
Abstract
Background
The efficacy of topical corticosteroids is limited in chronic rhinosinusitis (CRS) due to rapid clearance from the nasal cavity and insufficient drug delivery to inflamed sinonasal passages. LYR-210 is an implantable corticosteroid matrix designed to provide up to 24 weeks of treatment to patients with CRS by locally delivering mometasone furoate (MF) to the sinonasal mucosa. In a randomized, controlled, dose-ranging LANTERN study, LYR-210 (7500 µg) achieved clinically relevant improvement in CRS cardinal symptom composite scores, the 22-item Sinonasal Outcome Test (SNOT-22), ethmoid opacification, and the need for rescue treatment at 24 weeks.
Objective
As the plasma MF concentrations of LYR-210 (2500 µg) and LYR-210 (7500 µg) were evaluated at weeks 4, 12, and 24 in the LANTERN study (data on file at Lyra Therapeutics, Inc.), this study aims to characterize the pharmacokinetic profiles of both doses of LYR-210 at earlier timepoints post-placement in patients with CRS.
Methods
Twenty-four surgically naïve adult patients with CRS were enrolled in an open-label, multicenter study and underwent in-office bilateral administration of LYR-210 (2500 µg) (n = 12 patients) or LYR-210 (7500 µg) (n = 12 patients) into the middle meatus. Plasma MF concentrations were determined pre-placement and 1-h post-placement (day 1), and on days 2, 3, 7, 14, 21, 28, 42, and 56 by liquid chromatography-tandem mass spectrometry.
Results
Both LYR-210 doses were well-tolerated with no serious adverse events. Systemic MF levels were dose-dependent and lower than reported values of other respiratory MF products. Plasma MF concentrations showed steady drug release from LYR-210 (2500 µg) and LYR-210 (7500 µg) that persisted through day 56.
Conclusion
LYR-210 achieved dose-dependent, continuous local MF delivery at a steady rate with low systemic exposure for months.
Keywords
Introduction
Topical intranasal corticosteroids are recommended by clinical practice guidelines as a mainstay treatment for patients with chronic rhinosinusitis (CRS).1,2 However, their efficacy is suboptimal due to rapid clearance rates from the nasal cavity, inconsistent drug delivery deep into the inflamed sinonasal passages, and poor patient compliance.3,4 It is estimated that 40% to 60% of patients with CRS do not achieve adequate symptom control from standard treatments. 5 While these patients may be considered candidates for functional endoscopic sinus surgery (FESS), many decline the procedure, resulting in chronic symptom persistence. 6
To overcome the challenges of existing therapies and improve medical management of CRS, there is a need for long-acting local treatments that deliver a targeted and consistent therapeutic dose of medication to the site of the disease while alleviating the need for daily adherence. One such investigational treatment is LYR-210, an implantable corticosteroid matrix that delivers mometasone furoate (MF) directly to inflamed sinonasal mucosa over a 24-week treatment period. The Phase 2, randomized, controlled, dose-ranging LANTERN study showed that LYR-210, administered to surgically naïve patients with CRS who failed previous medical management, was safe and well-tolerated, and achieved rapid, durable, clinically meaningful, and dose-dependent improvements in CRS cardinal symptom composite scores and SNOT-22 total scores. 7 LYR-210 (7500 µg) also reduced the need for rescue medications and showed evidence of a reduction in ethmoid sinus opacification on magnetic resonance imaging during the 24-weeks of treatment. 7 In the LANTERN study, plasma MF levels of LYR-210 (2500 µg) and LYR-210 (7500 µg) were evaluated at weeks 4, 12, and 24, and MF was detectable in the plasma through 24 weeks (data on file at Lyra Therapeutics, Inc.). To characterize the systemic exposure of MF from these 2 doses of LYR-210 at earlier time points, this clinical study was initiated with the primary objective of evaluating the plasma pharmacokinetics (PK) of LYR-210 in patients with CRS. Safety and symptoms (e.g., SNOT-22) were also assessed.
Methods
Investigational Products
Lyra Therapeutics, Inc. (Watertown, MA, USA) designed and formulated the investigational product LYR-210 to gradually release up to 2500 µg [LYR-210 (2500 µg)] or 7500 µg [LYR-210 (7500 µg)] of MF, a potent, topically active anti-inflammatory corticosteroid, over 24 weeks. MF is embedded in a tubular mesh composed of bioresorbable polymers. The unique engineered elastomeric properties allow LYR-210 to dynamically expand and conform to the middle meatus, allowing continuous contact with the surrounding mucosa over the treatment duration (Figure 1).
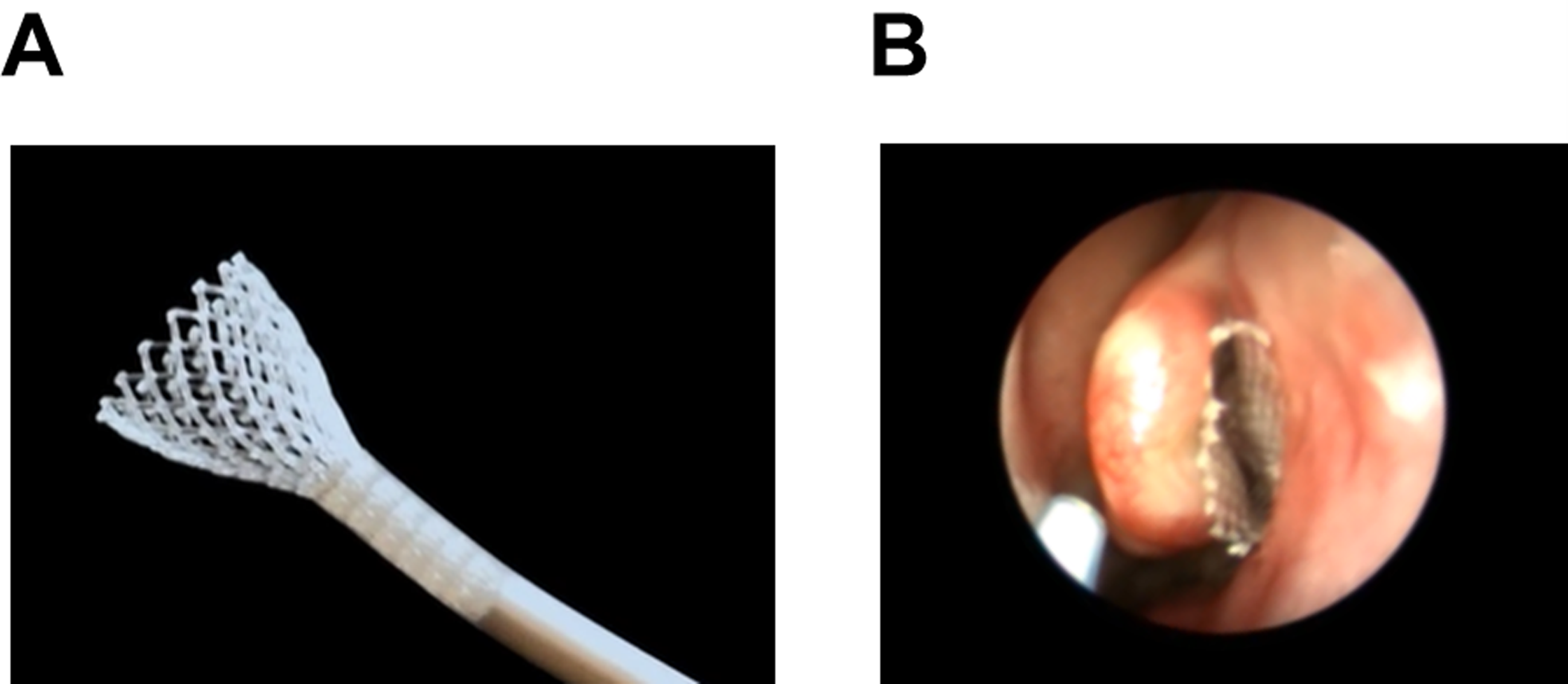
(A) LYR-210 self-expanding when deployed from the applicator. (B) LYR-210 placed in the middle meatus.
Study Design
The primary objective of this prospective, open-label, multicenter study was to evaluate the PK profiles of LYR-210 (2500 μg) and LYR-210 (7500 μg) for 56 days in adults with CRS who have not undergone FESS. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol and patient-informed consent were reviewed and approved by a central ethics committee in accordance with the regulatory requirements. All patients signed informed consent before participating in the study. Immediately following the initial screening visit, patients underwent a washout period of 7 to 14 days. During the washout, patients did not receive active treatment for CRS except for daily nasal saline irrigation, which they were instructed to use throughout the study.
Patients were enrolled in the study from 4 sites in the United States and received bilateral administration of either LYR-210 (7500 μg) or LYR-210 (2500 μg). Anesthesia was left to the discretion of the investigator and consisted of topically nebulized and locally administered decongestants and anesthetics. LYR-210 was administered into the middle meatus by an otolaryngologist under endoscopic visualization with a provided single-use applicator. Patients returned to the clinic for follow-up visits on days 2 or 3, 7, 14, 21, 28, 42, and 56 for PK, safety, and symptom assessments. Drugs containing MF, drugs known to be potent CYP3A4 inhibitors, monoclonal antibodies, allergy immunotherapy, and oral anti-fungal medications were not permitted during the study. Rescue medications for sinus infections, severe acute nasal blockage, acute allergic symptoms, and worsening CRS symptoms were permitted at the discretion of the study investigator. On day 56, LYR-210 matrices were removed by the otolaryngologist using standard nasal instruments. Figure 2 illustrates the study design.
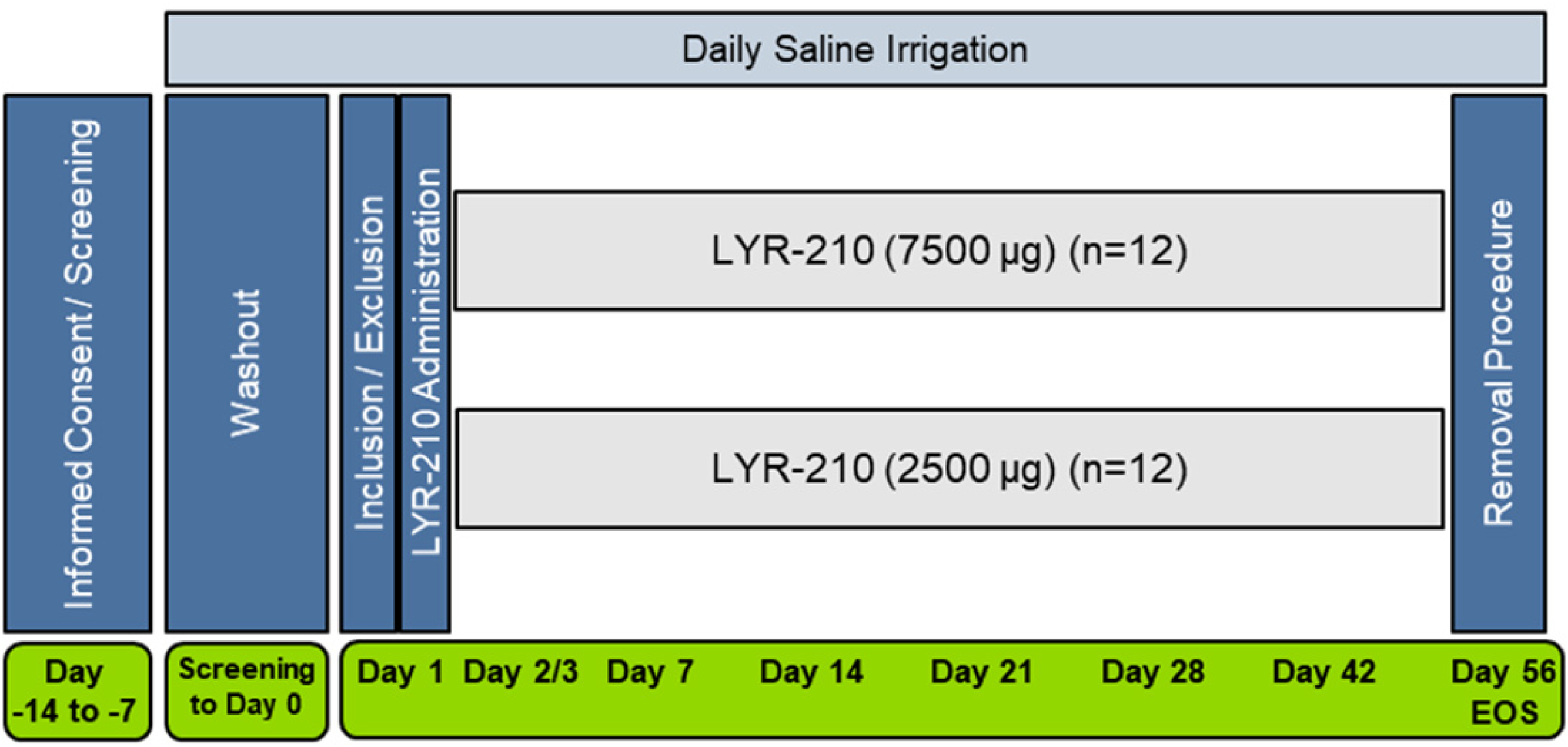
LYR-210 PK study design schematic. EOS = End of Study.
Inclusion and Exclusion Criteria
Adults (≥ 18 years of age) with CRS who had not undergone FESS were considered for participation in this study. Patients were included if they had CRS symptoms for 12 weeks or longer and one or more of the following findings on computed tomography or endoscopy: evidence of inflammation within paranasal sinuses or osteomeatal complex or nasal polyps; and the ability to tolerate topical anesthesia. Small, non-obstructive middle meatal polyps were allowed. Any woman of childbearing age was required to have a negative pregnancy test at screening and to use highly effective contraceptive methods for the study duration.
Patients were excluded if they used any medication containing MF during the washout period or if they had the previous FESS, including balloon sinuplasty or any endonasal surgery except for septoplasty or surgical manipulation to the nasal turbinates more than 6 months prior to the screening visit. Patients with endoscopic evidence of middle meatal obstruction preventing proper placement and/or visualization of LYR-210, evidence of mucosal erosion or ulceration, ongoing nasal infection, or evidence of nasal septal perforation were excluded. Patients were also excluded if they had structural, non-inflammatory-related CRS, sinus disease that extended into the orbital or intracranial space, evidence of mycetoma/fungal ball, allergic fungal rhinosinusitis, or sinus mucocele.
Plasma MF Concentrations
Plasma MF concentrations were measured on day 1 pre-placement and 1-h post-placement, and on days 2, 3, 7, 14, 21, 28, 42, and 56. Patients within each dose arm were evenly distributed for blood collection on day 2 or day 3. All patients had PK blood collection on day 1 and at timepoints subsequent to day 3. A liquid chromatography system equipped with tandem mass spectrometry (LC-MS/MS) detection was used to measure the concentration of MF in plasma with a lower limit of quantitation (LLOQ) of 0.25 pg/mL.
Safety Assessment
To evaluate the safety, physical examinations, including vital signs and laboratory, hematology, blood chemistry, and nasal endoscopy assessments were completed at screening and day 56 or end of the study. Adverse events (AEs) were recorded throughout the study and were assessed for their relationship to the study drug or procedure and severity.
Symptom Assessment
Symptom severity and social/emotional impact of CRS were evaluated via the SNOT-22, which was collected at baseline (day 1, pre-placement procedure) and on days 14, 28, 42, and 56.
Data Analysis
Patient enrollment size was not based on statistical power considerations because the primary study objective was to measure plasma MF concentrations. The safety analysis set consisted of all patients who received the study treatment or treatment attempt on day 1. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA). The number and percentage of patients with incidence of the categorical safety endpoints were presented by treatment group. No formal statistical tests comparing treatments on safety endpoints were performed, and there was no imputation of missing data. The intention-to-treat analysis set, which consisted of all patients who received the study treatment or treatment attempt on day 1 and had post-day 1 PK or symptom assessments available, was the primary analysis set for PK and symptom score analyses. Maximum concentration (Cmax) and time to reach maximum concentration (Tmax) were taken directly from the observed data. Total drug exposure up to the last measured concentration (AUC0−last) and the 12-h average drug exposure (AUC0−12h average) were calculated from the plasma concentrations versus time profiles. Descriptive statistics of these parameters were reported. Steady-state concentrations (Css) were calculated by AUC0−inf/τ (τ = 55 days). The mean SNOT-22 total score was compared between baseline and each time point post-placement using a two-sided paired t-test. The last observation carried forward was used to impute any missing SNOT-22 data.
Results
Patient Characteristics
Twenty-four adult patients with CRS who had not undergone FESS were enrolled between October 2020 and December 2020 in an open-label, multicenter study to evaluate the PK of 2 doses of LYR-210. Twelve patients were administered bilateral LYR-210 (2500 µg) and 12 were administered bilateral LYR-210 (7500 µg) into the middle meatus. Successful in-office placement of all 48 LYR-210 matrices was achieved, resulting in zero enrollment failures. Figure 3 illustrates the disposition of patients.
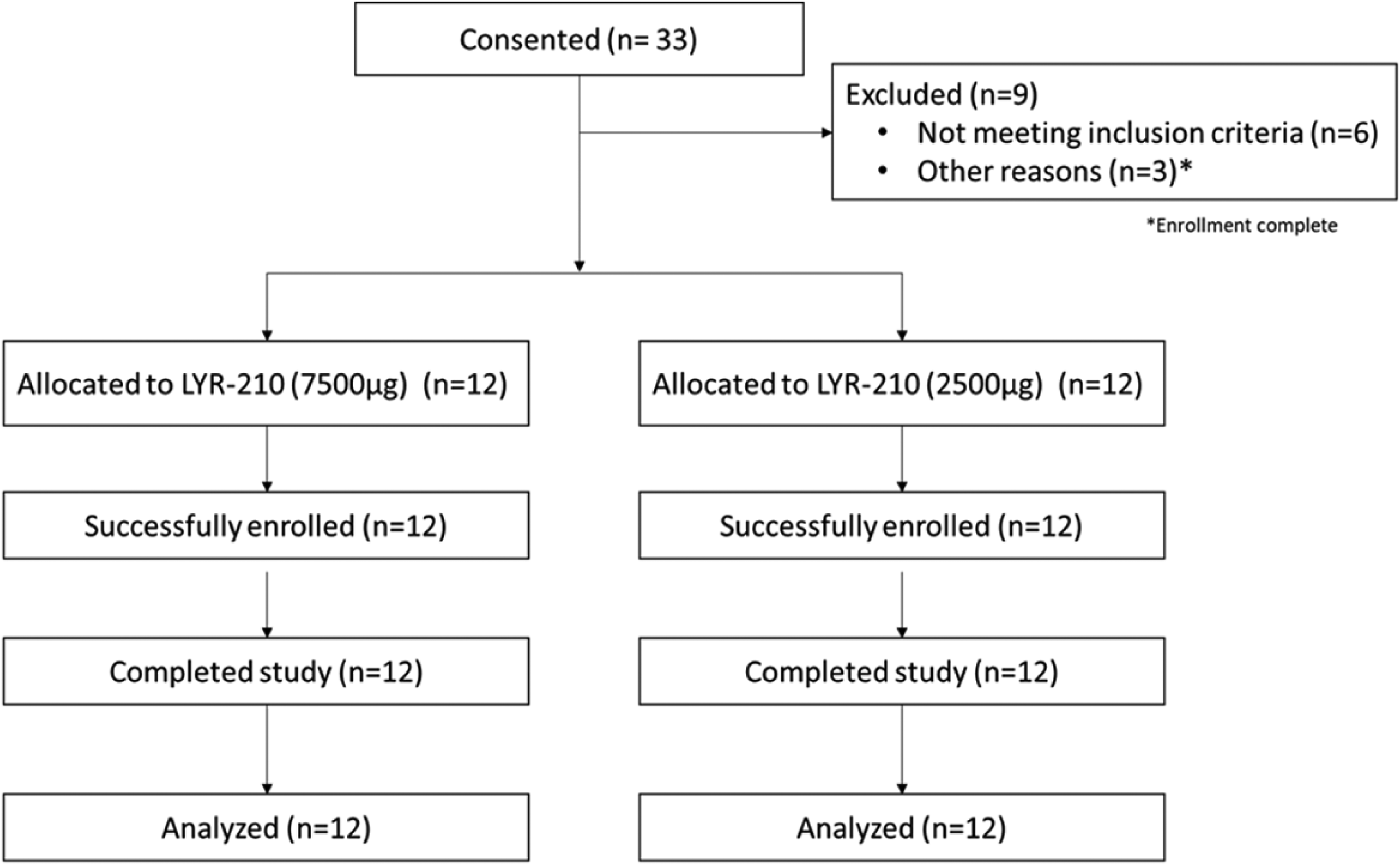
Disposition of patients (CONSORT) diagram.
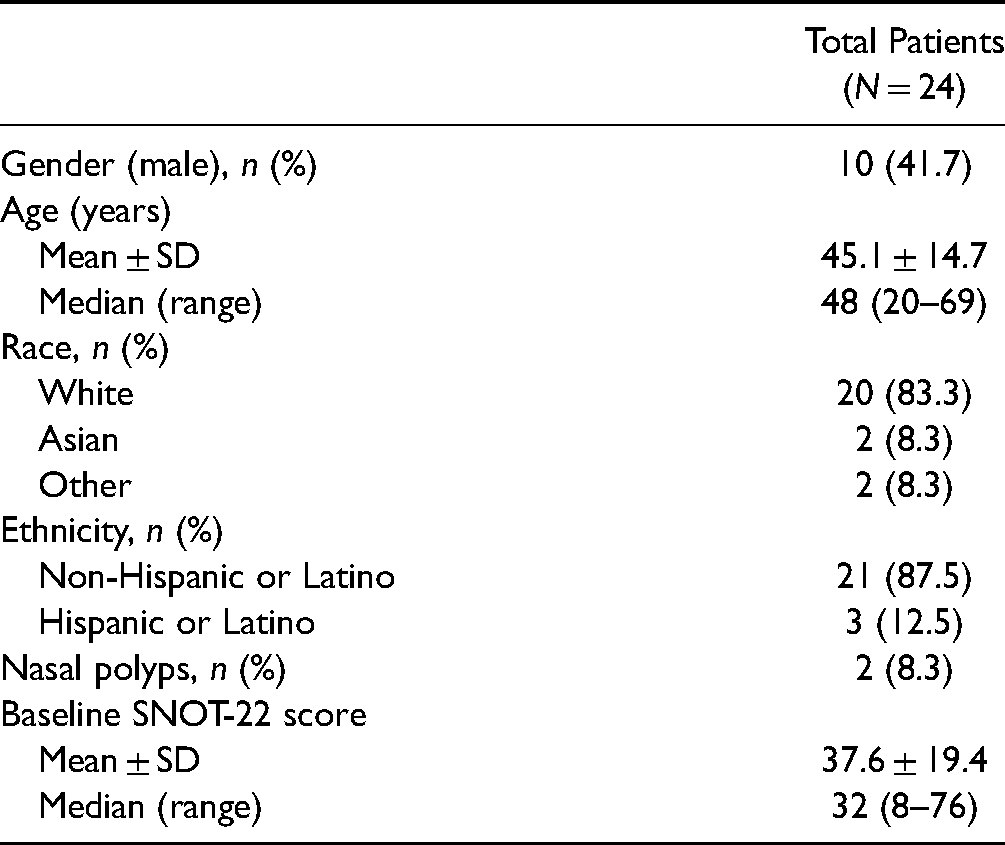
Of the 24 enrolled patients, 10 (41.7%) were male, 20 (83.3%) were white, and 3 (12.5%) were Hispanic or Latino. Two (8.3%) patients in the study were diagnosed with bilateral nasal polyps by endoscopy. Patient demographics are summarized in Table 1.
Patient Demographics.
Safety
All 24 patients completed the 56-day study. The matrix retention rate at day 56 was 98%. There were no serious adverse events in this study. The AEs reported by more than 1 patient in the study are summarized in Table 2. The most common AEs reported in the LYR-210 (2500 µg) arm were parosmia, sinusitis, and COVID-19 (each reported in 2 patients); and in the LYR-210 (7500 µg) arm were procedural pain, headache, and nasal odor (each reported in 2 patients). Most AEs were mild or moderate in severity based on physician assessment.
Adverse Events Summary.
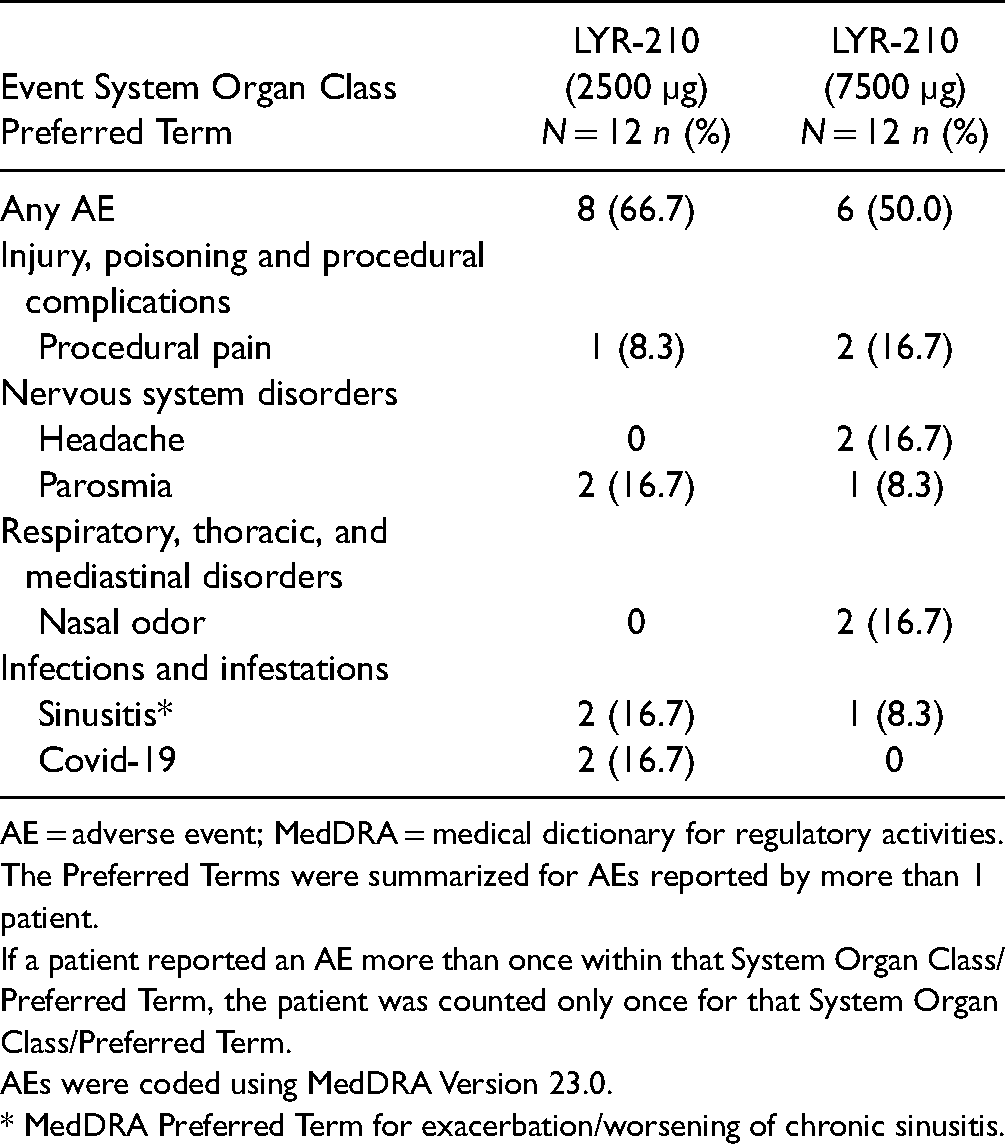
AE = adverse event; MedDRA = medical dictionary for regulatory activities.
The Preferred Terms were summarized for AEs reported by more than 1 patient.
If a patient reported an AE more than once within that System Organ Class/Preferred Term, the patient was counted only once for that System Organ Class/Preferred Term.
AEs were coded using MedDRA Version 23.0.
* MedDRA Preferred Term for exacerbation/worsening of chronic sinusitis.
LYR-210 Pharmacokinetics
Plasma MF concentrations were evaluated from patients administered bilateral LYR-210 (2500 µg) or LYR-210 (7500 µg) on day 1 pre-placement and 1-h post-placement, and on days 2, 3, 7, 14, 21, 28, 42, and 56. Figure 4 shows constant and dose-dependent MF plasma concentrations throughout the 56-day study duration. The plasma MF concentrations peaked on Day 2 (Cmax = 21.5 pg/mL) and Day 3 (Cmax = 58.9 pg/mL) for the LYR-210 (2500 µg) and LYR-210 (7500 µg) arms, respectively. The plasma MF concentrations dropped slightly and plateaued after Day 7. For this 56-day study, LYR-210 delivered a consistent daily dose of MF, achieving steady-state concentrations (Css) of 12.2 pg/mL and 41.2 pg/mL for LYR-210 (2500 µg) and LYR-210 (7500 µg), respectively.
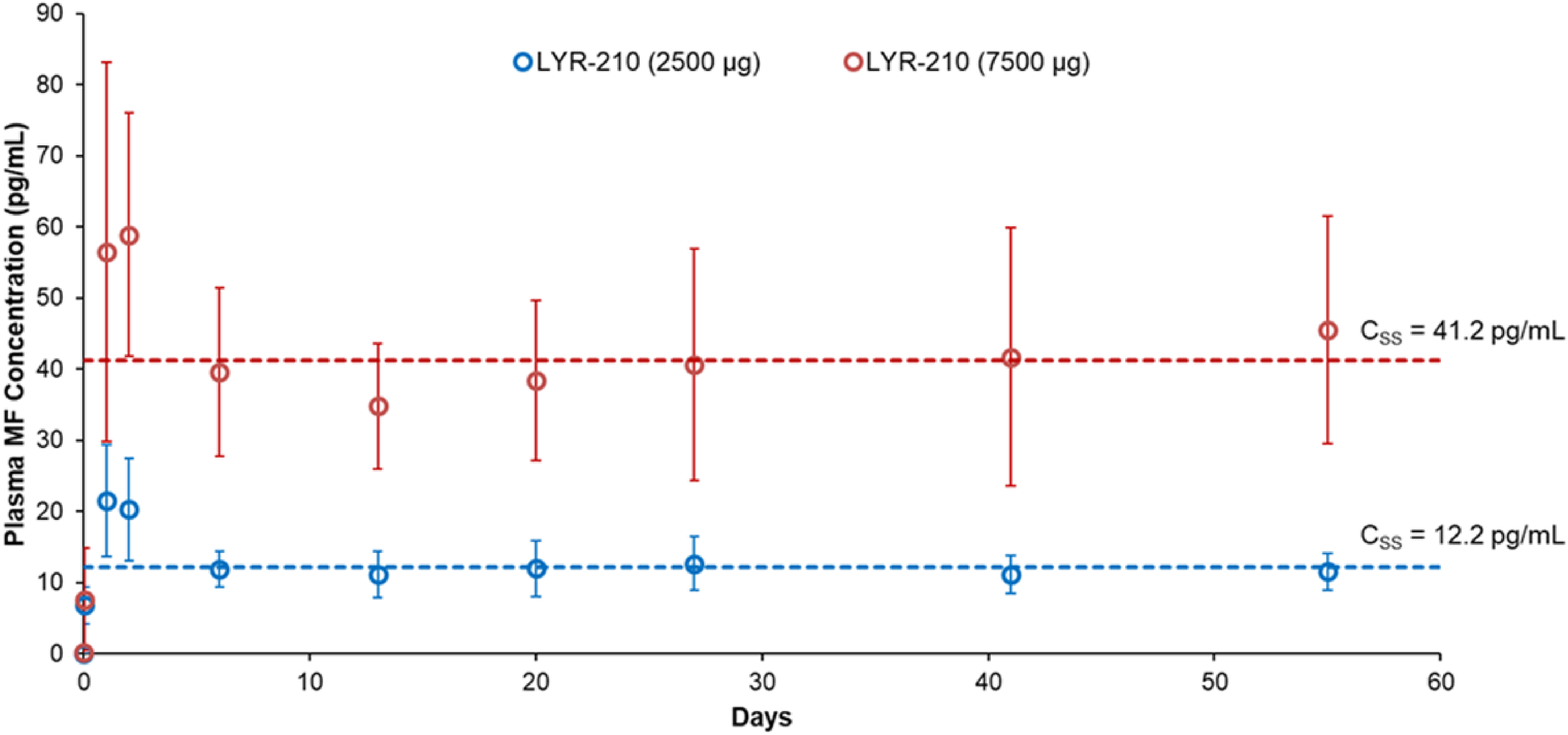
Plasma MF concentration-time profiles for LYR-210 (2500 µg) and LYR-210 (7500 µg) through 56 days. Data are represented as mean and standard deviation. Css = steady-state concentration.
Symptom Scores
The improvement in symptom scores was comparable between LYR-210 (2500 µg) and LYR-210 (7500 µg), however the study was an open-label study and the sample size was not powered for statistical comparison between the 2 doses. Thus, the symptom scores of patients administered LYR-210 (2500 µg) and LYR-210 (7500 µg) were grouped together. The mean SNOT-22 total score of patients at baseline was 37.6 ± 19.4 (median 32, range 8−76). As shown in Figure 5, by day 14, LYR-210 reduced the average SNOT-22 score of patients to 26.2 ± 12.9. The reduction in SNOT-22 scores persisted throughout the study, resulting in a mean score of 17.9 ± 13.3 at day 56. LYR-210 achieved statistically significant improvement at each timepoint post-placement compared to baseline.
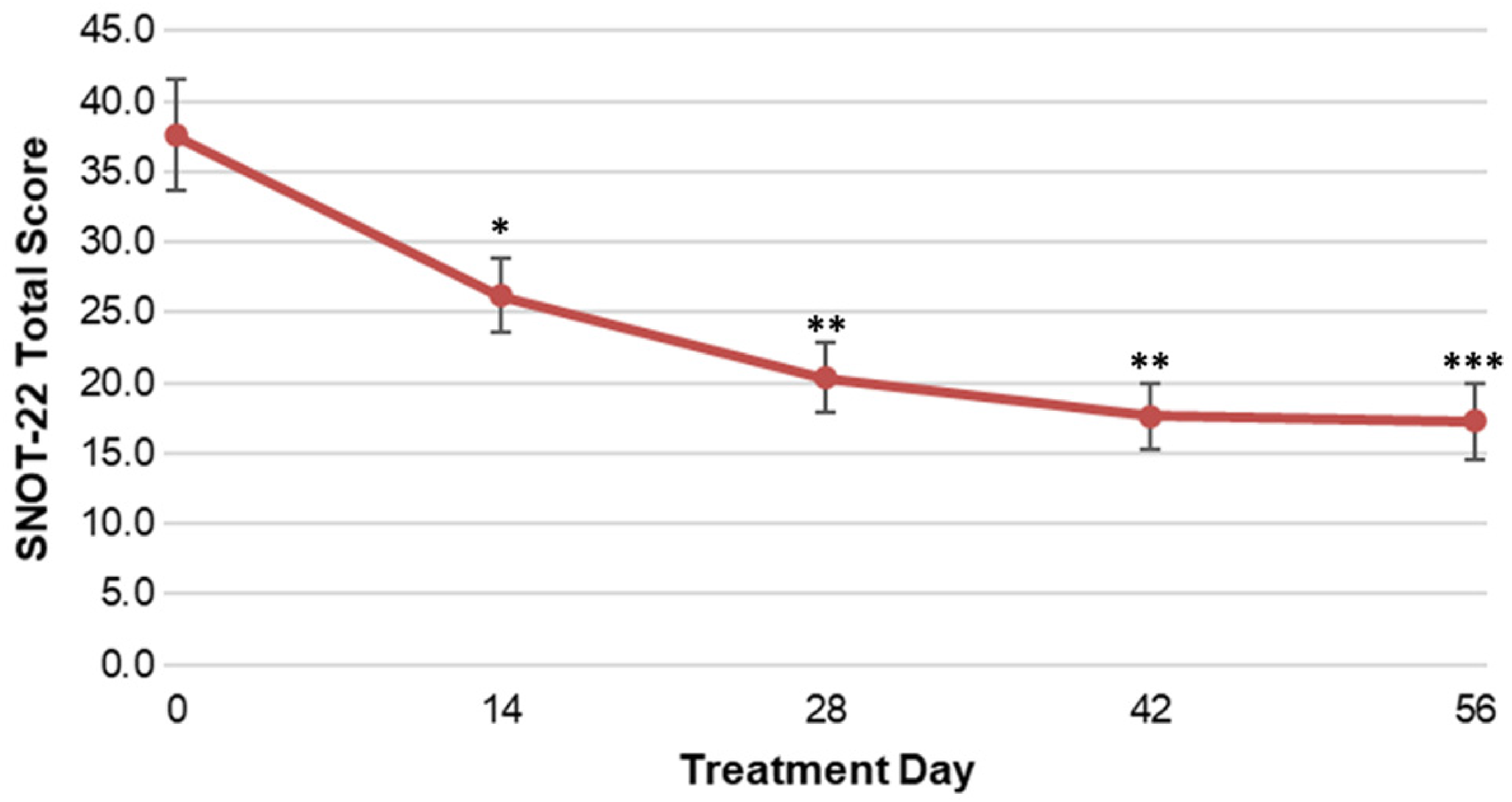
SNOT-22 total scores of patients administered LYR-210 (2500 µg) and LYR-210 (7500 µg) grouped together through treatment day 56. Data are represented as mean and standard error. *p < 0.01; **p < 0.001; ***p < 0.0001 (p-values obtained from a 2-sided paired t-test comparing each timepoint during treatment to baseline).
Discussion
This study shows that a single administration of a sinonasal treatment achieved continuous drug delivery to the middle meatus in patients with CRS for the duration of the 2-month study. Both doses of LYR-210 were safe and well-tolerated throughout the study. There was 100% placement success of LYR-210 matrices into the middle meatuses of all 24 patients. The systemic levels of MF detected in this study were lower than Asmanex® Twisthaler® (400 µg BID), which has a reported Cmax of 114 pg/mL of MF at day 28 (steady state) in patients with asthma, 8 supporting the systemic safety of the LYR-210 (7500 µg) and LYR-210 (2500 µg). Plasma MF concentrations of MF nasal sprays are generally undetectable at 100 to 200 µg/day in reported assays using an LLOQ of 50 pg/mL in adults and children.9–11 Thus, it is challenging to characterize and compare systemic levels of MF nasal sprays to LYR-210. Importantly, neither dose of LYR-210 demonstrated treatment-related serious adverse events, clinically significant increases of intraocular pressure, or significant decreases of morning serum cortisol levels in this study nor in the 24-week treatment period in the LANTERN study. 7 Furthermore, adverse events were comparable between groups in the 24-week post-treatment follow-up period in the LANTERN study after LYR-210 matrices were removed (data on file at Lyra Therapeutics, Inc.).
LYR-210 achieved dose-dependent MF dosing at a steady rate over the 56-day study duration. The plasma MF concentrations measured in this study support the ability of LYR-210 to deliver constant levels of MF to patients for months. The mean plasma MF concentrations at week 4 (day 28) for each dose of LYR-210 in this study (40.7 pg/mL for LYR-210 (7500 µg) and 12.7 pg/mL for LYR-210 (2500 µg)) are consistent with those in the LANTERN study at week 4 (45.4 pg/mL for LYR-210 (7500 µg) and 13.6 pg/mL for LYR-210 (2500 µg)), which further validates the findings in the present study. As LYR-210 is designed to release MF over 24 weeks, it does not exhibit drug dumping, which may be observed with other steroid-eluting implants.12,13
The patient population in this study consisted of a broad range of surgically naïve CRS patients, as there were no inclusion/exclusion criteria related to disease burden or severity. As described by Toma and Hopkins, SNOT-22 total scores can be stratified into mild (8–20), moderate (>20–50), or severe (>50) CRS symptoms. 14 According to these parameters, the enrolled patients in this study, on average, exhibited moderate CRS symptoms at baseline (mean SNOT-22 score, 37.6). This contrasts with the LANTERN randomized, controlled study, in which patients reported, on average, more severe CRS symptoms at baseline (mean SNOT-22 score, 68.2). 7 Despite the small sample size and more moderate baseline CRS symptoms, rapid and clinically relevant improvement in SNOT-22 scores was observed on day 14, which persisted throughout the duration of the study. On day 56, 62.5% of patients reported a SNOT-22 total score of <20, which is considered “mild” CRS disease. 14 Moreover, 37.5% of patients reported a SNOT-22 total score that was less than 8 at the end of this study, which has been shown to be consistent with reported scores by individuals with no sinus disease and is considered “normal.” 15 In the LANTERN study involving more severe patients, LYR-210 (7500 ug) demonstrated significant improvement in SNOT-22 total score compared to control, achieving approximately a 30-point change from baseline at week 8 that continued to improve through the 24-week treatment duration. 7 Thus, LYR-210 may be a promising alternative for patients with CRS of varying disease severities that are seeking a new treatment.
Limitations of this study include the absence of a control or placebo arm. Patient enrollment in this study was not powered to evaluate the efficacy or treatment effect of LYR-210 in CRS, however, LYR-210 achieved statistically significant improvement in SNOT-22 total scores at each timepoint post-placement compared to baseline. LYR-210 is designed to provide consistent dosing of MF for up to 24-weeks of treatment. However, since the primary objective of this study was to understand the PK profile of 2 doses of LYR-210 during the early days post-placement of LYR-210, the study duration was 56 days. While there were only 2 patients in this study that had nasal polyps, it may be representative of the real-world CRS patient population, wherein 70%–90% of patients are non-polyp and 10%–30% have polyps. 16
While intranasal corticosteroid sprays (INCS) are the mainstay treatment for CRS and avoid systemic exposure, they exhibit rapid clearance rates from the nasal cavity that consequently lends to multiple daily administrations that require patient compliance. Penetration of INCS deep into the paranasal sinuses is negligible, as a large proportion is deposited in the anterior nasal cavity or is swallowed. 17 Local steroid-eluting sinonasal implants have only been approved for post-surgical nasal polyp patients and do not address CRS without nasal polyp patients nor those that have not had an FESS, which represent the vast majority of CRS patients. The treatment duration of these implants is limited (up to 90 days), and the steroid eluted over the intended duration to the sinonasal mucosa is inconsistent.12,18
LYR-210 is intended for CRS patients who have failed previous medical management and is designed to be placed into the middle meatus that has not been impacted by FESS using endoscopic visualization and topical anesthesia by an otolaryngologist in a routine in-office visit. LYR-210 could address the shortcomings of INCS and other therapies by providing targeted and persistent dosing of MF to the inflamed sinonasal mucosa for up to 24 weeks in a single administration with minimal systemic exposure and elimination of patient adherence. LYR-210 would improve upon the currently available steroid-eluting implants with a >2x longer drug elution profile and more consistent daily dosing. It could also address a large unmet medical need, particularly in CRS patients without nasal polyps, and represent a potential alternative for patients who have failed medical management and are facing surgery as their next option.
Conclusion
LYR-210 achieved dose-dependent, continuous local MF delivery with minimal systemic exposure at a steady rate for months from a single administration. Moreover, LYR-210 demonstrated clinically relevant and persistent improvement in SNOT-22 scores despite the small sample size and moderate baseline symptoms. Thus, LYR-210 may be a promising new long-acting treatment that addresses a significant unmet need for CRS patients.
Footnotes
Acknowledgments
We gratefully acknowledge the patients who participated in the PK study, the clinical investigators, and the study coordinators for their contributions.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RAO, SS, VK, and JM were investigators in this study. YK is a former employee and CY, AP, LB, and JS are employees of and have stock options at Lyra Therapeutics, Inc., the sponsor of this study and manufacturer of LYR-210 (2500 µg) and LYR-210 (7500 µg) used in this study. RAO is a consultant to Lyra Therapeutics, Aerin Medical, Intersect ENT, and Optinose; and a speaker for GlaxoSmithKline, Sanofi-Regeneron, and Optinose. RMN was the medical monitor of this study and is a member of the Medical Advisory Board for Lyra Therapeutics, GlaxoSmithKline, Sanofi, Regeneron, Ismed, and AstraZeneca.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article. This work was supported by the Lyra Therapeutics Inc.
Ethical Approval
The clinical study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol and patient-informed consent were reviewed and approved by a central ethics committee in accordance with the regulatory requirements.
Informed Consent
All human subjects signed informed consent before participating in the clinical study.
Trial Registration
Not applicable, because this article reports data from a Phase 1 pharmacokinetic clinical trial.
Supplemental Material
Not applicable.