
Abstract
Among the greatest challenges facing organisms is that of detecting and effectively responding to life-threatening environmental changes that are intimately associated with metabolic fluctuations and certain forms of stress. These conditions have been linked to the onset of many human pathologies, including cancer. Over the past decade, members of the Sir2 family, or sirtuins, have been described as major players in sensing and coordinating stress response. Evidence has imputed mammalian sirtuins in carcinogenesis, although the mechanisms involved seem to be more diverse and complex than previously anticipated. Some sirtuins, such as SirT2 and SirT6, seem to work as tumor suppressors, but others, such as SirT1, are apparently bifunctional: operating as both tumor suppressors and oncogenic factors depending on the context and the study conditions. The mechanisms underlying these apparently contradictory activities are not well understood, although recent findings suggest that they might actually be two sides of the same coin. In this review, the authors summarize current knowledge on the functional implications of sirtuins in cancer and discuss possible explanations for their functional duality.
Over the past decade, the members of the Sir2 family, or sirtuins, have garnered tremendous attention in biomedical research. The founder member of the family, yeast Sir2p, was originally identified in Saccharomyces cerevisiae as a factor involved in rescue of mating deficiency. 1 Sir2p is involved in the epigenetic silencing of mating-type loci, nucleolar rDNA, and telomeres,2,3 through establishment of a heterochromatin-like compact structure in which the N-terminal tails of histones H3 and H4 are hypoacetylated.4,5 The significance of Sir2p function is reflected by the established link between Sir2p, longevity, and genome stability. 4
Sirtuins are present from bacteria to humans. 6 Although they have diversified and acquired new functions throughout evolution, their main functions seem to be to detect changes in the redox state of the cell resulting from stress (whether oxidative, metabolic, or genotoxic) and to coordinate an adequate response. Sirtuins are NAD+-dependent protein deacetylases and mono-[ADP-ribosyl]transferases.7-9 The ability of sirtuins to sense energy fluctuations in the cell is linked to their requirement of NAD+ as a cofactor for enzymatic activity. Sirtuins are defined by their homology to the catalytic domain of Sir2p, which spans approximately 250 residues. Sirtuins differ in their specificity and catalytic activity. For example, some seem to show ADP-ribosyltransferase activity, yet not all of them have detectable deacetylase activity. Although most sirtuins seem to have a broad range of histone and nonhistone protein substrates, some of them are strictly specific histone deacetylases (HDACs), whereas others seem to target nonhistone proteins.3,10-13 Mammalian sirtuins, also referred to as class III HDACs, comprise 7 members (SirT1-7) that differ widely in their localization, activity, and functions. SirT1, 6, and 7 are mainly nuclear; SirT2 and SirT3 are mainly cytoplasmic and mitochondrial, respectively, although both are present in limited levels in the nucleus; and SirT4 and 5 are strictly mitochondrial.3,12-14 Some sirtuins can relocalize in function of cell or tissue type, developmental stage, metabolic status, and certain stress conditions, suggesting that localization is important for regulating their function. Mammalian sirtuins perform myriad functions that can be classified according to 4 processes: chromatin regulation, cell survival under stress, metabolic homeostasis regulation, and developmental and cell differentiation. 15 The consequences of energy imbalance have been consistently associated with the onset and/or development of many human pathologies. Consistent with sirtuins playing an important role in this response at both the cellular and organism levels, they have been linked to cancer, diabetes, cardiovascular diseases, and neurodegenerative diseases, among other maladies.
Sirtuins apparently play contradictory roles in cancer, as illustrated in recent findings.14,16,17 On one hand, some sirtuins help protect DNA (from damage and oxidative stress), maintain genomic stability, and limit replicative life span, all of which suggest that they would protect organisms against cancer. On the other hand, some data suggest that promotion of cell survival under stress conditions by sirtuins could be directly involved in tumorigenesis, as it would inhibit senescence and allow unchecked cell division. As we explain in the coming sections, sirtuins have developed an elaborate network of interactions and targets that are directly involved in cell growth, cell cycle progression, genome integrity, and cell death, which probably enables them to finely modulate cellular physiology under stress conditions.
Sirtuins and Genome Stability
Genomic instability features in the onset of most cancers. In fact, an unstable genome is a hallmark of nearly all solid tumors and adult-onset leukemias. 18 The various factors that contribute to genome destabilization include different forms of stress, such as oxidative stress and genotoxic stress. Given their role in stress response, sirtuins are closely related to the regulation of chromatin dynamics in these compromising situations. Thus, they help stabilize the genome at two different levels: regulation of chromatin structure and expression and modulation of DNA repair (Figure 1).
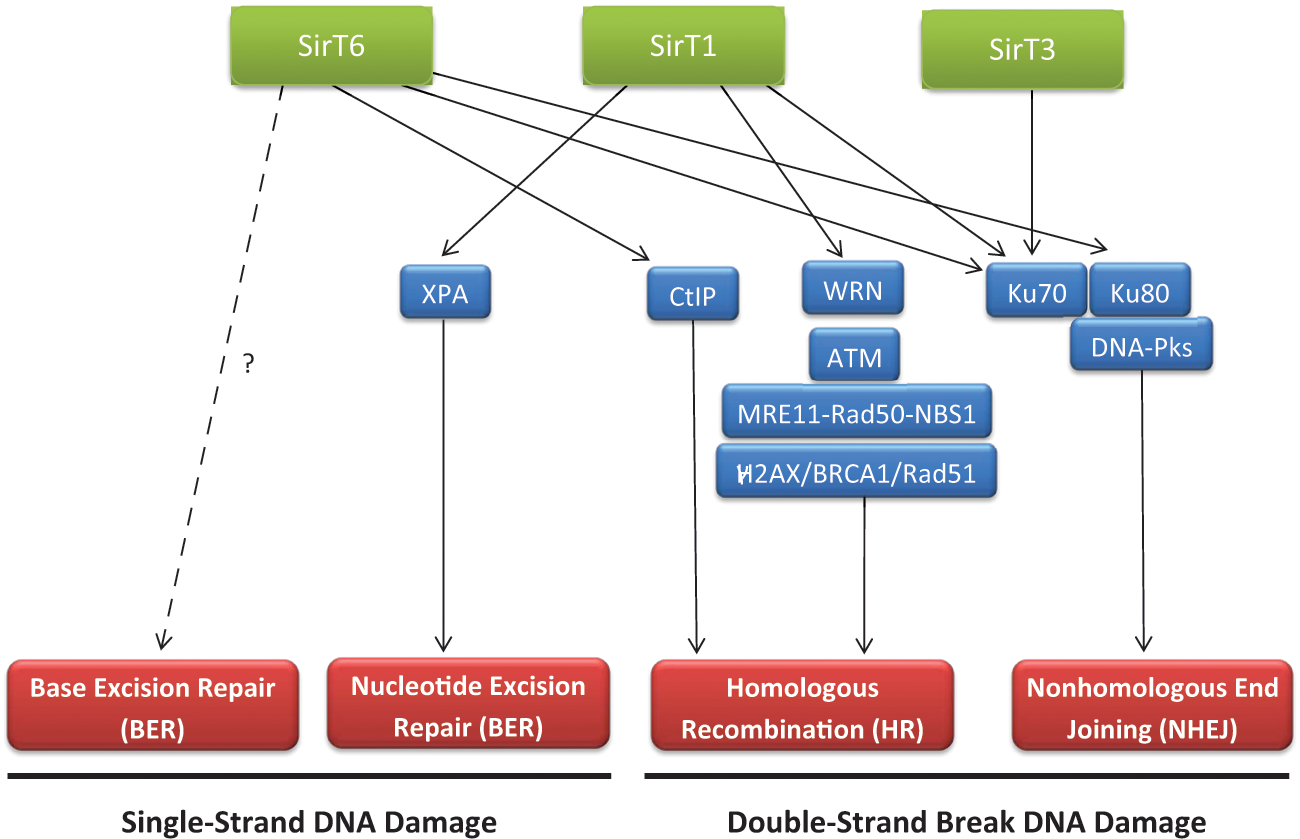
Sirtuins and DNA repair. Sirtuins are involved in DNA damage signaling and different DNA repair pathways (in red). The factors and pathways, through which they exert this function, are indicated. The link between SirT6 and BER has been established, although whether it is direct or not is still under discussion.
Sirtuins and Chromatin Regulation
Despite having adopted various functions over the course of evolution, sirtuins have remained intimately connected to chromatin regulation through a functional link to the regulation of two histone posttranslational modifications that are crucial for chromatin structure and epigenetic phenomena: H4K16Ac and H3K9Ac. 6 Among mammalian sirtuins, SirT1, 2, 6, and 7 have been shown to exert most of their function via chromatin regulation; the case of SirT3 is more complicated. However, only SirT1-3 and 6 help regulate chromatin through deacetylation of histones and nonhistone proteins.
SirT1 function is the best characterized among the mammalian sirtuins. It is intimately linked to formation of both types of heterochromatin (facultative heterochromatin [FH] and constitutive heterochromatin [CH]). SirT1 coordinates FH formation by deacetylating H4K16Ac, H3K9Ac, and H1K26Ac and by promoting H3K9me3 methylation through a close functional relationship with the histone methyltransferase Suv39h1, a keystone of chromatin organization.19-21 Suv39h1 methylates H3K9me3, which is recognized by the heterochromatin structural protein HP1 through its chromodomain, establishing the basis of heterochromatin structure. 22 Formation of FH by SirT1 is crucial in cellular response to stress. A clear example is the inhibition of ribosomal gene expression upon oxidative stress. Under these conditions SirT1, Suv39h1, and nucleomethylin together induce FH formation in rDNA, silencing the expression of ribosomal genes and, consequently, decreasing the cellular production of proteins. 23
Formation of FH by SirT1 is also important in the context of development in differentiation: SirT1 is part of PRC4, a complex that contains the polycomb H3K27me3 HMT Ezh2. This complex, which preferentially targets H1K26me3, seems to be involved in early development, in conditions of undifferentiation. This agrees with a general role for SirT1 in the inhibition of differentiation.24-27 Moreover, this might have important implications in cancer: first, because both SirT1 and Ezh2 protein levels are high in undifferentiated cells and decrease drastically upon differentiation and, second, because high levels of both factors correlate to cancer progression in a mouse model of prostate cancer. 28
The interaction of SirT1 with CH is more complex, and evidence suggests that it occurs at two different levels. First, some authors have claimed that SirT1 may be present in both pericentromeric and telomeric regions of CH.29,30 This would support a role for SirT1 in either the establishment or the maintenance of said regions. Indeed, loss of SirT1 has been associated with a loss of pericentromeric CH foci and desilencing of the γ-satellites in mice. 31 Regarding telomeres, SirT1 was recently reported to contribute to genomic integrity via positive regulation of telomere length in vivo. 30 SirT1 overexpression in mice decreases the rate of telomere erosion, whereas SirT1 deletion increases telomere shortening; the authors of the aforementioned study suggest that SirT1 overexpression affects the telomerase pathway. 30 However, the levels of SirT1 in these regions remain unknown, particularly in the case of pericentromeric CH: although chromatin immunoprecipitation experiments have detected SirT1 in these regions, immunofluorescence studies do not indicate any local SirT1 enrichment and, moreover, do not reveal any localization. 21 These seemingly contradictory results suggest that SirT1 levels are very limited, thereby challenging the view that SirT1 involvement is similar in pericentromeric and telomeric CH formation as in FH formation. Other evidence strongly suggests a direct role for SirT1 in CH formation and/or maintenance that would entail regulation of the available pool of nuclear Suv39h1. 31 These studies have shown that the oxidative/metabolic stress response includes upregulation of Suv39h1 levels through a SirT1-dependent mechanism. SirT1 decreases Suv39h1 degradation by inhibiting Suv39h1 polyubiquitination by MDM2. This increase in Suv39h1 levels enhances Suv39h1 turnover in CH, which in turn seems to accelerate renewal of the heterochromatin structure. This accelerated renewal correlates with greater genomic integrity during stress response. These observations reflect the first direct link between stress response and structural maintenance of CH and also support a view of chromatin as a dynamic entity under constant adaptation to environment. 31
In contrast to the role of SirT1 as a genome protector, through the mechanisms we have explained above, other evidence suggests that SirT1 might actually cause the opposite effect. For example, Fraga et al. 32 have described loss of global H4K16Ac and H4K20me3 as a hallmark in human tumors. Considering that SirT1 and SirT2 are the only sirtuins that seem to control global levels of H4K16Ac and that of these, only SirT1 seems to be upregulated in certain cancers, suggests that H4K16Ac loss in cancer may at least partially derive from SirT1 activity. Moreover, SirT1 is responsible for directly silencing certain tumor suppressor genes, and it has been detected in tumor suppressor promoter regions that correlate with hypoacetylation of H4K16 and H3K9 and to dense hypermethylation of 5′CpG islands. SirT1 inhibition leads to reexpression of these genes without affecting DNA methylation levels. 33 Taken together, these observations suggest that the SirT1 overexpression described in some cancers could help silence tumor suppressor genes, which in turn would promote cancer development.
SirT6 was recently described to have histone deacetylase activity; since then, two of its targets have been identified: H3K9Ac and H3K56Ac.34-36 Evidence suggests that SirT6-mediated histone deacetylase activity promotes a specialized telomere chromatin structure required for genomic stability,18,37 which suggests that SirT6 plays an important role in cancer and aging. In addition, global hyperacetylation of its target H3K56Ac has been described in various human cancers, including skin, thyroid, breast, liver, and colon cancers. Interestingly, loss of this modification correlates with tumor grade. 38 This body of evidence suggests that SirT6 acts as a tumor suppressor by maintaining genome stability.
Another two sirtuins have been reported to participate in chromatin regulation: SirT2 and SirT3. SirT2, whose main role is in cell cycle regulation, regulates global levels of H4K16Ac. 39 H4K16Ac deacetylation occurs specifically during the G2/M transition, when SirT2 is shuttled to the nucleus, enabling chromatin compaction during metaphase. 40 However, this process is not well understood; therefore, future work is required to elucidate the role of SirT2 in chromatin regulation and cell cycle control.
Although SirT3 is localized chiefly to the mitochondria, where it is apparently the main protein deacetylase, 41 it also has a small nuclear population. This is consistent with its specificity for H4K16Ac and H3K9Ac as deacetylation substrates. 42 However, unlike knockdown of SirT1 or SirT2, knockdown of SirT3 does not result in global H4K16 hyperacetylation, 42 which suggests that SirT3 might be involved in the regulation of limited regions of the genome (i.e., a limited number of genes). However, more studies are required to determine the role of SirT3 in chromatin regulation.
Sirtuins and DNA Repair
DNA repair is essential for preserving the fidelity of genomic information. Sensing and removing damage generated by environmental events (e.g., stress or cell metabolism) or by the inevitable errors that arise during normal cell division is a critical challenge for all organisms, given that these phenomena are paramount in tumorigenesis. 18 In hereditary cancers, genomic instability results from mutations in DNA repair genes, whereas in sporadic (nonhereditary) cancers, the molecular basis of genomic instability remains unclear. SirT1 was recently reported as playing an important role in DNA repair and in maintaining genome stability: specifically, SirT1−/− embryos showed more chromosomal aberrations and impaired DNA repair than did wild-type (WT) embryos. 16 As we described above, in yeast, one of the main functions of Sir2p is to silence the mating-type loci HML and HMR. 2 However, during DNA damage, the Sir2p complex dissociates from HM loci and relocalizes to sites of DNA breakage,43-46 which results in transient expression of genes related to DNA repair in HM loci as well as in chromatin remodeling around DNA break sites to facilitate recruitment of repair factors.47,48 Recruitment of SirT1 to double-strand breaks (DSBs) is similar to that of yeast Sir2p and, together with other histone-modifying enzymes, induces epigenetic changes around the break site that result in chromatin remodeling.48,49 At DSBs, SirT1 may deacetylate histones and/or DNA repair factors. SirT1 is important for modulating γ-H2AX, BRCA1, Rad51, and NBS1 foci formation upon γ-irradiation, through direct recruitment of these proteins to the DNA damage site. 16 Accordingly, SirT1 directly interacts with and deacetylates NBS1, a checkpoint factor that is involved in detection and activation of DNA repair and that is part of the DNA damage sensor complex MRN (MRE11-RAD50-NBS1). 50 In related work, Oberdoerffer et al. 29 showed that the relocalization of SirT1 to DSBs depends on ATM-mediated signaling through H2AX phosphorylation, which parallels the situation in yeast, whereby recruitment of Sir2p to DSBs requires DNA damage signaling via MEC1, the ATM ortholog. Other groups have reported that SirT1 regulates repair of certain DSB events independently of the signaling kinase pathway. 51 In particular, SirT1 is involved in homologous recombination (HR) through a Rad51-independent mechanism that involves the WRN helicase. 51 This functional relationship between SirT1 and WRN is supported by previous studies that reported that deacetylation of WRN by SirT1 increases WRN activity and function in DNA repair. 52 Corroborating a role for SirT1 in HR, SirT1-tg mice, which contain an extra copy of the SirT1 gene, exhibit higher frequencies of sister chromatid HR events at telomeres, centromeres, and chromosomes arms, thereby indicating that SirT1 overexpression may affect DNA repair efficiency. 30
Another interesting link between SirT1 and DNA repair is Ku70, which is involved in DNA repair of DSBs by nonhomologous end joining (NHEJ). 53 Ku70 is mainly located in the nucleus, where it participates in DNA repair, and a small fraction is located in the cytoplasm, where it regulates apoptosis. In response to DNA damage, Ku70 is acetylated at multiples lysines, which results in a Ku70-BAX dissociation that permits translocation of BAX to the mitochondria to promote apoptosis. Deacetylation of Ku70 by SirT1 blocks the conformational change in BAX and its translocation to the mitochondria, consequently inhibiting mitochondrial apoptosis and inducing Ku70-dependent DNA repair.54,55 Apart from SirT1, SirT3 has also been described as promoting cell survival through Ku70 deacetylation in response to genotoxic agents, suggesting that SirT3 also could be involved in Ku70-dependent DNA repair signaling. 56
SirT1 is also involved in repairing damaged single-strand DNA. It has been imputed in UV-induced DNA repair through the nucleotide excision repair (NER) pathway. UV irradiation stimulates the xeroderma pigmentosum group A (XPA) protein, a NER core factor, which interacts with SirT1 and leads to decreased levels of acetylated XPA, 57 which is required for optimal NER pathway function.
Among the remaining sirtuins, SirT6 is also important in DNA repair. Loss of SirT6 in mice correlates with accumulated mutations and decreased genomic stability. SirT6−/− mouse embryonic fibroblasts (MEFs) and SirT6−/− mouse embryonic stem cells (mESCs) are associated with a reduced proliferative rate and DNA damage hypersensitivity, as well as harbor multiple chromosomal defects, including fragmentation, detached centromeres, gaps, and translocations.14,17 SirT6 was initially thought to play a role in base excision repair (BER) for two main reasons: first, hypersensitivity of SirT6−/− cells to DNA damage has been remedied by overexpression of the isolated dRP lyase domain of DNA polymerase β, which catalyzes the rate-limiting step in BER 17 ; second, BER and maintenance of genomic stability require that the deacetylase domain of SirT6 be intact. 17 However, the role of SirT6 in BER has not been unequivocally demonstrated: in DNA damage experiments, SirT6 has not been found to physically interact with BER factors or to co-localize with these factors at the damage sites.17,37 Recent biochemical and functional studies of SirT6 in human cells have revealed that SirT6 helps enable efficient DNA DSB repair. 58 Furthermore, SirT6 has been reported to interact with proteins involved in the NHEJ pathway of DSB repair, including DNA-PKcs and Ku70/80. 58 Consistent with this finding, SirT6 has been shown to dynamically associate with chromatin in response to DSBs, promote a global decrease in H3K9Ac levels following DNA damage, and stabilize the DNA damage-dependent association of DNA-PKcs with chromatin. This suggests that SirT6 is required for changes in chromatin structure at DSBs that enable efficient recruitment of DSB repair proteins. Kaidi et al. 59 recently reported that, in addition to its function in DNA repair, SirT6 also promotes DNA end resection—a crucial step in DSB repair by HR—by binding to and deacetylating CtIP (C-terminal binding protein interacting protein).
This body of evidence suggests that SirT1, SirT6, and possibly SirT3 have important and diverse roles in DNA repair and supports a role for each as a tumor suppressor via their respective functions in repairing DNA damage, maintaining genome integrity, and inhibiting tumorigenesis 60 (Figure 1).
Sirtuins and Stress Response
Throughout evolution, from early eukaryotes onwards, sirtuins have helped organisms adapt to oxidative, metabolic, or genotoxic stress by activating the stress response pathway through various factors, controlling chromatin integrity and expression, signaling DNA damage and repair, and modulating cell metabolism. However, sirtuins are not always committed to cell survival: under certain extreme conditions, such as chronic stress, SirT1, SirT2, and SirT3 can protect the organism by inducing cell senescence or apoptosis61-63 (Figure 2). Herein lies what may be a cornerstone for understanding the contradictory nature of sirtuins in cancer. SirT1 best illustrates this contradiction: whereas its overexpression inhibits oncogene-induced senescence 64 and its inhibition by Sirtinol results in a senescence-like cell growth arrest with attenuated Ras-MAPK signaling in human cancer cells,65,66 SirT1−/− MEFs are more resistant to senescence than are WT MEFs. 61
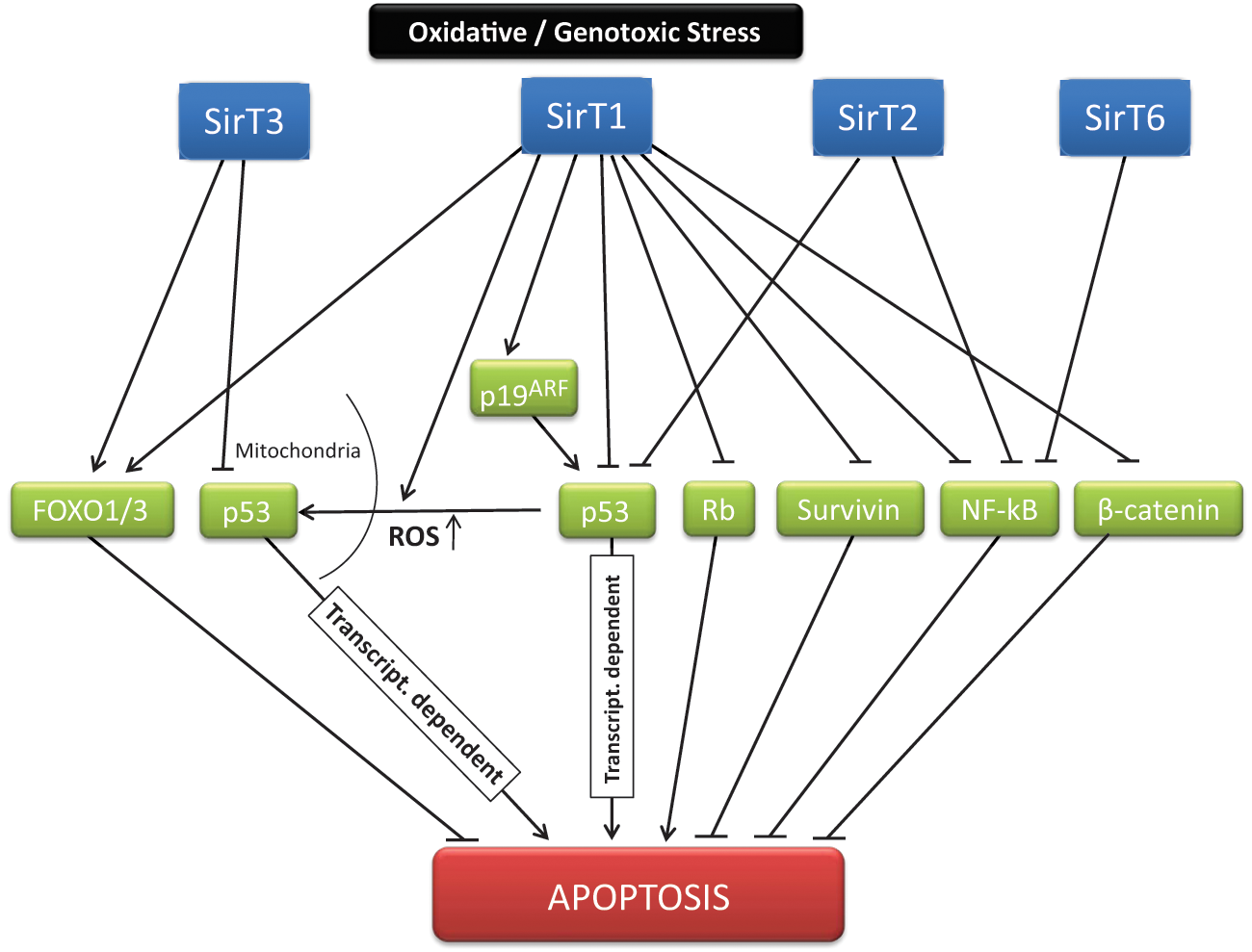
Sirtuins and cell survival. Under different forms of stress, sirtuins control cell fate through, among other mechanisms, modulation of apoptosis. The decision process before a particular situation is based as a result on a complex net of interactions and targets established by different sirtuins. The main described mediators and pathways of this sirtuin-dependent signaling are indicated.
SirT1 has been linked to stress response through its regulation of many important factors in cancer. Among the most important of these is p53, which is critical for cell cycle checkpoint regulation, apoptosis, and tumor suppression.67,68 In addition, more than half of all human cancers are related to p53 mutations, and a strong body of evidence suggests that cancers in which p53 is not mutated exhibit some alteration in its pathway.69-71 SirT1 apparently regulates both types of known p53-mediated apoptosis (p53-transcriptional dependent and p53-transcriptional independent). SirT1 regulates p53 in various ways—chiefly, via deacetylation of p53, which induces inactivation of p53 and associates with inhibition of p53-dependent apoptosis.67,68 Accordingly, SirT1−/− mice exhibit lower levels and hyperacetylation of p53 and higher levels of radiation-induced apoptosis than do WT mice. 72 This observation has led researchers to hypothesize that SirT1 activity may elevate cancer risk in mammals by inhibiting p53-induced apoptosis.73-76 However, other evidence suggests that SirT1 could regulate p53 in a more complex fashion. For instance, SirT1 could drive activation of p53 via regulation of p19ARF. In fact, SirT1−/− MEFs show lower levels of the tumor suppressor p19ARF, which positively regulates p53. 61 p19ARF does not appear to be acetylated, and despite the lack of understanding of how SirT1 regulates expression of p19ARF, this regulation appears to have major implications for how SirT1 regulates senescence through p53. 61 Thus, SirT1 seems to limit replicative life span by regulating p53 via p19ARF, but in response to acute DNA damage, SirT1 promotes DNA repair and survival by deacetylating p53, which inhibits p53-induced apoptosis.61,72 Another mechanism by which SirT1 regulates p53 is regulation of the subcellular localization of the latter, as part of the mitochondrial-dependent apoptotic response. 77 Thus, deacetylation of p53 K379 by SirT1 in mESCs inhibits nuclear localization of p53.72,77 When intracellular reactive oxygen species (ROS) are high, SirT1 deacetylates p53 and blocks its nuclear translocation, leading to accumulation of p53 in both the cytosol and the mitochondria. This in turn results in a transcription-independent p53-induced apoptosis.
However, the relationship between SirT1 and p53 is not as obvious as it might seem. In vivo studies have revealed that SirT1 does not seem to clearly affect p53-dependent functions, and none of the observed phenotypes in SirT1−/− background, which include hypersensitivity to radiation and apoptosis, seem to depend on p53 activity. 78 This contradiction between in vivo data and in vitro data may stem from functional redundancy among sirtuins. At least two other sirtuins have been shown to regulate p53: SirT2 and SirT3. SirT2 not only functions as a mitotic checkpoint in response to mitotic stress but also regulates cell death in response to certain conditions of DNA damage-induced stress.79,80 Matsushita et al. 79 observed that, compared to WT DT40 cells, SirT1- and SirT2-deficient DT40 cells exhibited significantly greater reporter activation by p53 and its related factor p73 in response to ionizing radiation. This suggests that SirT2 could downregulate p53 and p73 activity in response to DNA damage. Consistently, recent work suggests that downregulation of SirT2 causes apoptosis in cancer cell lines such as HeLa, but not in normal cells, through accumulation of p53, which results from p38 MAPK activation-dependent degradation of p300 and subsequent MDM2 degradation. 80 In the case of SirT3, recent reports suggest that it acts as a protein regulator of p53-induced senescence. 81 As we mentioned earlier, p53 executes some of its antiproliferative functions in the mitochondria. 82 SirT3 partially abrogates p53 activity to promote growth arrest and senescence. This inhibitory effect of SirT3 over p53 is blocked by interaction of p53 with BAG-2, a component of the CHIP ubiquitin ligase complex. 81 The researchers discovered a network in which sirtuins and p53 co-chaperones may coordinate cellular fate independently of transcriptional activity.
Other key players in the stress response regulated by sirtuins are the forkhead-box (FOXO) family of transcription factors, which are very important in both stress response and cancer because of their roles in cell cycle arrest, DNA repair, and apoptosis.83-86 FOXO proteins are tumor suppressors, and they were recently found as fusion proteins following chromosomal translocations in various cancers.87-93 In response to oxidative or genotoxic stress, FOXO proteins translocate from the cytoplasm to the nucleus, where they activate myriad genes involved in cell cycle arrest, DNA repair, and apoptosis.94-96 Acetylation of FOXO reduces its binding to DNA and enhances its phosphorylation and inactivation. 97 SirT1 deacetylates FOXO proteins, promoting transcription of FOXO targets involved in stress resistance and a decrease in transcription of apoptosis-related genes.94,96,98 For instance, FOXO3 deacetylation by SirT1 inhibits FOXO3’s ability to induce apoptosis after cellular stress and enhances its ability to induce cell cycle arrest; in contrast, in SirT1−/− cells, FOXO3’s ability to induce cell cycle arrest is diminished. 94 Moreover, SirT1 promotes survival after oxidative stress by inducing DNA repair in cooperation with FOXO1. SirT1 and FOXO1 are recruited to the manganese superoxide dismutase promoter, and SirT1 activity is required for transactivation of the antioxidant enzyme. 99 Supporting these findings, SirT1-dependent deacetylation of FOXO1 represses its transcriptional and pro-apoptotic activity in prostate cancer cells. 100 As in the case of p53, other sirtuins are also involved in FOXO activity regulation. In fact, SirT2 is the main deacetylase of cytosolic FOXO1. In response to stress, FOXO1 dissociates from SirT2 and is subsequently acetylated. The acetylated FOXO1 can then form a complex with Atg7, which in turn is critical for induction of autophagy, a process that has also been associated with inhibition of tumor development.101-104 In addition, Jacobs et al. 105 have suggested that FOXO3a is also a mitochondrial protein and that its mitochondrial function may be regulated by interaction with SirT3. In fact, they showed that SirT3 overexpression increases both the DNA-binding ability of FOXO3a and FOXO3a-dependent gene expression.
To further complicate this scenario, FOXO proteins and p53 share many transcriptional targets, and a report that FOXO proteins and p53 interact after oxidative stress suggests that SirT1 might regulate survival through both p53 and FOXO proteins. 94 In cells containing WT p53, FOXO proteins, and intact cell cycle checkpoints under normal conditions (i.e., no stress), SirT1 limits tumor formation by inducing cell senescence. In contrast, in conditions of DNA damage-inducing stress, SirT1 induces cell cycle arrest and DNA repair rather than apoptosis. However, loss of p53 and other tumor suppressors could result in increased SirT1 expression, which in turn could contribute to tumor formation; this is probably the reason that SirT1 depletion inhibits growth of certain tumors.66,106 Thus, further research is required to elucidate the complex functional relationship between sirtuins, p53, and FOXO proteins in tumor cells. 3
Another interesting link of sirtuins to stress is the transcriptional factor NF-κB (nuclear factor−κB). NF-κB plays a pivotal role in regulating gene expression programs related to aging, proliferation, and inflammation.107,108 SirT1 interacts with and deacetylates NF-κB, modulating its DNA binding and transcriptional activity.109-111 Treatment of cells with the SirT1 activator resveratrol correlates with a loss of NF-κB-regulated gene expression and sensitization of the cells to tumor necrosis factor α (TNFα)−induced apoptosis; these findings suggest that SirT1 activity augments apoptosis in response to TNFα via NF-κB inhibition. 109 Functioning of the NF-κB family members is also apparently regulated by SirT2 and SirT6. SirT2 has been reported to interact with p65 (an NF-κB family member) in the cytoplasm and to deacetylate p65 at K310 after TNFα stimulation. 112 Moreover, in SirT2−/− MEFs, p65 is hyperacetylated at K310, which correlates with an increase in Mpa2l gene expression. These data suggest that SirT2 deacetylases NF-κB and is an important regulator of TNFα-induced NF-κB-dependent gene expression. 112 SirT6 also plays a key role in transcriptional regulation via NF-κB. Following activation of NF-κB by TNFα, SirT6 is recruited to promoters of a subset of NF-κB target genes through a physical interaction with the NF-κB subunit RELA (p65). 113 At these promoters, SirT6 deacetylates H3K9Ac and destabilizes RELA-promoter interaction, thereby attenuating NF-κB signaling. Accordingly, SirT6-deficient human and mouse cells, as well as multiple types of SirT6-deficient mouse tissue, are associated with hyperactivation of certain NF-κB-dependent gene expression programs. 113 In addition, Kawahara et al. 113 reported that RELA heterozygosity partially rescues the premature lethality of SirT6−/− mice, attenuates some of their degenerative and metabolic defects, and reverses the excessive levels of NF-κB-driven gene expression observed. Recently, SirT6 was discovered to facilitate transcriptional regulation through HIF1α, a transcription factor important in cancer cell metabolism and cellular response to hypoxia.114,115 SirT6 was shown to interact with HIF1α and to associate with the promoters of a subset of glucose-regulatory HIF1α target genes in an HIF1α-dependent manner. 116 Accordingly, SirT6−/− mice show H3K9 hyperacetylation at these promoters and increased gene expression, which leads to increased glucose uptake and glycolysis in several tissues. 116 These data suggest that under normal nutrient conditions, SirT6 might compete with HIF1α to direct glucose flux away from glycolysis and toward oxidative phosphorylation, probably by inhibiting HIF1α- promoter association or by blocking HIF1α activity at its target genes. 37 The newly discovered roles for SirT6 as a transcriptional regulator suggest that modulation of NF-κB and HIF1α gene expression programs might be only the tip of the SirT6 iceberg, given that hundreds of genes are differentially expressed in SirT6−/− mouse cells compared with WT controls.37,113
Finally, we would like to underscore the central role of mitochondria in aging and carcinogenesis. Mitochondria are the principal site for production of the ROS that cause oxidative stress. 117 On the basis of proteomics results, researchers have recently suggested that 20% of mitochondrial proteins are acetylated—chiefly, those involved in life span and metabolism. 118 This suggests that mitochondrial sirtuins could play an important role in the altered energy metabolism and response to oxidative stress described in tumor cells. 3 Under stress conditions, such as DNA damage induced by etoposide and UV irradiation, the nuclear SirT3 population translocates to the mitochondria.41,42,119,120 Some reports have shown that SirT3 acts as a mitochondrial tumor suppressor. 121 Thus, SirT3−/− MEFs present abnormal mitochondrial physiology and increased stress-induced superoxide levels and genomic instability. Corroborating a role for SirT3 as a tumor suppressor, SirT3−/− MEFs infected with a single oncogene become immortalized and transformed. Furthermore, some human cancer specimens (e.g., some types of breast cancer) exhibit abnormally low SirT3 levels, and SirT3−/− mice develop estrogen receptor (ER)/progesterone receptor (PR)–positive mammary tumors. 121 Last, superoxide dismutase expression prevents this transformation and reverses both the tumor- permissive phenotype and the stress-induced genomic instability.
Pathways of Sirtuin Regulation in Cancer
Sirtuins have also been functionally linked to cancer-related pathways other than those described above. For example, SirT1 is functionally linked to the regulation of the tumor suppressor retinoblastoma (Rb), a nuclear protein that regulates the G1/S transition by interacting with the E2F transcription factors. 122 The activity of Rb is regulated by phosphorylation and acetylation at multiple residues. Deacetylation of Rb by SirT1 inhibits Rb-dependent apoptosis. SirT1 and Rb form a complex in which the pocket domain of Rb undergoes SirT1-dependent deacetylation, resulting in apoptosis regulation. 123
Another link of sirtuins to cancer is their functional relationship to the β-catenin pathway and, consequently, to WNT signaling. β-Catenin is the principal effector in the WNT signaling pathway, which controls the maintenance, development, and carcinogenesis of stem cells. 124 Constitutive activation of the β-catenin pathway has been found in 90% of colorectal cancers, and alteration of this pathway has been reported in many other cancers, such as breast and ovary cancers and melanoma.125,126 Two recent studies have associated increased WNT signaling to accelerated aging and have stated that calorie restriction (CR) attenuates this phenotype.127,128 Interestingly, sirtuin function has been linked to the health benefits of CR. CR induces a 2-fold increase of SirT1 expression in the intestine of rodents, which significantly reduced tumor formation, proliferation, and animal morbidity in APCmin/+ mice. 129 SirT1 deacetylates β-catenin, which suppresses localization of β-catenin to the nucleus and reduces the ability of β-catenin to activate transcription, suggesting that SirT1 functions as a tumor suppressor. 129
Another important role for SirT1 in cancer is its suppression of the apoptosis inhibitor Survivin in breast cancers associated with the tumor suppressor BRCA1, which is particularly relevant to breast and ovarian cancers and is involved in many different pathways, including genome stability protection, cell cycle control, and apoptosis. Survivin is overexpressed in cancers. In fact, BRCA1-associated breast cancers show abnormally low levels of SirT1 and high levels of Survivin. In normal cells, BRCA1 is located at the SirT1 promoter, where it increases expression of SirT1, 130 which goes on to inhibit Survivin expression by deacetylating histone H3 in nucleosomes at the Survivin promoter. 130
The oncogene B cell lymphoma 6 protein (BCL6) is another interesting SirT1 target. It is a transcriptional factor expressed in mature B cells that is required for formation of germinal centers, through repression of genes involved in differentiation and apoptosis. 131 BCL6 is involved in the pathogenesis of diffuse large-cell lymphoma and Burkitt lymphoma.131,132 Different studies have suggested that SirT1 binds to and deacetylates BCL6, thereby decreasing the oncogene’s activity.67,132
Modulation of Sirtuins in Cancer
Expression of various sirtuins is altered in many types of cancer. For example, SirT1, 4, and 7 have been described as being upregulated in certain cancers,60,133-136 whereas SirT2 is downregulated in gliomas and gastric carcinoma,137,138 as well as in melanomas, in which a mutation in the catalytic domain of SirT2 (P199L) that eliminates its enzymatic activity has been described. 139 Evidence suggests that SirT2 acts as a tumor suppressor and that its loss compromises the mitotic checkpoint, contributing to genomic instability and tumorigenesis.137,140,141 The case of SirT3 is more complex since it has been found to be upregulated or downregulated in different types of breast cancer.121,136 However, SirT5 and SirT6 levels are not known to be altered in cancer.
The mechanisms through which these sirtuins seem to be upregulated are quite diverse. Regulation of SirT1 expression in cancer is perhaps the most interesting case, as it involves multiple factors that are highly relevant to cancer. The reasons for SirT1 overexpression in cancer are unknown, and whether this overexpression actually causes tumorigenesis or is simply a consequence thereof remains uknown.60,76,133-135
SirT1 overexpression partly occurs at the transcriptional level, following loss of SirT1 promoter repressors. Among the best characterized of these factors is the tumor suppressor hypermethylated in cancer 1 (HIC1). 142 Both SirT1 mRNA and protein levels are abnormally high in HIC1−/− MEF cells. Accordingly, hypermethylation of the HIC1 promoter, which has been described in certain cases of tumor formation, results in SirT1 upregulation.72,73,143,144 HIC1 forms a transcriptional repression complex with SirT1 that, upon binding to the SirT1 promoter, inhibits SirT1 expression. 73 Accordingly, SirT1 is downregulated during aging, and cells lacking HIC1 do not exhibit this regulation, making them resistant to replicative senescence after oxidative stress and vulnerable to transformation if mutations are propagated. In addition, HIC+/− mice are tumor prone and show a p53- and SirT1-dependent block in apoptosis induction in response to DNA damage.73,144
Interestingly, a common feature of SirT1 regulation is that SirT1 apparently establishes a feedback loop with most of the factors that regulate its expression. SirT1 interacts with and deacetylates HIC1, thereby repressing its expression and regulating the activity of HIC1.73,145 Another example is the transcription factor Aiolos, a member of the Ikaros family that is involved in B cell differentiation. Aiolos negatively regulates SirT1 expression, and its loss, which is associated with lymphoma development, leads to SirT1 upregulation. 146
Another major regulatory factor in SirT1 expression is p53. As we mentioned above, SirT1 also regulates p53 activity, suggesting the existence of a regulatory feedback loop between these two proteins. Nemoto et al. 147 described two p53 binding sites in the SirT1 promoter that normally repress SirT1 expression. This could explain the SirT1 overexpression observed in tumors that have lost p53. Interestingly, a newly discovered alternative isoform of SirT1, SirT1-ΔExon8, and an autoregulatory loop between this isoform and p53 have recently been described. 148 SirT1 is alternatively spliced in a manner that is stress sensitive, p53 dependent, and conserved in mammals. SirT1 and SirT1-ΔExon8 differ in terms of stress sensitivity, RNA and protein stability, protein-protein interactions, and deacetylase activity. This new evidence suggests that the ability of SirT1 to regulate mammalian biology at different levels could be partially explained by the existence of alternate isoforms of SirT1. 148
In addition and related to p53-dependent SirT1 regulation, another (positive) feedback loop has recently been described between miR-34, p53, and SirT1. MicroRNAs (miRNAs) are small noncoding RNAs that inhibit target protein expression by posttranscriptional repression.149,150 They are essential in the development, physiology, and pathology of animals and plants and have been recently linked to tumorigenesis, tumor progression, and metastasis.151-155 p53 regulates the expression of the miR-34 family of miRNAs, which are involved in modulating cell cycle progression, senescence, and apoptosis.156-162 Interestingly, the 3′UTR of the SirT1 transcript contains a miR-34a-responsive element. Overexpression of miRNA34a due to p53 activity leads to a decrease in SirT1 protein levels, via posttranslational inhibition, and to a concomitant increase in p53 acetylation and p53-dependent apoptosis. 161 Furthermore, researchers have also found that the miR-34a/SirT1 cascade significantly contributes to chemoresistance in PC3 human prostate cancer cells—namely, ectopic miR-34a expression attenuates chemoresistance to camptothecin by inducing apoptosis.163,164 Likewise, Akao et al. 165 found that miR-34a expression was significantly downregulated in DLD-1/5FU (5-fluorouracil-resistant DLD-1) cells compared with the parent DLD-1 cells both under steady-state conditions and after 5-FU treatment. Introducing miR-34 into DLD/5FU cells significantly attenuates their resistance to 5-FU, which leads to a reduction in expression of SirT1 and the E2F proteins. This in turn suggests that the miR-34/SirT1/E2F cascade significantly contributes to resistance of DLD-1 cells to 5-FU. 165
Other regulatory feedback loops between SirT1 and different key players in cancer have been reported. These include a positive feedback loop between SirT1 and FOXO1, 166 whereby FOXO1 may regulate SirT1 expression by binding to the SirT1 promoter region. 166 A regulatory loop between SirT1 and the transcription factor E2F1, a tumor suppressor, and cell cycle and the apoptosis regulator has also been discovered. Under cellular stress or DNA damage, E2F1 binds to the SirT1 promoter and induces its expression. 167 E2F1 is acetylated by p300/CBP-associated factor (PCAF), which enhances E2F1 transcriptional activity. 168 SirT1 also deacetylates E2F1 and inhibits E2F1-induced transcription of target genes, including SirT1 itself; this may regulate induction of apoptosis in response to DNA damage. 167
Another interesting example is the recently described negative feedback loop between SirT1 and the oncogene c-Myc, which supports a tumor suppressor role for SirT1. 169 c-Myc binds to the SirT1 promoter and induces expression of SirT1, which in turn deacetylates c-Myc and negatively regulates c-Myc activity, consequently reducing c-Myc target gene expression and cellular transformation. 169 The authors of that study suggest that SirT1 blocks tumor initiation in premalignant cells by inhibiting c-Myc. 169
Another mechanism driving upregulation of SirT1 is stabilization of its mRNA mediated by HuR, a ubiquitous oncogenic RNA-binding protein that binds to 3′UTR of the SirT1 transcript. 170 HuR levels decrease dramatically as cells age and reach senescence, ultimately destabilizing SirT1 mRNA.170,171 Following oxidative damage, HuR is phosphorylated by the tumor suppressor Chk2, causing it to dissociate from SirT1 mRNA. The decrease in SirT1 levels makes cells more sensitive to apoptosis induction. 170 This is yet another mechanism in the fine balance between DNA repair and senescence: high levels of DNA damage and increased activation of Chk2 could decrease expression of SirT1, thereby shifting the balance toward apoptosis.
As we mentioned earlier, SirT1 is part of the complex PRC4. 28 Overexpression of PRC4 components such as EZH2, Suz12, Eed, and SirT1 has been reported in many cancer tissue types.28,172,173 Interestingly, overexpression of EZH2 results in overexpression of all the other PRC4 components through a currently unknown mechanism. 28 Furthermore, in stem cells, SirT1 has been directly associated with Suz12, and in breast and colon cancers, SirT1 is overexpressed, as are PcG proteins. 28
Apart from regulation of SirT1 expression, cancer and cell fate may also be linked to modulation of SirT1 activity. For example, SirT1 activity could be modulated by AROS (active regulator of SirT1), the tumor suppressor DBC1 (deleted in breast cancer 1), and the proapoptotic nuclear desumoylase SENP1. AROS enhances the activity of SirT1 by directly binding to its N-terminus, consequently inhibiting p53-mediated transcriptional activity. 174 In contrast, SENP1 desumoylates and inactivates SirT1 by binding to its C-terminus, thereby increasing p53 activity. DBC1, which is homozygously deleted in some breast cancer patients, inhibits SirT1 activity by directly binding to its catalytic domain. 75 Depleting DBC1 increases SirT1-mediated p53 deacetylation and inhibits p53-mediated apoptosis.74,75 Moreover, a recent study suggests that DBC1 disrupts the Suv39h1-SirT1 complex and inactivates both enzymes, thereby implying that DBC1 could help modulate SirT1 functioning in genome stability. 175 In contrast, recent DNA microarray data sets have detected overexpression of DBC1 in breast cancer. 176 In addition, DBC1 has been imputed in the modulation of ER-α and described as being a coactivator of androgen receptor (AR), suggesting that it may play a role in cancer cell survival.177,178 Furthermore, 2 recent studies suggest that the balance between DBC1 and SirT1 expression is lost in breast cancer and that this loss is associated with a poor prognosis for patients.179,180
Nicotinamide phosphoribosyltransferase (NAMPT) is yet another important enzymatic regulator of SirT1. It indirectly controls SirT1 activity by regulating NAD+ levels. Induction of NAMPT following certain forms of stress leads to an increase in NAD+. 181 This increase induces SirT1 activity, which triggers changes in gene expression that parallel those in cells that overexpress SirT1. 182 Inhibition of NAMPT induces premature senescence, whereas overexpression of NAMPT delays senescence and increases survival after oxidative stress in a SirT1-dependent manner. 183 NAMPT was very recently shown to be overexpressed in numerous human cancer cells along with SirT1. 184 In addition, NAD+ has been found to bind to tetrameric p53, consequently altering the conformation of p53 and preventing its binding to DNA. 185 Contrary to NAMPT function, activation of poly(ADP-ribose) polymerase (PARP) 1 after DNA damage depletes NAD+ and increases nicotinamide levels, consequently inhibiting sirtuin activity. 186
Surprisingly, a recent report indicates that although SirT1 is mainly located in the nucleus in primary cells, its primary localization in cancer/transformed cells (e.g., prostate cancer cell lines, lung and breast cancer cells, transformed cell lines, and prostate carcinoma tissue) is the cytoplasm. 187 As we mentioned earlier, localization is paramount in regulating sirtuin function. Byles et al. 187 suggest that this aberrant cytoplasm localization is one of the specific alterations of SirT1 in cancer cells and derives from greater cytoplasmic stabilization of SirT1 enabled by the PI3K/IGF-1R signaling pathway.
Sirtuins in Cancer: Tumor Suppressors, Oncogenic Factors, or Both?
The roles of sirtuins in cancer remain a subject of open discussion. Throughout this review, we have provided numerous examples that indicate that sirtuins—especially SirT1—have a dual role in cancer, operating as either tumor suppressors or as an oncogenic factor, depending on the scenario (Figure 3).
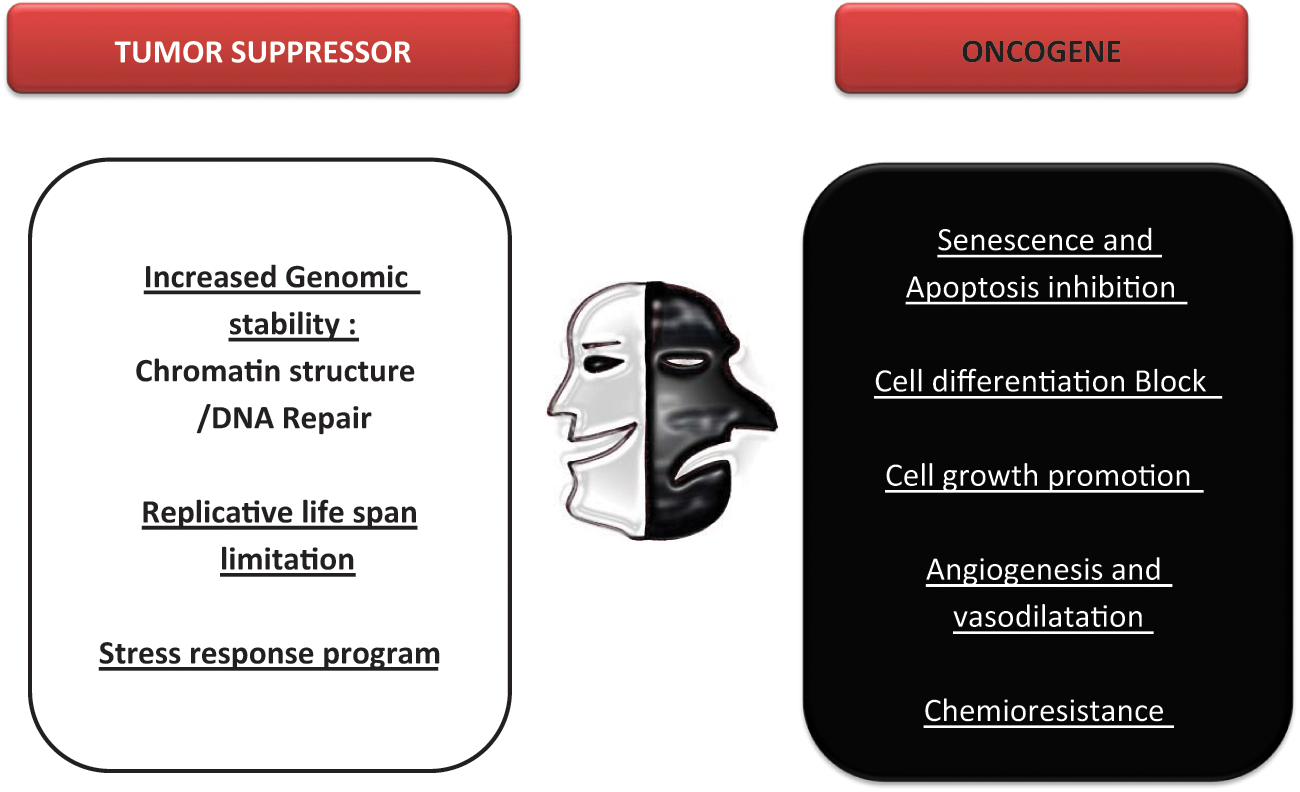
SirT1 as a tumor suppressor or/and tumor promoter. The evidence reported supports both an oncogenic and a tumor suppressor role for SirT1. Here, we indicate the different functions described for SirT1 that support one role or the other.
The possibility that SirT1 is a tumor suppressor is corroborated by its role in maintaining genome stability through chromatin regulation and DNA repair.3,57,60 Accordingly, SirT1−/− mouse embryos exhibit more chromosomal aberrations and impaired DNA repair relative to WT embryos. 16 Further evidence comes from mice containing a transgenic copy of SirT1 under its own regulatory elements (SirT1-tg mice), which exhibit global overexpression of SirT1 (at roughly 3 times normal levels). 188 At old age, SirT1-tg mice are healthier than WT mice; however, this effect is not sufficiently potent to extend longevity. Moreover, SirT1-tg mice also show lower levels of DNA damage and decreased expression of the aging-associated gene p16 (Ink4a) and are partially protected from diabetes, osteoporosis, and cancer. 188 In addition, SirT1-tg mice suffer less frequently from spontaneous carcinomas and sarcomas. Herranz et al. 188 developed a metabolic syndrome-associated liver cancer model in which SirT1-tg mice were less susceptible to liver cancer and exhibited greater hepatic protection from both DNA damage and metabolic damage relative to WT mice. These results support SirT1 tumor suppression activity in aging- and metabolic syndrome–associated cancer.
Alternatively, there is much evidence supporting a role for SirT1 in tumor initiation and progression—namely, in blocking senescence and apoptosis. First, SirT1 overexpression can block stress-induced apoptosis via chromatin structure modulation and via deacetylation of nonhistone proteins, including p53, FOXO, E2F1, Rb, BCL6, Ku70, and so on.67,132,167 However, and rather counterintuitively, transgenic mice overexpressing SirT1 do not exhibit a greater incidence of tumor formation.189-191 Furthermore, SirT1 is upregulated in various cancers.60,76,133-135 As we described earlier, SirT1 expression and activity are regulated by several key factors in cancer. Deregulation of these factors could result in SirT1 overexpression or activation, suggesting that increased levels of SirT1 could be a consequence, rather than a cause, of cancer. One explanation for this is that, given SirT1’s role in inhibition of senescence and apoptosis, some tumors could become addicted to SirT1, and therefore, SirT1 expression would be very important for tumor development. This scenario is supported by the observation that SirT1 is overexpressed in chemoresistant leukemia, neuroblastomas, osteosarcomas, and ovarian and breast cancer cells. Also, biopsies from cancer patients treated with chemotherapeutic agents exhibit higher SirT1 levels than do untreated samples. 192 Furthermore, ectopic SirT1 overexpression induces P-glycoprotein expression and makes cancer cells resistant to the chemotherapy drug doxorubicin, whereas depletion of SirT1 by siRNA partially reverses the drug-resistant phenotype. 192 Moreover, studies have shown that the sirtuin activator resveratrol has chemopreventive activity against various cancers, including leukemia, DMBA-induced mammary tumors (in rats), skin cancer, and prostate cancer.193-196
An oncogenic role for SirT1 is further supported by its link to tumor promotion, through its role in angiogenesis. Recent studies impute SirT1 as a key regulator of vascular endothelial homeostasis, which controls angiogenesis, vascular tone, and endothelial dysfunction. Potente et al. 197 found an aberrant postnatal neovascularization response of endothelial-restricted SirT1 mutant mice in which the deacetylase domain of SirT1 was lacking. In addition, SirT1-deficient zebrafish have exhibited vascular patterning defects and hemorrhages due to dysregulated endothelial sprouting and vessel navigation, related to regulation of the expression of multiple genes involved in vascular endothelial homeostasis and remodeling. 197 Potente et al. 197 determined that among these genes, the connection between FOXO1 and SirT1 is very important for regulation of postnatal angiogenesis. FOXO1 is highly expressed in the vascular endothelium and has been shown to be an essential negative transcriptional regulator of vessel formation.198-202 SirT1 can act as a repressor of FOXO1-dependent transcriptional activity in endothelial cells, and knockdown of FOXO1 partially rescues the inhibitory effects of SirT1 gene silencing on the angiogenic activity of endothelial cells; together, these findings indicate that SirT1 regulates endothelial angiogenic functions by modulating the transcriptional activity of FOXO1. 197 Moreover, SirT1 has been shown to interact with and deacetylate endothelial nitric oxide synthase (eNOS), a major factor in maintaining vascular homeostasis, leading to enhanced nitric oxide (NO) production. 203 eNOS-derived NO is essential for endothelial-dependent vasorelaxation and survival, migration, and postnatal neovascularization.204-207 Moreover, FOXO1 and FOXO3 have been shown to repress eNOS expression, suggesting crosstalk among SirT1, FOXO, and eNOS. 200
Much remains to be established on how SirT1 exerts its multiple functions in cancer and on how these functions affect tumorigenesis. Current knowledge suggests (Figure 4) that under normal conditions, in response to stress or to DNA damage, SirT1 might promote cell survival via cell cycle arrest, DNA repair, or inhibition of apoptosis. If the stress signal becomes chronic or the levels of damage cross a certain threshold, then SirT1 could induce cell senescence. This scenario is supported by the finding that SirT1−/− MEFs are resistant to senescence. However, following chronic stress or DNA damage, the loss of a tumor suppressor or of any other checkpoint-related factor could cause an imbalance in these regulatory processes and induce SirT1 overexpression beyond a critical limit. This in turn would agree with the drastic reduction in the very high levels of SirT1 protein in undifferentiated cells as differentiation progresses. As cancer development is involved in dedifferentiation of cells, it could imply restoration of SirT1 protein to predifferentiation levels. The aberrant overexpression of SirT1 would in turn contribute to transformation and tumor formation by promoting cell growth and inhibiting apoptosis.73-75,83,122
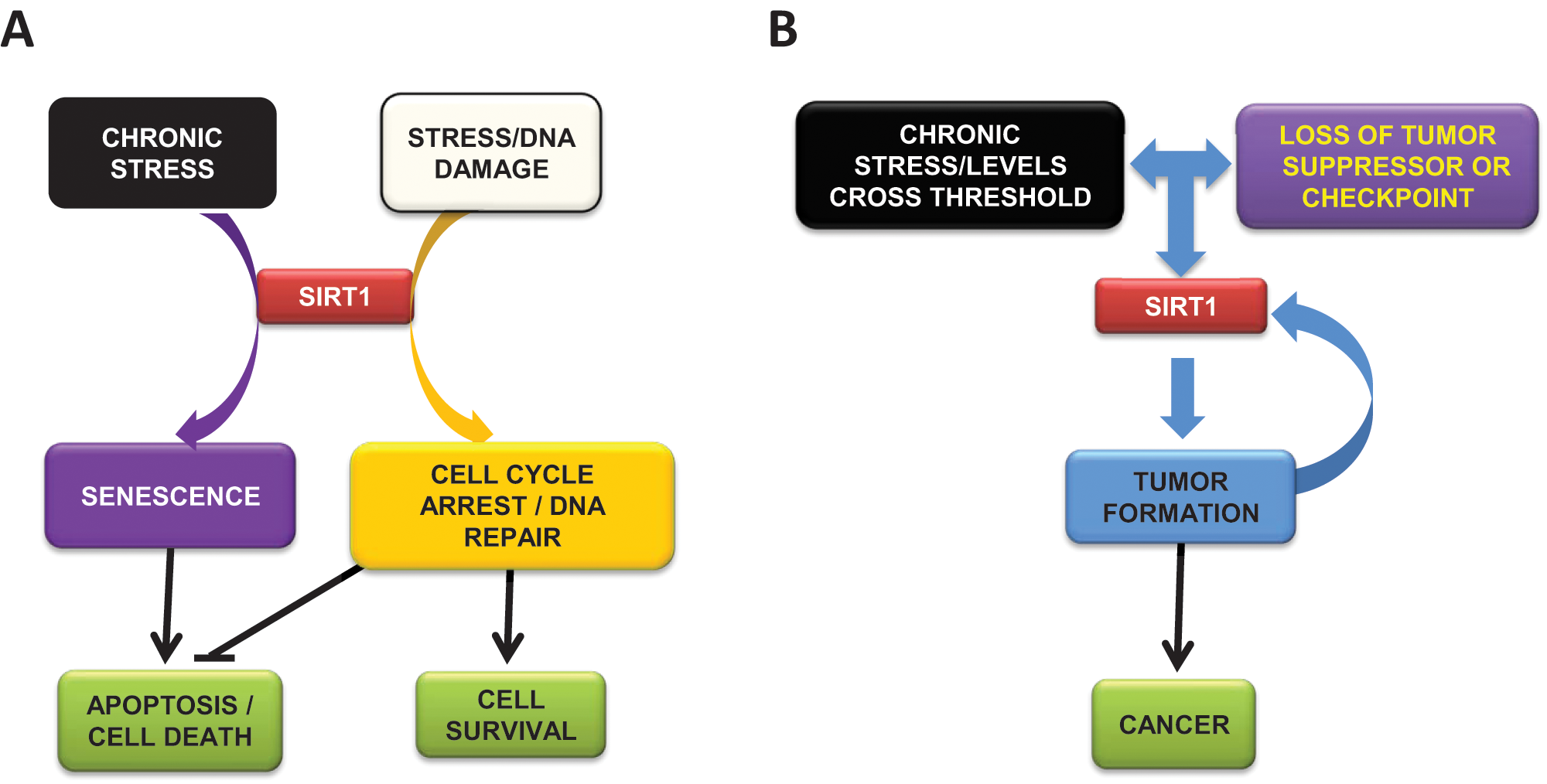
Model for SirT1 apparent duality in cancer. (A) Under stress conditions, SirT1 promotes cell cycle arrest, DNA repair, and, ultimately, cell survival. In chronic stress conditions or under certain massive levels of DNA damage, SirT1 induces senescence and apoptosis. (B) Under chronic stress and loss of tumor suppressors or checkpoints, SirT1 promotes tumor formation and cancer. A feedback between tumor progression and SirT1 levels is established, resulting in reinforced dedifferentiation, cell growth, and cell survival.
Although the roles of other sirtuins in cancer have not been extensively investigated, all current data suggest that SirT2 works as a tumor suppressor: it is downregulated in some cancers, and its loss compromises the mitotic checkpoint, contributing to genomic instability and tumorigenesis.137,140,141 Like SirT1, SirT3 apparently plays a dual function in cultured cells: under normal conditions it promotes apoptosis,56,62,208 whereas under stress conditions, it seems to promote cell survival.
The mitochondrial sirtuins SirT4 and SirT5 do not seem to play an important part in cancer; nonetheless, this area requires further research.
As for SirT6, all evidence suggests that it acts as a tumor suppressor, given its major role as a guardian of genome stability. In addition, the SirT6 chromosomal locus (19p13.3) is a frequent breakage site in human acute myeloid leukemia. 209 Moreover, the shift from aerobic respiration to glycolysis observed in SirT6-deficient mouse cells resembles the Warburg effect, in which cancer cells switch from oxidative phosphorylation to aerobic glycolysis.115,116
Finally, SirT7, which is localized in the nucleolus, seems to interact with and activate RNA polymerase I. 210 However, SirT7 does not deacetylate RNA polymerase I; in fact, no substrates have yet been identified for this sirtuin. Given this finding, and the fact that SirT7 knockdown in human cells induces cell cycle arrest and apoptosis,208,211 further studies are definitely needed to determine its role in cancer.
Final Remarks
During the past decade, the members of the Sir2 family, known as sirtuins, have emerged as principal factors in sensing and regulating cellular response to oxidative and metabolic stress, compromising conditions that have been directly linked to tumorigenesis and tumor development. Although myriad questions regarding the roles of sirtuins in cancer remain unanswered, there is a growing body of evidence indicating that this research area should be prioritized. Future studies should indicate whether modulation of sirtuins could have implications for cancer treatment.
Footnotes
Acknowledgements
The authors thank Lourdes Serrano and members of the Vaquero group for fruitful discussions.
The authors declare that there are no potential conflicts of interest with respect to the authorship and/or publication of this article.
The Chromatin Biology group is supported by Spanish Ministry of Science and Innovation grant SAF2008-00923 and the Catalonian government agency AGAUR 2009SGR914. Alejandro Vaquero is an ICREA Researcher.