaging analyses revealed that PMA triggers the dissociation of lysosomal MEK1 from WOX1 (see the schematic graph in Fig. 7C). MEK1 then relocates to the lipid raft and WOX1 to the mitochondria. The functional role of MEK1 in the lipid raft is unknown. Under apoptotic stress, Tyr33-phosphorylated WOX1 translocates to the mitochondria to induce apoptosis both in vitro and in vivo.15,16,30,31 U0126 blocks the dissociation and thereby prevents relocation of WOX1 to the mitochondria for inducing apoptosis. DN-WOX1 blocks the activation of WOX1 (or Tyr33 phosphorylation) and p53 and their apoptotic function.24,25 DN-WOX1 suppresses PMA-induced ERK phosphorylation and blocks the activated WOX1-regulated apoptosis of Jurkat cells. We also verified the effect of WOX1 knockdown by siRNA for reducing the protective effect of U0126 against PMA-induced apoptosis of Jurkat cells. We do not exclude other possibilities, which may cause U0126 protection of Jurkat cells from apoptosis by PMA, as there are many binding proteins of WOX1 in cells.16-20 U0126-mediated inhibition of ERK phosphorylation by PMA is probably not associated with its protection of Jurkat cells from apoptosis. PMA induces ERK phosphorylation in every test cell line, which is resistant to PMA-mediated apoptosis. U0126 suppresses the phosphorylation but fails to provide protection against death by PMA.
Indeed, PMA causes an unusual event of apoptosis by inducing initial cell membrane damage, which leads to limited cell swelling, release of cytosolic granules, mitochondrial polarization-depolarization, lipid raft aggregation (data not shown), rupture, and DNA fragmentation. No phosphatidylserine (PS) exposure (data not shown), cell shrinkage, and membrane blebbing were observed. Apoptosis is a process of programmed cell death, which involves initial PS exposure onto the cell surface, membrane blebbing, loss of adherence, shrinkage, nuclear breakup, and chromosomal DNA fragmentation. 44 The process is caspase dependent. However, in caspase-independent death or a death process called necroptosis, no PS exposure and nuclear and DNA fragmentations are shown.44,45
Our data support the notion that MEK1 blocks the apoptotic function of WOX1 via a direct interaction. MEK1 physically interacts with both the WW and SDR domains of WOX1, and PMA effectively increases the binding for MEK1 with the SDR domain in COS7 cells. The increased binding correlates with the intrinsic resistance of COS7 cells to PMA-induced apoptosis. In contrast, PMA triggers the dissociation of WOX1 from MEK1, which allows WOX1 to relocate to mitochondria and probably nuclei for causing apoptosis. In previous studies, we have shown that WOX1 exerts its apoptotic function in the mitochondria and nuclei.15,24,25,28-32 MEK1 possesses a proline-rich loop domain. 46 Whether the WW or SDR domain interacts with the proline-rich loop in MEK1 is unknown and remains to be established.
WOX1 participates in multiple signaling networks.16,18-20 Crosstalk of the WOX1/MEK1 signaling with other signal pathways is likely. For example, WOX1 physically interacts with transcription factor c-Jun both in vivo and in vitro.31,32,47 Mitogen-activated protein kinase kinase kinase 1 (MEKK1) phosphorylates c-Jun, which leads to the binding of WOX1 with c-Jun. UV radiation also promotes the binding of WOX1 with c-Jun. Depending on the cell lines, WOX1 may enhance or suppress the trans-activation of activating protein 1 (AP-1) via c-Jun. 47 MEKK1 activates downstream MEK1 and JNK1 in the MAP kinase pathways. 48 MEKK1 is also activated in response to DNA damaging agents such as irradiation, etoposide, and cisplatin. 49 However, whether stress-induced MEKK1 activation regulates the interaction between MEK1 and WOX1 is unknown. The Raf/Ras/MEK/ERK signal pathway promotes the transcription factor AP-1 activation via activating c-Fos and c-Jun, respectively. 50 AP-1 binds to the TPA-responsive elements (TRE) for gene induction. Therefore, AP-1 is also considered the terminal signal molecule conducted by PMA. Our observations provide a new functional role of WOX1 for interacting with MEK in the Raf/Ras/MEK/ERK signal pathway for controlling gene transcription.
Human and mouse WWOX/WOX1 plays a critical role in immune cell differentiation. Ablation of the Wwox gene in mouse revealed that Wwox participates in organogenesis, bone metabolism, steroidogenesis, and postnatal survival.19,20 Notably, the Wwox knockout mice develop leukopenia, B cell lymphomas, and splenic atrophy,19,20 suggesting an essential role of the Wwox gene in control of the development of the murine immune system. In humans, alteration of the WWOX gene has been shown in B and T cells in AIDS patients.51,52
In summary, we have shown a critical role of WOX1/MEK1 as a molecular switch in controlling apoptosis of T cells. Dissociation of WOX1/MEK1 leads to cell death. The cell death is probably associated with relocation of WOX1 to the mitochondria. Whether WOX1/MEK1 can act as a molecular switch for apoptosis in other types of cancer cells remains to be established. Inhibition of the Raf/Ras/MEK/ERK signal pathway by targeting Raf and MEK has been used as strategies in cancer therapies.53,54 We believe that failure of the therapeutic effects could be due to the drugs’ inability to cause dissociation of WOX1/MEK1.
Materials and Methods
Cell lines and chemicals
Cell lines used in these studies were human leukemia lymphoblast cell lines and Jurkat and Molt-4 T lymphocytes.33,34 These cells were routinely grown in RPMI-1640 medium (Sigma, St. Louis, MO) containing 10% heat-inactivated fetal bovine serum (Gibco, Carlsbad, CA), according to the instructions of the American Type Culture Collection (ATCC; Manassas, VA). Cells were grown at 37°C in an incubator with humidified atmosphere plus 5% CO2. Additional cell lines used were adherent human breast cancer MCF7, MDA-MB-231, and MDA-MB-435S cells; human colon cancer HCT116 cells; human lung cancer NCI-H1299 cells; human neuroblastoma SK-N-SH cells; human skin squamous cell carcinoma SCC4, SCC9, and SCC15 cells; mouse L929 fibrosarcoma and melanoma B16F10 cells; monkey kidney fibroblast COS7; and nonadherent THP-1 monocytic cells. PMA, 1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenylthio) butadiene (U0126), propidium iodide, caspase 8 inhibitor IETD-fmk, and staurosporine were from Sigma. 4′,6-Diamidino-2-phenylindole (DAPI) and pan-caspase inhibitor zVAD-fmk were from Calbiochem (San Diego, CA); MitoTracker Red, LysoTracker Red, and Vybrant Lipid Raft labeling kit from Molecular Probes/Invitrogen (Carlsbad, CA).
DNA fragmentation assay
Analysis for internucleosomal DNA fragmentation was performed as described.15,25 Briefly, Jurkat and Molt-4 T cells were pretreated with or without U0126 for 1 hour, followed by exposure to PMA in a dose-dependent manner (2.5-40 µM). After 16 to 24 hours, cells were washed once with phosphate-buffered saline (PBS) and then lysed in a cold lysis buffer (10 mM Tris [pH 8.0], containing 150 mM NaCl, 5 mM EDTA, and 0.5% Triton-X 100). The lysates were incubated on ice for 30 to 60 minutes. Total DNA was extracted with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1), followed by precipitation with 0.1x volume of 3 M sodium acetate, pH 5.2, and 2 volumes of ethanol. The pellets were dissolved in sterilized MilliQ H2O and treated with 0.8 µg/µL RNase A (Sigma) for 1 hour at 37°C. The DNA preparations were analyzed by 2% agarose gel electrophoresis, stained with ethidium bromide, and visualized under a UV illuminator. The images were photographed by an image system (Alpha Innotech, Santa Clara, CA) and quantified by Adobe Photoshop software or NIH Image J.
Cell cycle analysis
Jurkat and Molt-4 T cells were pretreated with or without U0126, followed by exposure to PMA in a dose-dependent manner. After 16 to 24 hours, cells were precipitated by centrifugation at 2000 rpm and gently washed once with PBS and fixed in 70% ethanol for 30 minutes.25,29 Following fixation with ethanol and precipitation (centrifugation at 2500 rpm in microfuges), the cells were stained with propidium iodide solution (2 µg/mL, containing 10 µg/mL RNase A) for 30 minutes at room temperature. The DNA contents were analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ).25,29
PMA induction of Molt-4 T cell differentiation
The less differentiated Molt-4 T cells were stimulated with low doses of PMA (1-10 nM) for 2 days, followed by culturing in medium only for 1 day. The PMA stimulation cycle was repeated 3 times. During stimulation, morphological changes and sizes in diameters of the cells were photographed and measured using a Nikon Eclipse TE2000-5 microscope (Nikon, Tokyo, Japan). At day 9, these cells were harvested and challenged with high doses of PMA (2.5-40 µM) to induce cell death.
Antibodies, Western blotting, and co-immunoprecipitation
We have generated specific rabbit antibodies against the N-terminal amino acid sequence of WOX1 and its first WW domain with Tyr33 phosphorylation.15,25 Both antibodies are pan-specific for human, mouse, and rat WWOX/WOX1. Commercial antibodies used in Western blotting were against ERK (Transduction Laboratories, Lexington, KY), MEK, phospho-MEK (p-MEK), phospho-ERK (p-ERK) (Santa Cruz Biotechnology, Santa Cruz, CA), and α-tubulin (Sigma). Co-immunoprecipitation was performed as described.15,24-25,27-32
Dominant negative WOX1 and effect on PMA-induced apoptosis
Expression constructs for wild-type WOX1 and dominant negatives (DN-WOX1) were made as described. 25 For the dominant negatives, both the full-length WOX1 and the WW domain constructs were altered at Lys28 and Asp29 to Thr28 and Val29, respectively, in the first WW domain (DN-WOX1). 25 These constructs were made in pEGFP-C1 vector (Clontech, Mountain View, CA). Jurkat T cells were electroporated with the indicated expression constructs in the presence of 400 µg/mL albumin (10 µg DNA/3 × 106 cells; 200 volt and 60 msec; BTX ECM 830 Square Wave Electroporator, Genetronics, San Diego, CA). These cells were cultured for 48 hours and then exposed to PMA for 16 to 24 hours. Subsequently, DNA fragmentation and cell cycle analysis were performed. Expression of EGFP-tagged proteins was confirmed by fluorescence microscopy.
Förster (fluorescence) resonance energy transfer (FRET)
Binding of MEK1 with WOX1 was analyzed by FRET as described.29,32 The full-length and the N-terminal WW domains of mouse WOX1 were constructed in frame with EGFP and human MEK1 in frame with DsRed, respectively. Additional constructs were SDR and SDR/D3 domains of WOX1 (Fig. 7B). Human WWOX was also constructed in the pDsRed2-C1 vector. COS7 fibroblasts were cultured on cover slips overnight and transfected with the MEK1 and the full-length or the WW domain of the WOX1 construct by liposome-based Genefector (Venn Nova, Pompano Beach, FL). Following 24 to 48 hours in culture, cells were stimulated with an excitation wavelength of 455 nm for EGFP, and FRET signals were detected at an emission wavelength of 600 nm for DsRed (Nikon Eclipse TE-2000U inverted fluorescence microscope). The background fluorescence and spectral bleed-through of FRET images were corrected from an area free of cells. The spectrally corrected FRET concentration (FRETc) was calculated by Youvan’s equation (using Image-Pro 6.1; Media Cybernetics, Bethesda, MD) to normalize the FRET signals to the expression levels of fluorescent proteins: FRETc = (fret – bk[fret]) – cf[don]*(don – bk[don]) – cf [acc]*(acc – bk[acc]), where fret = fret image, bk = background, cf = correction factor, don = donor image, and acc = acceptor image.
Time-lapse fluorescence microscopy
Time-lapse fluorescence microscopy was carried out to measure morphological changes in Jurkat and Molt-4 T cells in response to PMA. 25 The cells were labeled with red fluorescent cholera toxin for lipid raft or JC-1 for mitochondria (Molecular Probes/Invitrogen) and then challenged with PMA (10- 100 µM). Videos were taken per frame at 1- or 10-minute intervals.
Statistical analysis
All experiments were carried out 3 to 5 times. Where indicated, statistical analysis was carried out using Student t test analysis (Microsoft Excel; Microsoft, Redmond, WA).
Footnotes
The authors declared no conflicts of interest regarding the authorship and/or publication of this article.
This research was supported by the Department of Defense, USA (W81XWH-08-1-0682); the National Science Council, Taiwan (NSC96-2320-B-006-014, 97-2628-B-006-041-MY3, and 97-2628-B-006-045-MY3); the National Health Research Institute, Taiwan (NHRI-EX97-9705BI); and the National Cheng Kung University Landmark Projects (C0167 and R026).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.