
Abstract
The coding region determinant binding protein, CRD-BP, is a multifunctional RNA binding protein involved in different processes such as mRNA turnover, translation control, and localization. It is mostly expressed in fetal and neonatal tissues, where it regulates many transcripts essential for normal embryonic development. CRD-BP is scarce or absent in normal adult tissues but reactivated and/or overexpressed in various neoplastic and preneoplastic tumors and in most cell lines. Its expression has been associated with the most aggressive form of some cancers. CRD-BP is an important regulator of different genes including a variety of oncogenes or proto-oncogenes (c-myc, β-TrCP1, GLI1, etc.). Regulation of CRD-BP expression is critical for proper control of its targets as its overexpression may play an important role in abnormal cell proliferation, suppression of apoptosis, invasion, and metastasis. Molecular bases of the regulatory mechanisms governing CRD-BP expression are still not completely elucidated. In this article, we have identified c-myc as a novel transcriptional regulator of CRD-BP. We show that c-myc binds to CRD-BP promoter and induces its transcription. This induction of CRD-BP expression contributes to the role of c-myc in the regulation of translation, increase in cell size, and acceleration of cell cycle progression via a mechanism involving upregulation of β-TrCP1 levels and activities and accelerated degradation of PDCD4.
Introduction
CRD-BP, the coding region determinant binding protein, also known as insulin-like growth factor 2 mRNA-binding protein 1 (IMP-1 or IGF2BP-1), is a multifunctional RNA binding protein involved in different processes such as mRNA turnover, translation control, and localization. It belongs to a conserved family of RNA binding proteins consisting of 2 RNA recognition motifs and 4 K-homology domains. 1,2 CRD-BP expression is temporally and spatially regulated. It is abundantly expressed in fetal and neonatal tissues. Its expression is crucial in the regulation of many transcripts essential for normal embryonic development. 3-5 While scarce or absent in normal adult tissues, CRD-BP is found to be de novo activated and/or overexpressed in various neoplastic and preneoplastic tumors and in most cell lines. 4-11 Its expression has been associated with the most aggressive form of some cancers. 4,8,12
CRD-BP was shown to upregulate the expression of different genes including c-myc, 13 β-TrCP1, 10 MDR1, 14 and GLI1. 15 It was also shown to be necessary for proper cell adhesion, cytoplasmic spreading, and invadopodia formation through its binding to the 3′UTR of CD44 mRNA and stabilization of the mRNA. In addition, CRD-BP participates in the posttranscriptional regulation of other transcripts such as ALCAM, AMIGO2, CD24, collagen V, α1, dysadherin, keratin 19, lumican, MMP1, MCAM, and synCAM. These transcripts encode proteins involved in cellular adhesion, invasion, and extracellular matrix remodeling. 16 Regulation of CRD-BP expression appears critical for proper control of its targets as its overexpression may play an important role in abnormal cell proliferation, suppression of apoptosis, invasion, and metastasis. Mechanisms regulating CRD-BP expression are not completely elucidated. CRD-BP was found to be regulated by gene amplification in some cancers. 6,8 Epigenetic modifications have been suggested to be responsible for its silencing in adult tissues. 17 CRD-BP was also shown to be a direct target of the Wnt/β-catenin signaling pathway, 10,18 and more recently, it was found to be regulated by the microRNA let-7. 19
In this article, we have identified c-myc as a novel transcriptional regulator of CRD-BP. We show that c-myc binds to the CRD-BP promoter and induces its transcription. We demonstrate that this induction of CRD-BP expression contributes to the role of c-myc in the regulation of translation, increase in cell size, and acceleration of cell cycle progression via a mechanism involving upregulation of levels and activities of β-TrCP1 (the substrate recognition subunit for SCFβ-TrCP E3 ubiquitin ligase) and accelerated degradation of PDCD4.
Results and Discussion
c-Myc and Max interact with 4 sites on the CRD-BP promoter in a c-myc–dependent manner
The c-Myc protein has been shown to possess at its carboxyl terminus a sequence-specific DNA binding activity. It forms a heterodimer with its partner Max 20,21 and binds to specific DNA sequences containing the core hexanucleotide 5′-CACGTG-3′. 22,23 Recognition of the 5′-CACGTG-3′ consensus sequence (also known as E box) in gene promoters has led to the identification of gene targets regulated transcriptionally by c-myc. 24-27 The human CRD-BP gene contains 4 consensus sequences 5′-CACGTG-3′ in its promoter region, suggesting potential c-myc binding in this region (Fig. 1A). These putative binding sites of c-myc are conserved between human and mouse (Suppl. Fig. S1). To test whether the c-Myc–Max heterodimer interacts with the putative binding sequences identified in the CRD-BP promoter, we performed chromatin immunoprecipitation (ChIP) assay coupled with real-time qPCR. We observed an interaction between Max and DNA fragments of the CRD-BP promoter containing each of the 4 putative binding sites of c-myc, and those interactions were significantly increased (7- to 53-fold) when c-myc was overexpressed (Fig. 1B and Suppl. Fig. S2). We also observed an interaction between c-Myc and the DNA fragments of the CRD-BP promoter containing each of the 4 putative binding sites, and the interactions were significantly increased as well (3- to 17-fold) when c-myc was overexpressed (Fig. 1C and Suppl. Fig. S2). These interactions are specific as no interaction was observed when normal rabbit IgG or no antibody control was used in the ChIP reactions (Suppl. Fig. S2A-D). In addition, the gene used as a negative control was detected only in the inputs (Suppl. Fig. S2E). Overall, the use of Max- and c-Myc–specific antibodies showed that both Max and c-Myc interact with DNA fragments of the CRD-BP promoter containing each of the 4 putative sites, and these interactions are significantly increased (albeit to different extents) when c-myc is overexpressed.
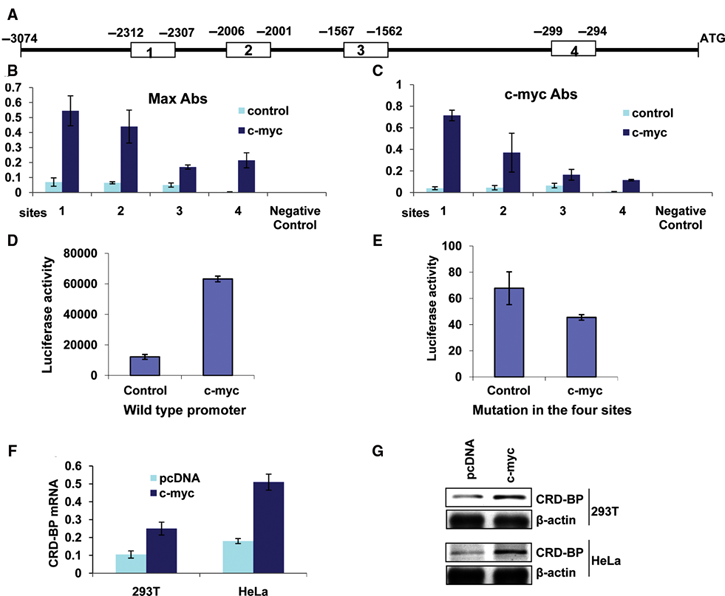
Interaction of c-Myc and Max with the CRD-BP promoter: c-myc binds to the CRD-BP promoter and induces its expression. (
c-myc induces CRD-BP promoter–driven transcription
Since c-Myc binds to the CRD-BP promoter, we sought to determine whether it regulates transcription driven by the CRD-BP promoter. To address this question, we subcloned the CRD-BP promoter region from −3074 to +488 upstream of the luciferase gene into the pGL3 basic vector and assessed the ability of human c-myc to induce CRD-BP promoter–driven luciferase activity. We observed a 5-fold increase in CRD-BP promoter–driven luciferase activity in Hela cells when cotransfected with the c-myc expression vector and the luciferase construct (Fig. 1D and Suppl. Fig. S3D). Mutations in the 4 sites (5′-CACGTG-3′) by site-directed mutagenesis (to generate 5′-CAGCTG-3′) drastically decreased the basal levels of luciferase activity (~179 times lower than that of the wild type [Fig. 1D and 1E]) and rendered the luciferase gene insensitive to the induction by c-myc (Fig. 1E and Suppl. Fig. S3D). The overall luciferase activity from constructs bearing mutations in single sites was significantly reduced (~60 times lower than that of the wild type [Fig. 1D and Suppl. Fig. S3A]), and the difference in luciferase activity between the control and cells overexpressing c-myc was also reduced (Suppl. Fig. S3A and S3D). Mutations in any 2 (Suppl. Fig. S3B and S3D) or 3 sites (Suppl. Fig. S3C and S3D) lowered further the luciferase activity to a level comparable to the one of the construct with mutations in all the 4 sites. These results suggest that each c-myc binding site in the CRD-BP promoter contributes to the regulation of CRD-BP transcription. Analyses of CRD-BP RNA and protein expression showed a significant increase in CRD-BP mRNA (Fig. 1F) and protein (Fig. 1G) in 293T and HeLa cells when c-myc was overexpressed. These results imply that c-Myc binds to the CRD-BP promoter and induces its transcription and expression.
c-Myc downregulates PDCD4 in a CRD-BP–dependent manner
CRD-BP is a multifunctional RNA binding protein that was shown to bind and stabilize several mRNAs 13,14 including the mRNA encoding the F-box protein β-TrCP1. 10 Overexpression of CRD-BP was demonstrated not only to stabilize β-TrCP1 mRNA but also to elevate the steady-state levels of its mRNA and protein, resulting in an increase in the activities of the SCFβ-TrCP1 E3 ubiquitin ligase and in an accelerated turnover of its substrates. 10 PDCD4 was recently identified as one of the substrates of SCFβ-TrCP1 E3 ubiquitin ligase. 28 We hypothesized that c-myc–dependent upregulation of CRD-BP expression would induce an increase in β-TrCP1 levels and activities. As a result of this increase in β-TrCP1 activities, PDCD4 degradation is expected to be accelerated. Indeed, ectopic expression of c-myc in 293T and HeLa cells led to a significant increase in β-TrCP1 levels (Fig. 2A). This increase in β-TrCP1 levels appears to be CRD-BP dependent as the inhibition of CRD-BP by specific shRNA attenuated the induction of β-TrCP1 expression by c-myc (Fig. 2A). We also found that overexpression of c-myc significantly decreased the steady-state levels of PDCD4 in several cell lines (Fig. 2A and 2B). This reduction in PDCD4 expression could be alleviated by cotransfecting these cells with specific CRD-BP shRNA or β-TrCP1 shRNA (Fig. 2A and 2B). Our data also showed that PDCD4 protein was destabilized by c-myc in 293T cells (Fig. 2C); however, cotransfection of the cells with specific CRD-BP shRNA or specific β-TrCP1 shRNA abolished c-myc–induced destabilization of PDCD4 in those cells (Fig. 2C).
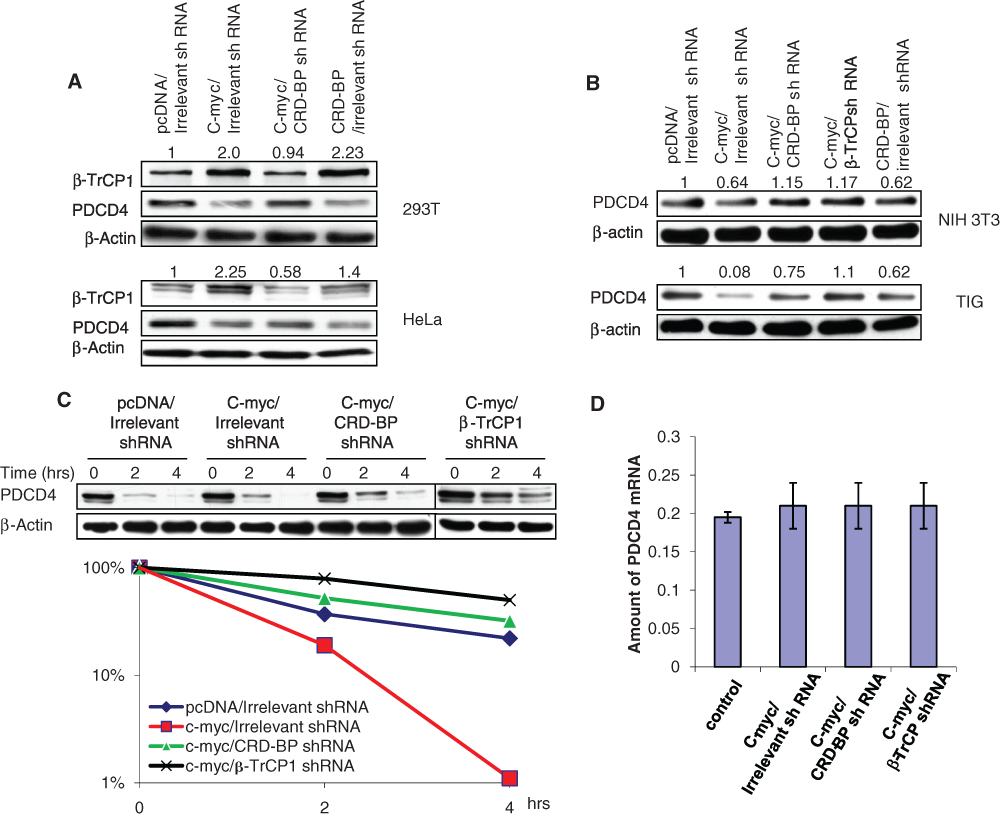
c-Myc induces β-TrCP1 expression and accelerates PDCD4 turnover in a CRD-BP–dependent manner. (
mRNA expression of PDCD4 was not affected by c-myc (Fig. 2D). Inhibition of CRD-BP or β-TrCP1 by specific shRNAs did not affect PDCD4 mRNA expression in cells overexpressing c-myc either (Fig. 2D). This confirms that PDCD4 regulation by c-myc, CRD-BP, and β-TrCP1 is independent of transcription. Our data suggest that upregulation of CRD-BP expression by c-myc leads to increased β-TrCP1 expression and activities, which in turn results in accelerated degradation and decreased expression of PDCD4.
CRD-BP–mediated regulation of PDCD4 contributes to the regulation of translation by c-Myc
c-myc is an important regulator of a wide array of cellular processes necessary for normal cell growth and differentiation. Its deregulation is one of the hallmarks of many cancers. c-myc was shown to promote cell proliferation by 1) activating the cyclins (such as cyclin D1, cyclin D2, cyclin E1, cyclin A2), CDK4, and cdc25A; 2) repressing the transcription of cell cycle checkpoint genes (including the growth arrest– and DNA damage–inducible proteins 45 and 153 [GADD45, GADD153]); and 3) inhibiting the function of cyclin-dependent kinase inhibitors (p21 and p27). It was also shown to regulate cell growth, metabolism, and protein synthesis by inducing the expression of its targets such as lactate dehydrogenase (LDH), carbamoylphosphate synthetase-2, aspartate transcarbamylase, dihydroorotase (CAD), ornithine decarboxylase (ODC), ribosomal proteins, eukaryotic translation initiation factor 4E (eIF4E), and eukaryotic translation initiation factor 2A (eIF2A) (reviewed in Meyer and Penn 29 ).
Additionally, c-myc was reported to increase protein synthesis independently of the known cell cycle targets that it transcriptionally regulates. 30,31 Transcription-independent roles of c-myc have been implicated in protein synthesis by modulation of mRNA cap methylation 32,33 and by setting the number of origins in DNA replication. 34 These roles are important for protein synthesis, cell cycle progression, and cell proliferation.
On the other hand, the tumor suppressor PDCD4 was shown to repress protein synthesis via inhibition of RNA helicase activity of the translation initiation factor eIF4A. The unwinding of the secondary structure at the 5′UTR of messenger RNAs by eIF4A is required for cap-dependent translation. 35 We hypothesized that CRD-BP–mediated inhibition of PDCD4 contributes to c-myc upregulation of translation. To test this hypothesis, we used a luciferase reporter construct with a secondary structure in the form of a stem loop inserted in the 5′UTR of the luciferase gene. 36,37 We observed that in the Burkitt lymphoma cells, ramos, which constitutively overexpress c-myc, the luciferase activity was significantly reduced when c-myc was knocked down by specific shRNA (Fig. 3A). This reduction in the luciferase activity was also observed when CRD-BP or β-TrCP1 was knocked down by their specific shRNAs (Fig. 3A). The inhibitory effect of c-myc shRNA could be reversed by overexpression of CRD-BP or β-TrCP1 (Fig. 3A). Conversely, overexpression of c-myc increased luciferase activity, whereas knocking down of CRD-BP or β-TrCP1 by specific shRNA almost completely abolished the effect of c-myc in myc-null fibroblasts (HO 15.19) (Fig. 3B) and in 293T cells (Fig. 3C). However, the luciferase activity remained unchanged whether c-myc, CRD-BP, or β-TrCP1 was overexpressed or knocked down when using the luciferase gene construct with no secondary structure in the 5′UTR (Fig. 3A-C). Using 293T cells, we also observed an accelerated disappearance of a synthetic double-strand RNA in a helicase assay when c-myc was overexpressed, whereas knocking down of CRD-BP or β-TrCP1 by specific shRNAs attenuated the effect of c-myc in those cells (Suppl. Fig. S6), suggesting an increase in the helicase activity by c-myc. These results are likely to be underappreciated in comparison to the reporter assay as cell lysates inherently contain RNases that even in the presence of RNase inhibitors influence the helicase assay. Overall, our results suggest that inhibition of c-myc decreases CRD-BP and β-TrCP1 expression and activities and therefore increases expression of PDCD4 and inhibits the RNA helicase activity of eIF4A, resulting in cap-dependent translation repression. On the contrary, overexpression of c-myc increases CRD-BP and β-TrCP1 expression and activities and therefore accelerates degradation of PDCD4 and induces high RNA helicase activity of eIF4A, resulting in an increase in translation.
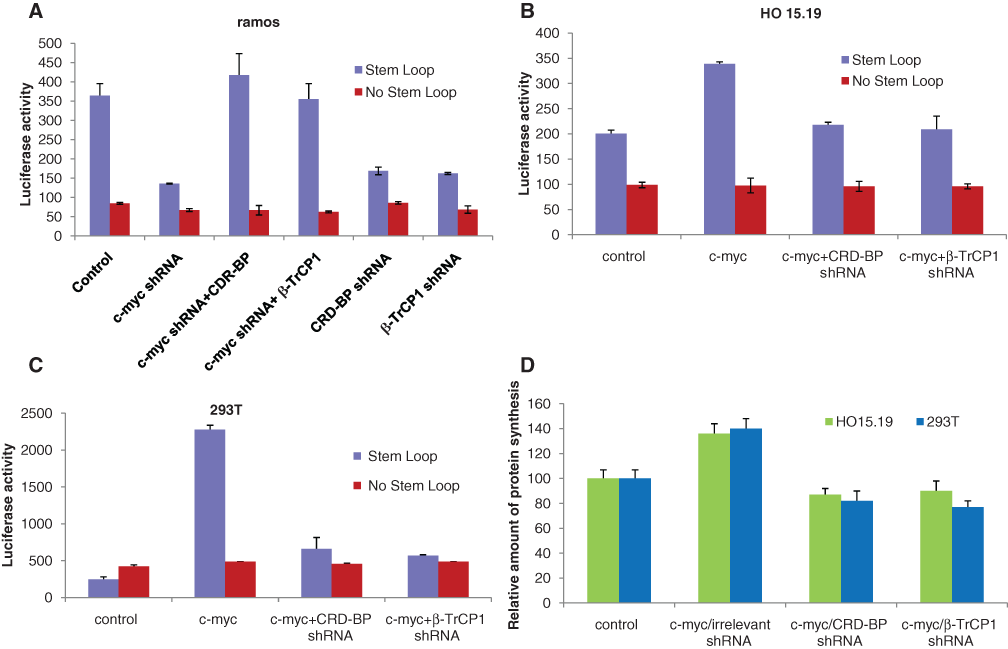
CRD-BP- and β-TrCP1–mediated regulation of PDCD4 contributes to the regulation of translation by c-myc. Luciferase activity in (
We also noticed that the increase in total protein synthesis in HO 15.19 and 293T cells by c-myc depends on CRD-BP and β-TrCP1 as CRD-BP- and β-TrCP1–specific shRNAs abrogated c-myc–induced elevation in protein synthesis (Fig. 3D). A previous study showed that Myc-overexpressing cells have augmented cap-dependent translation. 30 With the knowledge that PDCD4 prevents cap-dependent translation by inhibiting eIF4A activity, our data suggest that CRD-BP- and β-TrCP1–mediated downregulation of PDCD4 contributes to the regulation of translation by c-myc.
CRD-BP–mediated downregulation of PDCD4 contributes to the regulation of cell size growth and cell cycle progression by c-Myc
An increase in protein synthesis induces an increase in cell size as proteins constitute the large portion of cells, and this increase in protein synthesis could facilitate cell cycle progression. c-myc was shown to augment cell size 30,31 and to accelerate the cell cycle progression independently of known cell cycle targets that it transcriptionally regulates. 30 PDCD4, on the contrary, was reported to reduce cell size and to slow down the cell cycle progression. 28 It is conceivable that CRD-BP- and β-TrCP1–dependent downregulation of PDCD4 contributes to the increase in cell size and the acceleration of cell cycle progression by c-myc. To test this hypothesis, we measured the size of cells when c-myc was either inhibited or overexpressed. We observed a decrease in cell size when c-myc was knocked down by specific shRNA in ramos cells (Fig. 4A). This reduction in cell size was also observed when CRD-BP or β-TrCP1 expression was inhibited by their specific shRNA (Fig. 4A). The decrease in cell size as a result of inhibition of c-myc could be reversed by overexpression of CRD-BP or β-TrCP1 (Fig. 4A). Conversely, ectopic expression of c-myc in HO 15.19 cells induced an increase in cell size (Fig. 4B). Knockdown of CRD-BP or β-TrCP1 by specific shRNA abolished the effect of c-myc on cell size (Fig. 4B). These data suggest that effects of c-myc on cell size depend at least in part on the expression of CRD-BP and β-TrCP1. Analysis of cell cycle distribution showed that 53% of the cells were in S phase in HO 15.19 cells when c-myc was overexpressed compared to 21% in the control cells (Fig. 4C and Suppl. Fig. S4A and S4B). This is in line with a previous study that showed that ectopic expression of c-myc in normally quiescent cells can potentiate entry into S phase. 31 Eµ-Myc/+ cells that overexpress c-myc under the control of the Ig heavy chain enhancer also showed a markedly increased number of cells in S phase compared to wild-type cells. 30 In our study, the increase in the number of cells in the S phase induced by c-myc was significantly reduced by CRD-BP or β-TrCP1 shRNAs with a percentage of cells in the S phase of 33% and 34%, respectively (Fig. 4C and Suppl. Fig. S4C and S4D). These results suggest that CRD-BP and β-TrCP1 contribute to c-myc–induced accumulation of cells in S phase. Overexpression of c-myc also induced the growth of HO 15.19 cells, and this effect was dependent on the expression of CRD-BP or β-TrCP1 (Fig. 4D). Cell growth normally precedes cell division during cell cycle progression. The ability of c-myc to enhance protein synthesis and cell size might be a prerequisite for the increase in colony formation observed in cells overexpressing c-myc as these cells might rapidly reach a critical size that allows them to undergo division.
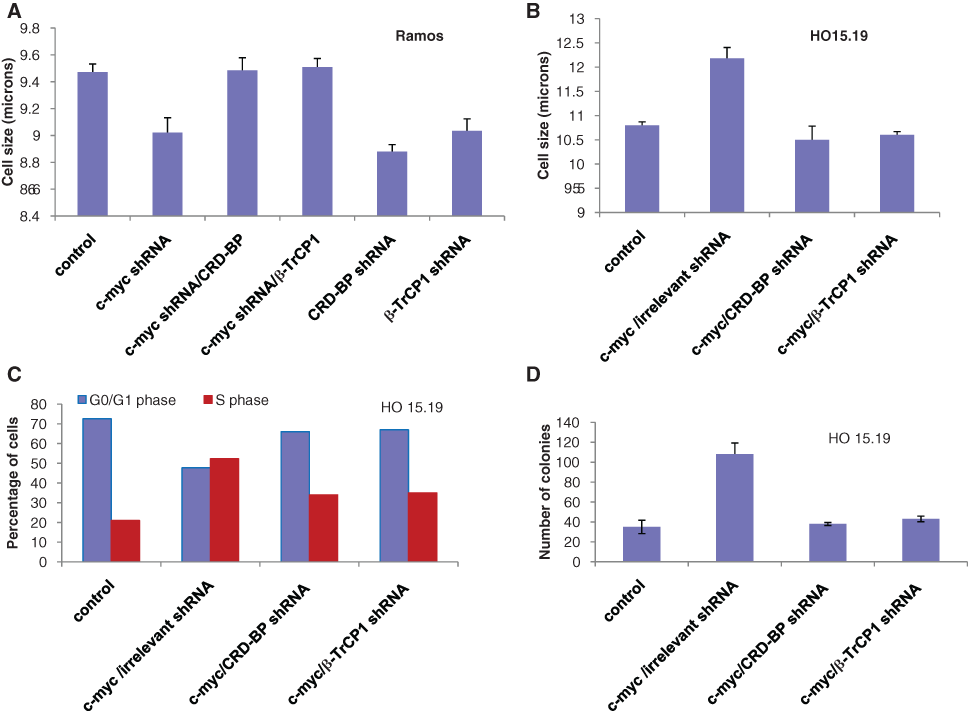
CRD-BP- and β-TrCP1–mediated regulation of PDCD4 contributes to the regulation of cell size, growth, and cell cycle progression by c-myc. (
Molecular bases of the regulatory mechanisms governing CRD-BP expression are still not completely elucidated. Here, we demonstrated a novel molecular mechanism by which c-myc, a previously unknown regulator of CRD-BP, binds to its promoter and induces its transcription. This transcription regulation of CRD-BP contributes to the role of c-myc in the regulation of translation, increase in cell size, and acceleration of cell cycle progression by a mechanism involving upregulation of β-TrCP1 and downregulation of PDCD4 expression and activity (Suppl. Fig. S7). Other targets of β-TrCP1 can potentially contribute to the changes in protein translation, cell cycle, and cell size observed in this study; however, the contribution of IκBα and β-catenin, which are 2 of the better-studied targets of β-TrCP1, is unlikely as their protein expression was not significantly affected in response to c-myc with or without downregulation of CRD-BP or β-TrCP1 (Suppl. Fig. S5).
CRD-BP was previously demonstrated to upregulate c-myc expression at the posttranscriptional level. 10,13,38 Together with that, our data on the transcriptional regulation of CRD-BP by c-myc suggest a positive feedback loop regulation between c-myc and CRD-BP. CRD-BP might therefore play an important role in conditions due to the deregulation of c-myc. Deregulation of c-myc activity is responsible for about 90% of Burkitt lymphomas. CRD-BP, with its role in the regulation of genes involved in cellular adhesion, invasion, and extracellular matrix remodeling as well as other oncogenes, might contribute at least in part to the role of c-myc in Burkitt lymphomas. Inhibition of CRD-BP might subvert the effects of c-myc in this condition. Targeting CRD-BP might be of a significant value in the treatment of Burkitt lymphomas in which c-myc is constitutionally activated and/or other conditions with deregulated c-myc activity and CRD-BP expression.
Materials and Methods
Expression vectors
The promoter region of the CRD-BP gene from −3074 to +488 was amplified by PCR using Pfu Turbo DNA polymerase (Stratagene, Santa Clara, CA) and cloned into pGL3 basic upstream of the luciferase gene. The β-TrCP1 and CRD-BP cDNAs were cloned each into pcDNA3.1 downstream of the T7 promoter. CRD-BP shRNAs were described before. 10 The c-myc expression vector and c-myc shRNA were described previously. 39 β-TrCP1 shRNAs were a gift from Dr. Fuchs. The luciferase reporter genes with and without the stem loop structure in the 5′UTR were previously described. 37 GFP expression vector (pmaxGFP) was obtained from Amaxa Biosystems (Gaithersburg, MD).
Tissue culture and transfections
The cell lines 293T, HeLa, and NIH 3T3 were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in accordance with the ATCC’s recommendations. Ramos cells were from the ATCC and were maintained in RPMI medium with 10% heat-inactivated FBS. HO 15.19 cells were a gift from Dr. J. Sedivy, and TIG normal human fibroblasts were a gift from H. Tahara. They were maintained in DMEM medium with 10% BCS. All our tissue culture media were supplemented with 1% penicillin and streptomycin. 293T cells were transfected by calcium phosphate transfection. HeLa, NIH 3T3, and TIG cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in accordance with the manufacturer’s recommendations. Ramos and HO 15.19 cells were electroporated using the Nucleofector Kit V from Amaxa. The amount of DNA in each transfection was kept constant by the addition of an appropriate amount of empty expression vector. To analyze the effect of c-myc on PDCD4 expression and activity in 293T cells, TIG cells, NIH 3T3 cells, and HO 15.19 cells, 24 hours after transfection, the cells were starved for 48 hours and then collected. Ramos cells were starved for only 24 hours.
Antibodies and immunotechniques
Antibodies against c-Myc, Max, and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) were purchased as well as the secondary antibody conjugated with horseradish peroxidase (Chemicon, Billerica, MA). Antibody against CRD-BP was a generous gift from Dr. J. Ross. β-TrCP1 antibody was obtained as described before 40 as well as PDCD4 antibody. 41 To obtain whole cell lysates for Western blot analysis, the cells were lysed using a denaturing RIPA buffer containing PBS (pH 7.4), 0.5% sodium deoxycholate, 0.1% SDS, 1% (v/v) IGEPAL, 100 mM sodium orthovanadate, and proteinase inhibitor cocktail (Sigma, St. Louis, MO). Immunoblotting procedures were performed as described previously. 10
Luciferase reporter assays
HeLa cells were transfected with the wild-type or mutant CRD-BP promoter constructs and different additional plasmids as indicated. Luciferase activity was estimated using luciferase reporter assay reagent (Promega, Madison WI), and β-galactosidase used for normalization was estimated by the β-galactosidase assay reagent (Pierce, Rockford, IL).
Ramos, HO 15.19, and 293T cells were transfected with the luciferase reporter construct bearing or not the stem loop structure in the 5′UTR and different additional plasmids as indicated in the figures. Luciferase activity was also estimated using luciferase reporter assay reagent (Promega), and β-galactosidase used for normalization was estimated by the β-galactosidase assay reagent (Pierce).
PCR
Real-time PCR for quantitative measurements of CRD-BP and PDCD4 was done using SYBR Green PCR Core reagents (Applied Biosystems, Carlsbad, CA). Primer sequences are in Supplementary Table S1. GAPDH was used as a reference gene. 10 Site-directed mutagenesis to generate different mutants of the CRD-BP promoter construct was performed according to the protocol described by Stratagene. Primer sequences are in Supplementary Table S2.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the EZ ChIP Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA) according to the manufacturer’s recommendations. 293T cells were transfected with pcDNA3.1 or c-myc expression vector. Forty-eight hours posttransfection, the cells were cross-linked, sonicated, and immunoprecipitated. Rabbit polyclonal c-Myc (sc-764) and Max (sc-197X) antibodies (Santa Cruz Biotechnology) or rabbit IgG (sc-2027) (Santa Cruz Biotechnology) were used. Real-time qPCR for detection of the binding sites of c-Myc on the CRD-BP promoter was done on DNA purified from the immunoprecipitate complexes using SYBR Green PCR Core reagents (Applied Biosystems). Primers for real-time qPCR detection are in Supplementary Table S3. For the negative control, we used the primers flanking a region of genomic DNA between the GAPDH and the chromosome condensation-related SMC-associated protein (CNAP1) gene. 10
Protein synthesis
HO 15.19 and 293T cells were cotransfected with pcDNA3.1 and irrelevant shRNA, c-myc expression vector and irrelevant shRNA, c-myc expression vector and CRD-BP shRNA, or c-myc expression vector and β-TrCP1 shRNA. Twenty-four hours after transfection, the cells were starved, and 35S methionine was added to the media without serum to a final concentration of 10 µci/mL. Twenty-four hours later, the cells were collected, and total proteins were quantified. Translation was assessed by measuring the 35S incorporation per 5 µg of total protein using β-scintillation.
Cell cycle distribution
HO 15.19 cells were cotransfected with GFP expression plasmid, pcDNA3.1, and irrelevant shRNA; GFP expression plasmid, c-myc expression vector, and irrelevant shRNA; GFP expression plasmid, c-myc expression vector, and CRD-BP shRNA; or GFP expression plasmid, c-myc expression vector, and β-TrCP1 shRNA as indicated. Twenty-four hours after transfection, the cells were starved for 48 hours and stained with Hoechst (Invitrogen). Cell cycle distribution was analyzed on GFP-positive cells by flow cytometry.
Colony formation
HO 15.19 cells grown in 100-mm plates were cotransfected with pTK-puro plasmid, pcDNA3.1, and irrelevant shRNA; pTK-puro plasmid, c-myc expression vector, and irrelevant shRNA; pTK-puro plasmid, c-myc expression vector, and CRD-BP shRNA; or pTK-puro plasmid, c-myc expression vector, and β-TrCP1 shRNA as indicated. Forty-eight hours after transfection, cells from each plate were seeded in five 100-mm plates and treated with puromycin (10 µg/mL) for 14 days. The colonies were counted under the light microscope.
Footnotes
Acknowledgements
The authors thank Drs. S. Fuchs, J. Ross, J. Sedivy, and H. Tahara for their generous gifts of reagents.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the National Cancer Institute (NCI) [grant CA121851] to V.S.S.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.