
Abstract
Melanoma is a highly metastatic cancer, and there are no current therapeutic modalities to treat this deadly malignant disease once it has metastasized. Melanoma cancers exhibit B-RAF mutations in up to 70% of cases. B-RAF mutations are responsible, in large part, for the constitutive hyperactivation of survival/antiapoptotic pathways such as the MAPK, NF-κB, and PI3K/AKT. These hyperactivated pathways regulate the expression of genes targeting the initiation of the metastatic cascade, namely, the epithelial to mesenchymal transition (EMT). EMT is the result of the expression of mesenchymal gene products such as fibronectin, vimentin, and metalloproteinases and the invasion and inhibition of E-cadherin. The above pathways cross-talk and regulate each other’s activities and functions. For instance, the NF-κB pathway directly regulates EMT through the transcription of gene products involved in EMT and indirectly through the transcriptional up-regulation of the metastasis inducer Snail. Snail, in turn, suppresses the expression of the metastasis suppressor gene product Raf kinase inhibitor protein RKIP (inhibits the MAPK and the NF-κB pathways) as well as PTEN (inhibits the PI3K/AKT pathway). The role of B-RAF mutations in melanoma and their direct role in the induction of EMT are not clear. This review discusses the hypothesis that B-RAF mutations are involved in the dysregulation of the NF-κB/Snail/RKIP/PTEN circuit and in both the induction of EMT and metastasis. The therapeutic implications of the dysregulation of the above circuit by B-RAF mutations are such that they offer novel targets for therapeutic interventions in the treatment of EMT and metastasis.
Keywords
Introduction
Cancer is a disease caused by uncontrolled cell growth and proliferation. When localized, the formation of the tumor by uncontrolled cell proliferation is easily treated by surgery and various therapeutic drugs; however, complications arise when the tumor metastasizes. Once metastasized, the cancer becomes deadly and is extremely difficult to treat due to accumulated resistance to chemotherapy, radiotherapy, and immunotherapy. When metastasizing, the tumor cells invade the basement membrane and circulatory system, extravasate through the endothelial lining of the blood vessels, and form colonies in the new tissue sites. The migration from the epithelial layer into the mesenchymal layer before reaching the basement membrane is known as “epithelial to mesenchymal transition” (EMT). During EMT, the polarized epithelial cells acquire certain attributes of mesenchymal cells and, thus, are able to penetrate the mesenchymal layer through the basement membrane and invade neighboring tissues. 1 Breaching of the mesenchymal layer leads to infiltration into the blood vessels and migration to other parts of the body. This review will focus on the role of B-RAF mutations in melanoma and the induction of EMT.
Metastasis in Melanoma
Melanoma is one of the most highly metastatic cancers. Melanoma is a type of cancer that arises from melanocytes, cells dedicated to melanin production and located in the basal layer of the epidermis. It represents 2% to 3% of malignant tumors in both the United States and Northern Europe. The incidence of melanoma has doubled in the last 20 years; a 4% increase every year is documented in the United States. 2 Whereas the incidence is continuously increasing, the mortality seems to be decreasing, most likely due to the increase of screening programs and early detection. Cutaneous melanoma is the most frequently occurring melanoma; however, melanoma can occur in other sites, such as the uvea. Occurrence of melanoma in the uvea accounts for the greatest percentage of malignant melanomas other than melanoma of the skin. 3 Other possible sites of melanoma include mucosal melanomas (female genitalia, head and neck, anorectal region); however, these occurrences are less common. 4 The main risk factors for cutaneous melanoma are related to sun exposure (UV), phenotype (Fitzpatrick Classification Scale), distribution of existing melanocytic nevous, type and location (lower limbs in females, posterior trunk in males), and family history. 5 Melanoma has the greatest potential for metastasis, even compared to other skin cancers such as basal cell carcinoma and squamous cell carcinoma. 6 However, if discovered in its early stages, the primary tumor can be removed surgically, and there is a high probability of complete recovery. 6 Approximately 80% of melanoma cases are detected at an early stage in which the melanoma is localized in the primary site and can be excised. 7 Melanoma tumors can be classified into 2 groups, those in the radial growth phase (RGP) and those in the vertical growth phase (VGP). Tumors classified in the RGP are associated with early stage melanoma and are confined, for the most part, to the epidermis with a low probability of dissemination. Once progressed to the VGP classification, the tumor cells are now tumorigenic, characterized by important dermal invasion, and become metastastic. 8,9
Metastatic melanoma is refractory to current therapies and has a very poor prognosis, with a median survival rate of 6 months. 10 Chemotherapy treatment of melanoma is not very effective due to the high refractory nature of this malignancy to most standard cytotoxic agents. In a prospective randomized controlled trial, adjuvant high-dose interferon was shown to increase relapse-free survival and overall survival (OS) when compared to patients with high-risk resected cutaneous melanoma. 11 Melanoma that has already spread to distant sites is rarely curable with standard therapy, although high-dose interleukin-2 (IL-2) has been reported to produce durable responses in a small number of patients. 12 However, the most promising treatments of melanoma seem to be targeted to mutations in transduction/survival pathways that are common in melanomas. Recent progress in the molecular understanding of the signaling pathways involved in melanomagenesis has led researchers to develop targeted therapies for this disease. These include selective inhibitors of the RAF and MEK kinases and inhibitors of the PI3K pathway. 7
Inhibitors of the RAF kinases serve as a targeted therapy for melanoma because a direct correlation has been found between the presence of mutated signaling protein B-RAF and the progressive stages of melanoma growth; there is a higher incidence of B-RAF mutations in VPG tumors compared to tumors of the RGP. 13 Therefore, studies of the role of mutations in B-RAF signaling and in cell survival are important to decipher the underlying mechanisms of melanoma tumorigenesis and its progression. This review will focus on B-RAF signaling and the gene products that play major roles in the regulation of EMT in melanoma and how these gene products work in concert, namely, the NF-κB/ Snail/RKIP/PTEN circuit. Evidence supports B-RAF mutations in the regulation of this above circuit and the induction of EMT in melanoma (see schematic diagram in Fig. 1).
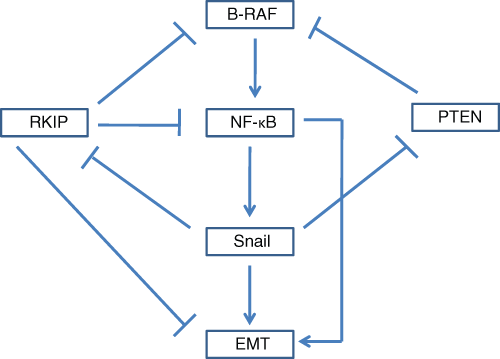
The role of the B-RAF/NF-κB/Snail/RKIP/PTEN circuitry in the regulation of EMT in melanoma. B-RAF induces EMT in melanoma through the constitutive activation of NF-κB and resulting in the up-regulation of the metastasis inducer, Snail. Inhibition of B-RAF by either up-regulation of RKIP or PTEN inhibits Snail expression. Expression of Snail down-regulates both RKIP and PTEN, thus providing a positive feedback loop for self-amplification and EMT.
B-RAF Mutations in Melanoma
B-RAF and B-RAF Mutations
B-RAF is part of a family of serine/threonine kinase proteins designated as the RAF proteins. RAF proteins are activated by RAS, the small membrane-bound G protein that is regulated upstream by extracellular signals. The RAF family has 3 functional proteins, namely, A-RAF, B-RAF, and C-RAF (also known as RAF-1); all 3 have a high degree of homology in 3 conserved regions, and their activation is regulated by various phosphorylation sites 14-16 (Fig. 2A). Two of the conserved regions of the RAF family of proteins, CR1 and CR2, are found in the N-terminus, while the third, CR3, is located in the C-terminus and codes for the proteins’ kinase activity. Several phosphorylation sites are conserved, while some are unique to each protein. The presence of both conserved and unique phosphorylation sites indicates that, although there are common mechanisms for regulating the RAF proteins, each can be regulated independently of the others. In tumor cells, as well as in normal cells, B-RAF is the protein providing the most signaling for the transduction pathway, whereas the role of A-RAF and C-RAF is to refine the signal and make small adjustments at the level of signaling. B-RAF is the protein used most often to transduce signals in cellular signaling pathways due to its elevated basal kinase activity in relation to its isoforms A-RAF and C-RAF. This elevated basal kinase activity is due to the presence of an aspartic acid just upstream of the conserved region CR3 that is not present in either A-RAF or C-RAF. Because of the negative charge provided by aspartic acid, B-RAF has a greater propensity towards activation (Fig. 2A). Both A-RAF and C-RAF proteins require not only phosphorylation but also additional activation factors such as activation by the tyrosine kinase Src; therefore, point mutations are not sufficient to induce activity. 17 Mutations in B-RAF easily result in hyperactivation due to its propensity towards activation by the aspartic acid and because only phosphorylation is required for activation. As a result, it is shown that the majority of activating mutations occur in B-RAF.
The most common B-RAF mutations are observed in the kinase domain of the protein, particularly within the activation segment. The most common mutation in the B-RAF protein consists of the substitution of glutamic acid for valine at codon 600 in exon 15 (Val600Glu; B-RAFV600E) (Fig. 2B). 18 This substitution leads to constitutive activation of the protein, which results in an increase in its basal kinase activity. In vitro, the kinase activity of mutant B-RAF is approximately 500-fold greater than that of its wild-type counterpart, and the transformation rates in vivo are 70 to 138 times more efficient than transformation by the wild-type B-RAF. 19,20 The effectiveness of B-RAF mutations is implicated in its common presence in various cancers. B-RAF has been found to be mutated in several human cancers including but not limited to colorectal (5%-22%), ovarian (30%), thyroid (36%-53%), and most commonly in melanoma (27%-70%). 20
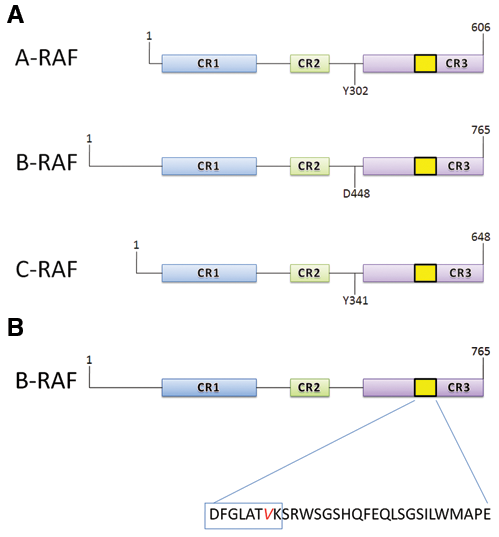
Conformations and mutations in RAF proteins. (
B-RAF mutations have been reported to be important in the development and progression of melanoma; however, B-RAF mutations alone are not sufficient to cause melanoma tumorigenesis. Observations show that 82% of benign nevi have high frequencies of B-RAF mutations relative to frequencies seen in RGP tumors. 21 This disparity in the frequency of activating B-RAF mutations between benign nevi and RGP tumors implicates that B-RAF mutations alone cannot induce melanoma but must cooperate with other signaling pathways such as the PI3K/AKT pathway and/or additional genetic alterations such as loss of p53 or PTEN. 22,23 In addition, the microenvironment has long been acknowledged as a facilitator of melanoma initiation and progression; recent studies have illuminated tumor-associated factors of the microenvironment, including hypoxia and the extracellular matrix, as important mediators of melanocyte transformation and transdifferentiation to melanoma. 24
The dynamic role of genetic and epigenetic changes, including B-RAF mutations in cooperation with other genetic alterations and the microenvironment, in the transition between melanocytic and melanoma cell phenotypes has been extensively reported. As a representative example, in the zebrafish-expressing constitutively active B-RAFV600E, but not wild-type B-RAF, dramatic patches of ectotopic melanocytes were formed, but no evidence of transformation was seen. When p53-deficient transgenic zebrafish were generated to express B-RAFV600E, not only were melanocyte lesions formed, but these lesions also rapidly developed into invasive melanomas. 25 In accordance, recent studies have determined that hypoxia may be essential for melanocyte transformation, and aggressive melanoma-conditioned extracellular matrices can epigenetically transdiffentiate normal melanocytes toward an invasive melanoma-like phenotype. 26,27
To summarize, activating B-RAF mutations constitute one epigenetic change, which plays a profound role throughout melanoma progression. However, only cooperation of B-RAF mutations with other signaling pathways and the microenvironment can actively promote the earliest steps of melanocyte transformation and later enhance aggressive phenotypes including invasion, vasculogenic mimicry, and angiogenesis. In the following sections, we further discuss how B-RAF signaling synergizes with key molecular pathways and the tumor microenvironment in the induction of EMT in melanoma (Fig. 3).
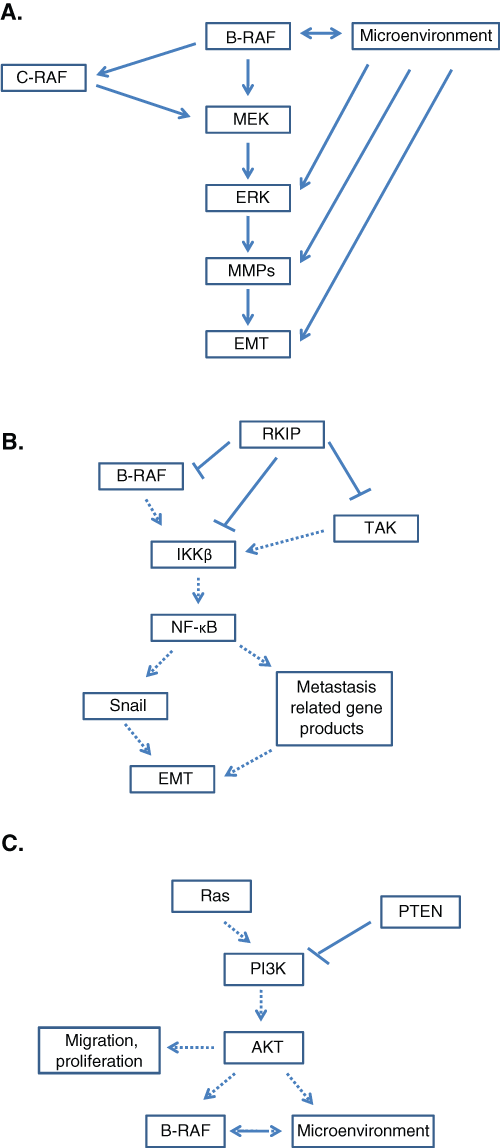
Regulation of survival pathways by mutant B-RAF. (
Cell Signaling with B-RAF Mutations and EMT
In cancer, the dysregulation of several regulatory pathways has been implicated to promote metastasis of tumor cells. The MAPK, NF-κB, and PI3K/ATK pathways are some of the most commonly dysregulated pathways. The dysregulation of MAPK, NF-κB, and PI3K/AKT pathways is common in melanoma; these 3 pathways are constitutively hyperactivated. 20,28,29 A mechanism of hyperactivation of the MAPK and NF-κB pathways, resulting in increased metastatic potential, is through mutation and constitutive activation of the upstream signaling protein, B-RAF. Deletion of PTEN accounts for the hyperactivation of the PI3K/AKT pathway, which collaborates with B-RAF to hyperactivate the MAPK and NF-κB pathways (Fig. 1). Mutated B-RAF has been shown to induce constitutive ERK signaling in NIH3T3 (mouse embryonic fibroblast cells) and induce tumor formation in vivo. 20,30 When B-RAF mutations were observed in LOX-IMVI and A375 melanoma cell lines, the same results as the NIH3T3 cells were observed. LOX-IMVI and A375 cells transfected with mutant B-RAF when injected into mice resulted in tumorigenesis. Inhibition of the mutant B-RAF in vivo led to inhibition of tumorigenesis and regression of tumor growth. 31 Oncogenic B-RAF has also been shown to increase IKK activation and simultaneous IκB degradation. Conversely, knockdown of B-RAF signaling resulted in a decrease of IKK activation and IκB degradation. 32 Loss of PTEN has been found in several kinds of solid tumors and plays an important role in tumorigenesis and angiogenesis. 33 From these above findings, B-RAF is a likely major factor in the regulation of EMT via the deregulation of the MAPK, NF-κB, and PI3K/AKT survival pathways as will be described below.
Activation of the MAPK Pathway by B-RAF Mutations
The mitogen-activated protein kinase (MAPK) pathway, also known as the Ras/Raf/MEK/ERK pathway, is a commonly studied pathway associated with mutant B-RAF. The MAPK pathway is a signal transduction cascade that relays extracellular signals from the plasma membrane to the nucleus through a series of kinase-induced phosphorylations. Studies have shown that the MAPK pathway is hyperactivated in the majority of human melanomas; the hyperactivation of this pathway leads to cell proliferation, survival, and the induction of EMT. 34 The signaling pathway of a normal cell through the MAPK pathway is first initiated by the activation of RAS and recruitment of cytosolic RAF to the cellular membrane. At the cell membrane, RAF becomes activated by tyrosine kinases, such as the Src-family kinases, and in turn activates the MAP kinases MEK 1 and 2. ERK 1 and 2 are then phosphorylated and translocated into the nucleus to bind promoters of genes involved in the induction of cell growth and proliferation (Fig. 3A). 35,36 Most commonly, when B-RAF is mutated, it is constitutively activated without the need for signaling from RAS. The downstream effect of constitutively activated B-RAF is continual translocation of ERK to the nucleus promoting cell proliferation and invasion. Other B-RAF mutations, such as D593V, are found to have a lower kinase activity, 0.3-fold activity of wild-type B-RAF, resulting in impaired signaling activity directed to the MAPK pathway. However, these mutations with lower kinase activation continue to regulate translocation of ERK through activation of endogenous C-RAF and, consequently, activation of MAPK. 16 Regardless of the type of mutation, the MAPK pathway is constitutively activated, and ERK is continuously translocated to the nucleus to promote cell growth, proliferation, and EMT. ERK has been found to be constitutively activated in several cancer types including 90% of melanomas 37 ; this finding has been correlated with high frequencies of activating B-RAF mutations in melanoma. However, B-RAF is only one factor in ERK1/2 activation; additional factors and mechanisms, mostly related to the tumor microenvironment, also contribute to ERK hyperactivation during melanomagenesis.
The MAPK pathway is negatively regulated by Raf-1 kinase inhibitor protein (RKIP). RKIP is a metastasis suppressor gene product and is a member of the phosphatidylethanolamine-binding protein (PEBP) family. 38 RKIP has not only been associated with cellular signaling but has also been shown to be involved in neurological functions and reproduction. 39 RKIP has been shown to regulate MAPK activity via inhibition of B-RAF activity. 38,40 This inhibition of B-RAF activity in normal cells by RKIP prevents uncontrolled proliferation and invasion by regulating MAPK signaling through B-RAF signaling. RKIP not only regulates MAPK but is also responsible for the regulation of the NF-κB pathway, another pathway hyperactivated in melanoma by B-RAF mutations. The MAPK regulates gene products involved in the induction of EMT (see below).
Activation of the NF-κB Pathway by B-RAF Mutations
The NF-κB pathway is an important cell survival signaling pathway in tumor cells that regulates the expression of antiapoptotic, proproliferative, and prometastatic genes. 32 The NF-κB pathway is activated through IKK (IκB kinase). IKK triggers the phosphorylation and degradation of IκBα by the proteasome leading to translocation of NF-κB to the nucleus. 41 NF-κB then promotes metastasis through the up-regulation of several metastatic genes such as COX-2, metalloproteinases, VEGF, and most importantly the metastasis inducer Snail. 42,43 Constitutive activation of NF-κB has become a hallmark of melanoma. One of the potential mechanisms by which the NF-κB pathway is constitutively active is through mutant B-RAF signaling. Mutant B-RAF activates the canonical NF-κB pathway through IKKβ, which promotes degradation of IκB and translocation of NF-κB to the nucleus. 28,44,45 Regulation of NF-κB is executed by RKIP. RKIP, as previously stated, is able to regulate B-RAF activity and therefore also regulates the downstream activity of NF-κB. RKIP regulates NF-κB activity using 2 different pathways: through direct RKIP inhibition of B-RAF activity and through direct inhibition of the NF-κB pathway via physical association and inhibition of the NF-κB–inducing kinase (NIK), TGF-beta–activated kinase (TAK), and IκB kinase (IKK) (Fig. 3B). 46 Thus, both the MAPK and the NF-κB pathways are regulated negatively by RKIP and positively by B-RAF. Up-regulation of B-RAF activity is necessary in order to overcome the negative regulation of RKIP, and this up-regulation of B-RAF is carried out by deregulation and constitutive activation of the PI3K/AKT pathway.
Activation of the PI3K/AKT by B-RAF Mutations
Another commonly deregulated pathway associated with B-RAF mutation is the PI3K/AKT pathway. The phosphatidylinositol-3 kinases (PI3K) are directly regulated by RAS. 47 Activation of PI3K produces phosphatidylinositol-3,4,5-triphosphate (PIP3) and phosphatidylinositol-3,4-biphosphate (PIP2). PIP2 and PIP3 are inducers of AKT; therefore, an increase in PIP2 and PIP3 induces the formation of AKT isoforms. AKT activity is antagonized by the gene product PTEN (phosphatase and tensin homolog deleted on chromosome 10) through the degradation of PIP2 and PIP3. 48 AKT activity stimulates cell cycle progression, survival, and migration. 23 The loss of PTEN results in constant up-regulation of AKT activity, leading to increased survival and migration of cells. Studies show that activation of AKT in mice does not induce metastasis unless concurrent with silencing of PTEN. 49 However, other than the role of AKT in cell survival and migration, AKT, specifically the isoform AKT3, also plays a role in regulating B-RAF (Fig. 3C). In normal cells, PTEN is strongly expressed but is subsequently less expressed in melanomas and completely lost in the metastatic lesions. 50 Decreased PTEN expression levels up-regulate AKT activity. The up-regulation of AKT activity is important for the regulation of B-RAF signaling. AKT3 inhibits B-RAF activity, which, counterintuitively, promotes cell proliferation. Mutant B-RAF, which expresses high levels of signaling, induces cellular defense mechanisms targeted against activated oncogenes, and the induction of these cellular defense mechanisms leads to cell senescence. Inhibition of B-RAF by AKT3 regulates B-RAF to levels high enough to promote proliferation, survival, and metastasis but low enough to prevent activation of cellular defense mechanisms and cell senescence. 51,52 Without dysregulation of the PI3K/AKT pathway, AKT3 expression would not be high enough to optimize mutant B-RAF activity, and neither the MAPK nor the NF-κB pathway would be hyperactivated.
Within each of the above mentioned pathways, several gene products are regulated by NF-κB, particularly important in the regulation of EMT, and include Snail, RKIP, and PTEN. The implication of these gene products and their association with B-RAF are important for understanding their role in transformation and the induction of EMT in melanoma tumors.
Cross-Talk between the Microenvironment and Genetic Alterations in Melanoma during the Induction of EMT
Tumor cells communicate bidirectionally with the surrounding microenvironment, which consists of elements as diverse as extracellular matrix components and growth factors (including VEGF, nutrients, and varying concentrations of oxygen). Collectively, these interfere with the genetic cell programming and supply signals that control cell survival, growth, differentiation, and invasion. The microenvironment has emerged as a major player in melanocyte transformation and transdifferentiation and in promoting melanoma cell invasion and metastasis. For EMT induction in melanoma, all genetic abnormalities including common mutations and/or deregulated expressions of B-RAF, N-RAS, and PTEN seem to synergize with several microenvironmental factors. Among these factors are the loss of cell-cell adhesions, resulting from 1) altered expression of cadherins, catenins, and integrins; 2) the interaction between melanoma cells and surrounding keratinocytes, fibroblasts, and immune cells; and 3) the tumor angiogenetic efficiency. 53 In addition, it has been shown that hypoxia promotes melanocyte transdifferentiation and melanoma migratory and invasive abilities through up-regulation of genes normally associated with the extracellular matrix remodeling and invasion; these genes include laminin 5 γ2 (Ln-5 γ2), Urokinase, and genes encoding matrix metalloproteinases. 54,55 Other studies have shown that constitutively active AKT, which is observed in a high percentage of melanomas, can transform melanocytes when oxygen levels are low. 26 Furthermore, different microenvironments may be selective for higher levels of both B-RAF–dependent and B-RAF–independent ERK1/2 activation; these microenvironments are critical for tumor cell proliferation and spread. Immunohistochemical studies in melanoma samples have shown that expression levels of phosphorylated ERK1/2 are not always correlated with the mutational status of B-RAF or N-RAS, 56 suggesting that other B-RAF–independent or B-RAF–complementary factors and mechanisms promote ERK1/2 activation. Among these factors are growth factor autocrine loops, such as the CSF-dependent activation of c-Kit, and extracellular signals, such as derived from epidermal and nerve growth factors. 57-61 On the other hand, Conner et al. have shown that adhesion-regulated ERK1/2 activation in melanocytes is by-passed by B-RAF mutations in malignant melanoma cells, although melanoma cells may still be dependent on integrin-activated PI3K and AKT signaling for survival and migration. 62 The above findings exemplify how the microenvironment can complement aberrant genetic changes such as mutations in B-RAF to promote melanomagenesis and to support an invasive cell phenotype. The contribution of the microenvironment in cell fate might further explain why RGP tumors are characterized by a relatively low frequency of B-RAF mutations; EMT is important for promoting invasion and transition to VGP cells. 13 In contrast to general conceptions that invasion and dissemination occur during the later vertical phase, recent findings show that early dissemination of tumor cells that have not yet fully progressed is a contributor to subsequent development of metastasis. This finding is in accordance with similar observations in breast cancer and suggests that genetic alterations are not autonomous in determining cell behavior and cell fate; however, with the support of microenvironmental factors, even the less “progressed” tumor cells with few genetic alterations may accumulate additional alterations that will favor ectopic growth. 63
The Regulation of NF-κB Activity and Downstream Snail, RKIP, and PTEN Gene Products by B-RAF Mutations and Induction of EMT
Evidence suggests a strong association of B-RAF mutations with NF-κB, Snail, RKIP, and PTEN in the regulation of EMT. Each of these has been individually shown to be directly associated in the regulation of EMT in melanoma.
Role of NF-κB Activation by B-RAF Mutations and Induction of EMT in Melanoma
Metastatic prostate cancer cells have elevated NF-κB activity and constitutively active IKK, which is responsible for the activation of NF-κB. 64 These findings are observed in several cancers including melanoma. 65 Knockdown of NF-κB by RNAi reversed the mesenchymal-like phenotype and suppressed the motility and invasion capacity of metastatic cancer cells. In vitro motility and invasion assays showed that both the motility and invasive potentials were reduced in cells in which NF-κB was inhibited. 66 In vivo, inhibition of NF-κB in Ras-transformed epithelial cells led to an actively suppressed EMT gene program. One mechanism of regulation of NF-κB activation is through the B-RAF signaling cascade via IKK activation and IκB degradation. Hyperactivation of NF-κB via mutant B-RAF greatly increases the metastatic potential. Several studies have provided evidence for potential underlying mechanisms by which activated NF-κB is able to induce metastasis. 67 Matrix metalloproteinases (MMPs) have been shown to be important for invasion and angiogenesis and are highly implicated in metastasis. 68,69 Regulatory elements within the 5′ flanking sequence of the human MMP-9 gene have consensus sequences for NF-κB. NF-κB activation has been shown to be necessary for inducing MMP-9 mRNA and protein expression. 69 In addition to these findings, NF-κB has also been shown to activate transcription of the metastasis-inducer Snail transcription factor.
Role of Snail Activation by B-RAF Mutations and Induction of EMT in Melanoma
Snail is a transcription factor that is a member of the zinc-finger protein family and has been linked to metastasis and resistance against apoptosis. Snail has been shown to induce metastasis by repression of E-cadherin. 70 E-cadherin is a cell adhesion molecule; loss of cell-cell adhesion by reduction of E-cadherin leads to EMT. Epithelial cells that overexpress Snail acquire metastatic and invasive potential and are correlated with down-regulation of E-cadherin expression. 71 Snail is also involved in E-cadherin–independent EMT through the down-regulation of claudins and occludins. Claudins and occludins are associated with establishing and maintaining cell polarity. 72,73 Loss of these molecules in tumor cells results in a deregulation of cell polarity and increased potential for EMT. Snail is also demonstrated to induce metalloproteinases; induction of metalloproteinases is associated with portal invasion, metastasis, and recurrence in human cancers. 74,75 B-RAF has been shown to up-regulate Snail and thus provides a potential mechanism for B-RAF–induced EMT. 76 Hyperactivation in mutant B-RAF results in overexpression of Snail and, therefore, increased metastatic potential of the melanoma. An important molecule antagonizing the metastatic effects of B-RAF and Snail is the metastasis suppressor, RKIP.
Role of the Inhibition of RKIP Expression by B-RAF Mutations and Induction of EMT in Melanoma
RKIP is a metastasis suppressor gene product. Studies have shown that overexpression of RKIP inhibited metastasis and down-regulation of RKIP increases the metastatic potential. In mice, RKIP that was exogenously expressed led to a decreased level of metastasis and invasion in transformed metastatic cells. 77 RKIP also plays a role as a proapoptotic factor in drug-resistant tumors; induced overexpression of RKIP in drug-resistant cells showed an increase in apoptosis-matching levels of apoptosis induced by the constitutively active proapoptotic factor t-Bid. 78 RKIP has been shown to be able to directly down-regulate the metastasis-inducing pathway, the NF-κB pathway. As discussed above, NF-κB activation requires the phosphorylation and degradation of IκB. RKIP has been shown to negatively modulate the activating phosphorylations of IKKα and IKKβ by upstream kinases. 46 RKIP is also able to down-regulate NF-κB activity via B-RAF signaling. Inhibition of B-RAF signaling by RKIP is achieved by down-regulation of B-RAF kinase activity; this inhibition of B-RAF kinase activity results in the down-regulation of all metastatic pathways associated with B-RAF signaling. 40 RKIP is not the only regulator of B-RAF; PTEN is also an important regulatory molecule in B-RAF–associated pathways and metastasis.
Role of Inhibition of PTEN Expression by B-RAF Mutations and Induction of EMT in Melanoma
Phosphatase and tensin homolog deleted on chromosome 10, also known as PTEN, is a tumor suppressor gene located at 10q23-24 on human chromosome 10. 47 PTEN is a member of the protein tyrosine phosphatase (PTP) family and has a relatively high constitutive phosphatase activity in vitro and in vivo. 79 The amino acid sequence of PTEN resembles that of 2 proteins, protein tyrosine phosphatases as well as lipid phosphatases. 80 Sequence similarities have also been found between PTEN and the cytoskeleton protein tensin, whose role involves interaction with integrins in integrin-signaling complexes. 81 Integrins are involved in forming complexes of signal transduction molecules such as MAPK, 82,83 Ras, 84 NF-κB, 85 and PI3K. 86 Because of the sequence similarity between PTEN and tensin, it also interacts with integrin-signaling complexes. 87,88 PTEN plays a significant role in cell cycle arrest, regulation of cell adhesion, migration, and proliferation. PTEN is demonstrated to be critical for embryonic development, and the expression of PTEN is ubiquitous in all human tissues. 89,90 Loss of PTEN and/or its function is found in many tumors as well as various other diseases. PTEN loss occurs through homozygous gene deletion or point mutation, resulting in loss of its expression. 91-93 Several mechanisms have been described for the mutation of PTEN; most mutations occur within the coding region of the gene and inactivate phosphatase activity. 79 The deletion of PTEN in melanoma results in hyperactivation of the PI3K/AKT pathway and overexpression of AKT3. Overexpression of AKT3 is necessary as a collaborating molecule in B-RAF signaling. Levels of AKT3 must be up-regulated in order to optimize B-RAF signaling levels; optimization of B-RAF signaling levels induces cell proliferation, survival, and metastasis via the B-RAF–associated pathways MAPK and NF-κB. 51,52
As discussed above, B-RAF mutations regulate NF-κB, Snail, RKIP, and PTEN activities in melanoma and metastasis. Noteworthy, when examined collectively, it appears that these activities are part of an NF-κB/Snail/RRKIP/PTEN circuit such that this circuit regulates EMT.
Dysregulation by B-RAF Mutations of the NF-κB/Snail/RKIP/PTEN Circuit and Induction of EMT
All 3 pathways associated with B-RAF mutations, namely, the MAPK, NF-κB, and PI3K/AKT pathways, result in the up-regulation of the metastasis inducer, Snail. The PI3K/AKT pathway has been shown to activate the NF-κB pathway via IKKβ. 94 The PI3K/AKT pathway is normally antagonized by PTEN; however, PTEN somatic mutations are found in 40% to 60% of melanoma cell lines and 10% to 20% of primary melanomas. 95 Therefore, NF-κB is up-regulated not only by B-RAF but also by the uninhibited PI3K/AKT pathway as well. Not only is PTEN found to be commonly mutated, but it is also down-regulated by the transcription factor Snail (Fig. 4A). 96 Thus, the NF-κB pathway is shown to be constitutively activated by the PI3K/AKT pathway due to the mutation/suppression of PTEN as well as Snail-induced inhibition of PTEN.
Regulation of the NF-κB pathway is also achieved through the MAPK pathway through MEK signaling. MEK has been shown to induce NF-κB activation through the IKK complex. 97 The MAPK pathway also mediates the NF-κB pathway via the translocation factor, ERK. ERK regulates downstream the translocation of NF-κB into the nucleus through up-regulation of Snail expression (Fig. 4B). 76 When B-RAF is mutated, the MAPK pathway is constitutively activated and, therefore, constitutively up-regulates Snail indirectly via NF-κB activation and directly by ERK translocation.
Snail has been shown as a negative regulator of RKIP 98 ; up-regulation of Snail through the PI3K/AKT and MAPK pathways inhibits both PTEN and RKIP, which then, in turn, amplifies the expression of Snail through constitutively active NF-κB (Fig. 4C). From the cross-talks among the different B-RAF–associated pathways, it is clear that the regulation of Snail via PTEN and RKIP is an important mechanism in the regulation of EMT.
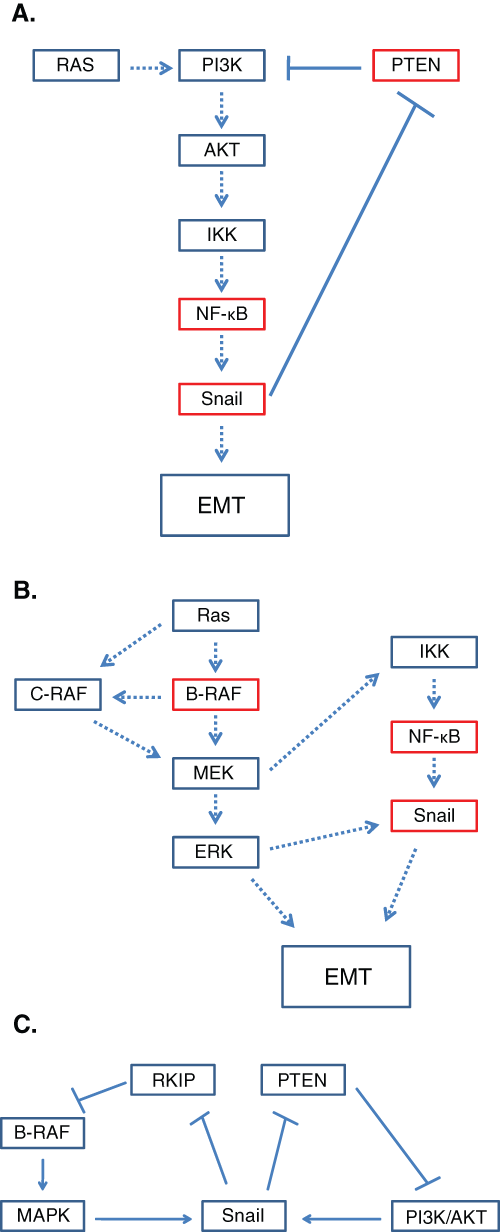
Activation of NF-κB by the PI3K/AKT and MAPK pathways. (
B-RAF Mutations in Melanoma and Therapeutic Targets Directed at the Dysregulation of the NF-κB/Snail/RKIP/PTEN Circuit to Prevent EMT
One RAF kinase inhibitor, sorafenib (BAY 43-9006), is an oral multikinase inhibitor that targets RAF kinase as well as the VEGF receptor. 99 Sorafenib has been observed to inhibit growth of melanoma xenografts in mice; however, no antitumor effects were seen in advanced melanoma patients. Sorafenib is well tolerated but has little effect in advanced melanoma patients as a single agent. 100 RAF kinase inhibitors could potentially be the most potent drugs; inhibition of the RAF kinase could inhibit B-RAF activity and all downstream signaling pathways. Other inhibitors currently in clinical trials are the MEK kinase inhibitors PD0325901 and AZD6244. 7 B-RAF–mutated tumors seem to be particularly sensitive to clinically available MEK inhibitors. 101 It appears that MEK inhibitors are more often agents that induce growth arrest rather than cell death in most B-RAF–mutated models. 18 Inhibition of MEK would result in inhibition of ERK translocation as well as inhibition of NF-κB translocation and, therefore, a decrease in the metastatic potential. The PI3K pathway is another potential target for chemotherapy; however, current trials using CCI-779 and RAD001 to target the PI3K pathway do not have a high specificity in targeting melanoma cells and could potentially, counterproductively, activate AKT. 102,103 If new PI3K inhibitors are developed, overexpression of AKT could be controlled, and overexpression of mutant B-RAF would induce tumor cell senescence. Current treatments with individual drugs have not yielded satisfactory results. These are, in part, due to the complicated interrelations among the pathways involved in metastasis, namely, the cross-talks among the PI3K/AKT, NF-κB, and MAPK pathways. Combinational treatments with the above drugs may lead to synergistic effects. Hopeful results have already been seen in the combination of sorafenib with carboplatin and paclitaxel. 104 PLX4032 is a novel, oral, small molecule that specifically targets B-RAFV600E. It has recently been tested in clinical trials and has yielded positive responses in patients with metastatic melanoma. In phase I trials, a response was seen in 78% of the patients. However, despite the positive results obtained in phase I trials, several challenges have been uncovered. Toxic side effects such as the development of squamous cell carcinoma occurred in 20% to 30% of treated patients and could, therefore, deter long-term usage of the drug. 105,106
Conclusions and Future Directions
Additional potential therapies that might prove useful in the treatment of malignant melanoma include the targeting of RKIP and Snail. Such potential drugs could include proteasome inhibitors, such as bortezomib and NPI-0052, which are shown to induce RKIP expression and inhibit the activity of NF-κB and the transcription of Snail. 107 Induction of RKIP would also inhibit metastasis through the MAPK pathway. Targeting for RKIP induction would be more efficient than targeting downstream effectors such as NF-κB and Snail. Drugs inducing the expression of PTEN could also prove effective. Overexpression of PTEN would suppress AKT expression and induce cell senescence caused by extreme hyperactivation of mutant B-RAF. Combinational therapies should continue to be pursued in order to concurrently inhibit all potential mechanisms of EMT and metastasis. Further research is required to identify other possible gene products that are needed to cooperate with mutant B-RAF for initiation, maintenance, and progression of melanoma. Discovery of additional genetic alterations that cooperate with B-RAF will provide future therapeutic tools for the treatment and diagnosis of malignant melanoma.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This study was supported by various donors and by the Jonsson Comprehensive Cancer Center at UCLA.