
Abstract
Background
Cystic fibrosis (CF) is a genetic disease that may result in multiple systemic disorders and potentially fatal severe respiratory compromise. However, the advent of CF transmembrane conductance regulator (CFTR) modulators has changed the management of CF for patients with select mutations. Although clinical trials have highlighted increased pulmonary function and decreased exacerbations as a result of these novel therapies, their effect on the sinuses has not been well-described.
Objective
Our objective is to review the CFTR modulators to provide otolaryngologists, physicians who frequently care for patients with CF, a basic understanding of these drugs and their effects on chronic rhinosinusitis (CRS) in patients with CF.
Methods
The clinically approved and available CFTR modulators and specific indications for their use are reviewed. Additionally, a systematic review of these therapies and effects on CRS in CF was performed.
Results
Four Food and Drug Administration approved CFTR modulators are available for patients with CF. Current drugs are approved for gating, residual function, or F508del mutations. Multiple reports describe CFTR modulators’ increase in transepithelial ion transport in nasal epithelial cultures; however, clinical studies regarding effects of these modulators on sinonasal health are limited to 5 studies that present new data of the effects of CFTR modulators in CRS.
Conclusions
CFTR modulators have changed management of CF. Initial studies of these medications demonstrate promising results in CF; however, there is a paucity of literature describing the effect of CFTR modulators on CF-associated CRS, although initial results are encouraging.
Keywords
Introduction
Cystic fibrosis (CF) is a genetic disease that can result in severe respiratory compromise and potentially respiratory failure or death. 1 Over 2000 mutations in the CFTR gene result in absent, partially functional, or nonfunctional CF transmembrane conductance regulator (CFTR) proteins which are critical cell surface chloride channels. CFTR is synthesized intracellularly and transported to the cell surface, where it regulates the transport of salt ions in and out of the cell. 2 In the presence of CFTR mutations, the defective ion transport leads to a dehydration of airway surface liquid volume leading to compromised mucociliary clearance. 2 Systemic CFTR deficiency manifests as pulmonary, endocrine, gastrointestinal, reproductive, and sinonasal disease. 3 The phenotypic disease expression varies widely and is partially driven by specific mutations.4–6
CFTR mutations have traditionally been divided into 6 classes (Figure 1).7,8 Class I mutations introduce premature termination codons, leading to severely reduced or absent CFTR protein. Class II mutations result in protein misfolding, degradation in the endoplasmic reticulum, and decreased protein biogenesis; this leads to a marked reduction in CFTR reaching the cell surface. Class III mutations lead to diminished CFTR protein channel opening probability and are known as “gating mutations.” Class IV mutations cause diminished channel conduction in response to stimulation. Class V mutations impact the abundance of CFTR protein by introducing promoter or splicing abnormalities. Class VI mutations diminish the protein conformational stability, decreasing the quantity of functional CFTR protein present at the plasma membrane.
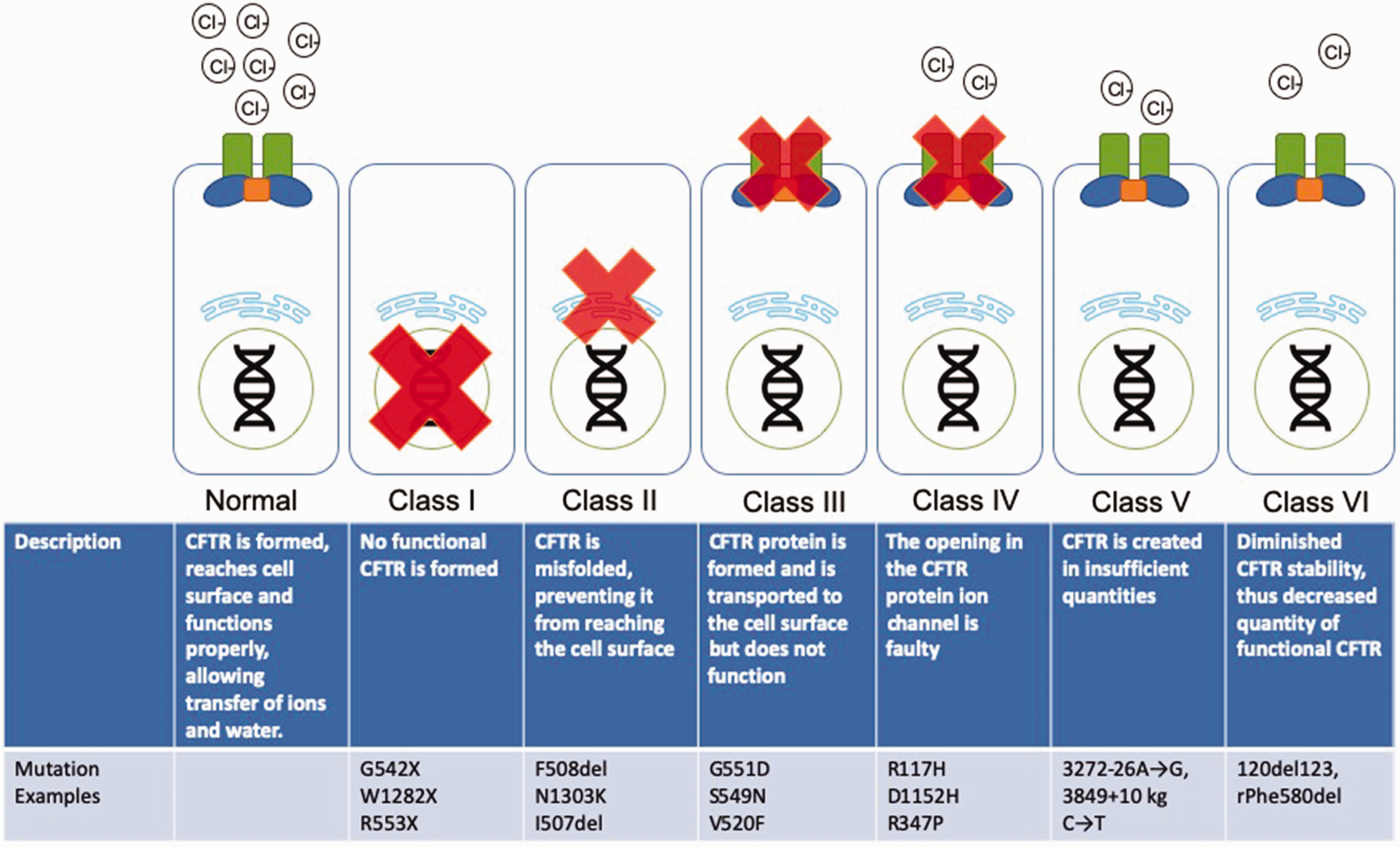
CF mutation classes. Adapted from Cystic Fibrosis Foundation 2017 Patient Registry Annual Data Report.9 CFTR, cystic fibrosis transmembrane conductance regulator.
The advent of CFTR modulators has radically changed the management of CF as patients with select mutations now have options for genetic-directed treatment.10–17 Current modulators are indicated for patients with specific mutations (Tables 1 to 3).
CFTR Mutations FDA Approved for Ivacaftor Monotherapy.

Studies have demonstrated that CFTR modulators result in improved sweat chloride levels, fewer pulmonary exacerbations, greater forced expiratory volume in 1 second (FEV1), as well as greater body mass.11,18,19 However, the pathology of CF is not limited to the pulmonary mucosa, and thus the benefit is not limited to pulmonary manifestations. The CFTR protein is also highly expressed in the reproductive tract, digestive tract, as well as in the sinonasal mucosa, leading to the systemic manifestations of disease.20–22 The sinonasal mucosa is exquisitely sensitive to CFTR dysfunction. Over 60% of adults have symptomatic chronic rhinosinusitis (CRS). 9 The CFTR mutations manifest with characteristic viscous mucous of the sinonasal cavity, impaired mucociliary clearance, as well as chronic inflammation and infection of the sinonasal cavity.22–24 Despite hypoplastic sinuses, radiographic evidence of sinus disease is present in >90% of adults with CF. 25
The Role of CFTR Modulator Therapies
Prior to the advent of these genetic-focused personalized therapies, the medical treatment of CF primarily emphasized augmenting pulmonary toilet with respiratory therapy, improving mucociliary clearance with saline or DNAse, and eradicating infection with topical and systemic antibiotic therapy. 15 In the last 10 years, medical management of CF has dramatically changed for patient with select mutations. As clinicians who take care of patients with CF, it is important we have an understanding of these new therapeutics. Current CFTR modulators can be classified as CFTR potentiators or CFTR correctors. Potentiators affect the CFTR protein at the plasma membrane, improving that ion transport function of mutated, dysfunctional ion channels (Figure 2). Correctors improve CFTR protein processing and trafficking to the plasma membrane (Figure 2).
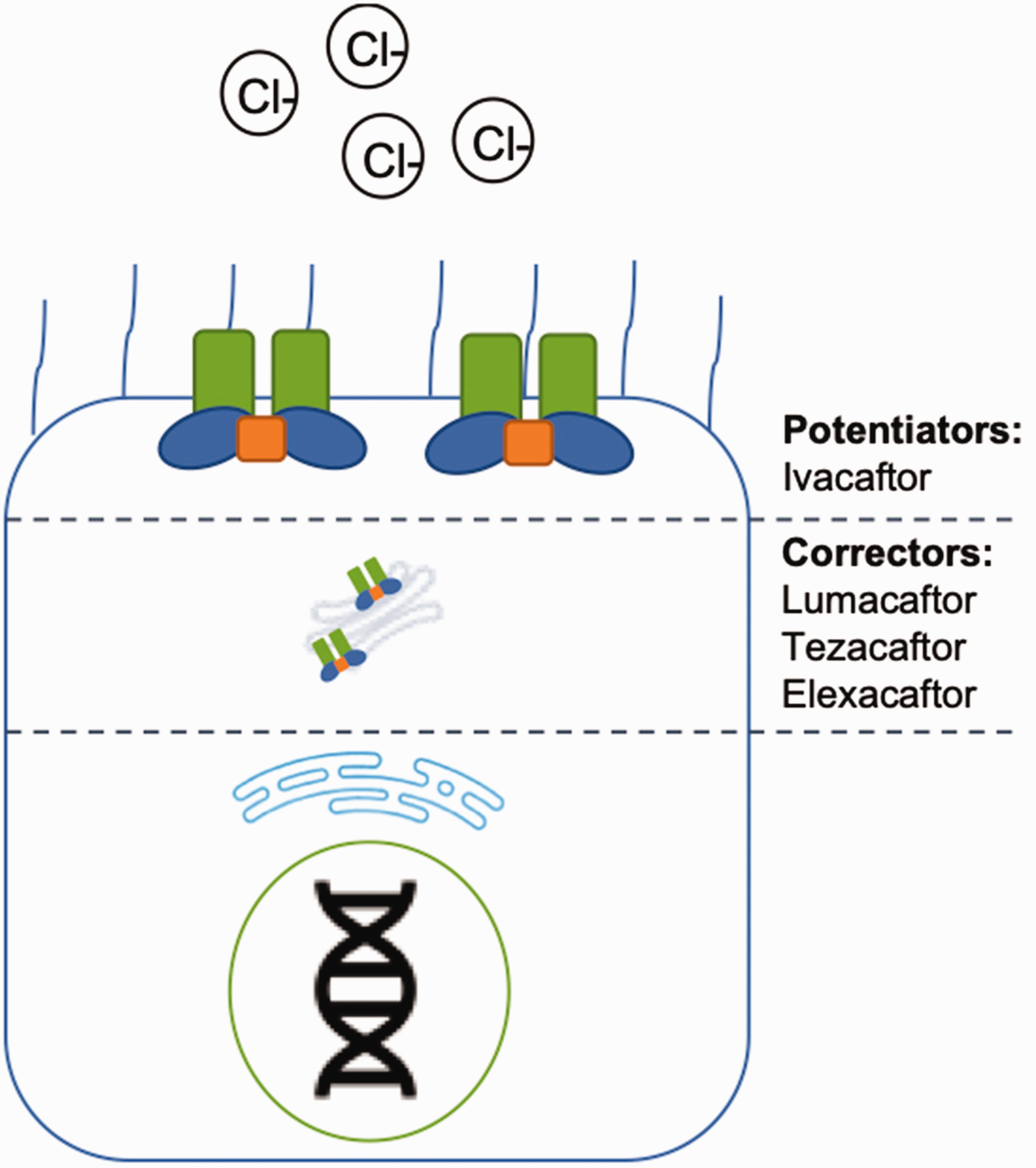
Effect location of CFTR modulators.
Potentiators
CFTR mutations can lead to ineffective ion channels at the plasma membrane that do not allow for adequate Cl− flux. In this circumstance, CFTR potentiators improve channel function by increasing Cl− transport. The only current potentiator on the market is ivacaftor (marketed under the trade name Kalydeco).
Ivacaftor
Ivacaftor increases chloride ion flux in epithelial cells expressing G551D gating mutation, potentiating dysfunctional CFTRs. 26 Further studies have demonstrated some quality-of-life benefit for other non-G551D class III gating mutations as well as some mutations with class III/IV cellular phenotypes, often termed “residual function mutations”; however, improvement to pulmonary function in this cohort is equivocal. Ivacaftor has been shown to potentiate 38 different CF-causing mutations.27,28 Ivacaftor monotherapy is approved in patients as early as 6 months of age. During the initial clinical trial, ivacaftor was shown to have a good safety profile. 18 Adverse events that occurred more frequently in the ivacaftor group were headache, upper respiratory tract infection, nasal congestion, rash, and dizziness—none of which were considered to be serious or led to discontinuation of the study drug. 18 Percentage predicted FEV1 (ppFEV1) was noted to improve by 10.4% in the ivacaftor group while ppFEV1 decreased by 0.2% in the placebo group at 24 weeks after treatment initiation. 18 At 48 weeks, the mean difference of ppFEV1 was 10.4%. 2 Fewer pulmonary exacerbations were seen in patients taking ivacaftor, which led to fewer hospitalizations as well as number of days hospitalized. 18 Subjective symptoms, weight gain, sweat chloride levels have been shown to improve as well. 2 It is currently recommended in patients ages 6 months to 6 years old with specific gating and residual function mutations (Table 1).
Correctors
Although ivacaftor is effective in certain CFTR mutations, it is effective only if the CFTR protein is expressed at the plasma membrane. If the CFTR is not expressed at the cell surface, ivacaftor will have no benefit. Class II mutations result in impaired CFTR transport to the plasma membrane. Corrector therapeutics help increase the amount of CFTR protein that is transported and inserted into the plasma membrane. Currently, 3 Food and Drug Administration (FDA) approved correctors are on the market, lumacaftor, tezacaftor, and elexacaftor. These drugs facilitate the improved folding and translocation of the CFTR protein to the plasma membrane; however, the CFTR is still defective and does not have normal ion transport properties. In combination with a CFTR potentiator such as ivacaftor, improved chloride movement is facilitated.
Lumacaftor
Lumacaftor is a CFTR corrector that has been shown to improve pulmonary function, sweat chloride levels, and body mass index. 29 It is used as a combination therapy with ivacaftor, marketed under the trade name Orkambi, and has been tested and approved only in homozygote F508del mutations for ages 2 and up. 19 Pulmonary function has been shown to improve, with the initial trial demonstrating ppFEV1 improvement in the treatment group of ∼5% at 24 weeks after treatment initiation. This dual therapy has been shown to increase weight gain and decrease sweat chloride levels. Furthermore, quality-of-life scores measured with the Cystic Fibrosis Questionnaire are improved, as well as dose-dependent decreased pulmonary exacerbations compared to placebo (Hazard Risk [HR] 0.61–0.70). 30 However, it has an increased side-effect profile, with the initial trial having adverse events that led to discontinuation of the study regimen (4.2% of patients) in patients including elevation of creatine kinase and transaminase levels, hemoptysis, bronchospasm, dyspnea, pulmonary exacerbation, and rash. There are associated longer term increases in blood pressure as well. 30 Because of the increased side-effect profile compared to tezacaftor-ivacaftor, it is primarily clinically used in patient age 2 to 6 with F508del mutations, the age-group tezacaftor-ivacaftor has not been approved in. 31
Tezacaftor
Tezacaftor is a CFTR corrector that improves plasma membrane expression for mutations with class II cellular phenotypes (ie, F508del).11,32 Tezacaftor is utilized as combination therapy with ivacaftor marked under the tradename Symdeko. The initial study demonstrated ppFEV1 improvements in tezacaftor–ivacaftor trial group of 6.8% compared to placebo, whereas ivacaftor monotherapy improved ppFEV1 by 4.7% at 24 weeks after initiation of therapy. Other benefits seen are improvements in quality-of-life scores measured using the Cystic Fibrosis Questionnaire, as well as decreased pulmonary exacerbations compared to placebo (HR 0.64). 30 The medication was well tolerated with no serious side effects except for slightly increased creatine phosphokinase level (3.7% vs. 3.1% placebo). Tezacaftor and ivacaftor act synergistically in heterozygotes of residual function mutations and F508del homozygotes.11,33 Currently, tezacaftor–ivacaftor dual therapy is recommended in all patients 6 years and older with the approved gating, residual function, or homozygous F508del mutations (Table 2).
CFTR Mutations FDA Approved for Tezacaftor–Ivacaftor Use.
CFTR Mutations FDA Approved for Elexacaftor–Tezacaftor–Ivacaftor Triple Therapy.

Elexacaftor
Elexacaftor is a next-generation CFTR corrector that is utilized with tezacaftor and ivacaftor as a new “triple therapy” for patients heterozygous or homozygous for F508del mutations, marketed under trade name Trikafta.32,34–37 Triple therapy for F508del heterozygotes increased ppFEV1 by 14.3% at 24 weeks. In a superiority trial, the addition of elexacaftor for F508del homozygote patients already receiving tezacaftor–ivacaftor increased ppFEV1 by 10% after 4 weeks of treatment, compared to those just receiving tezacaftor–ivacaftor. In vivo studies of patients with F508del/minimal function heterozygotes and F508del homozygotes also showed improvement in quality-of-life scores and sweat chloride concentration compared tezacaftor–ivacaftor. This triple therapy was FDA approved October 2019, and the additional benefit seen by F508del homozygotes and the extension to F508del/minimal function heterozygotes is projected to impact disease progression in approximately 90% of CF patients (Table 3). 9
CFTR Modulators for CRS
We performed a search for English literature on National Center for Biotechnology Information (NCBI) using the following strategy: “(Ivacaftor[tiab] OR Kalydeco[tiab] OR Lumacaftor[tiab] OR Tezacaftor[tiab] OR Symdeko[tiab] OR Trikafta[tiab] OR elexacaftor[tiab]) AND (sinus[tiab] OR sinusitis[tiab] OR rhinosinusitis[tiab] OR sinonasal[tiab]).” This resulted in 19 hits. Abstracts were reviewed by A. J. K. and S. E. L. After excluding 1 nonrelevant study, 7 preclinical studies (ie, animal models or in vitro studies) and 6 reviews, we identified 5 studies present new data of the effects of CFTR modulators in rhinosinusitis (Table 4). Of these 5 included studies, 3 were case reports. References from included studies and review articles were examined for any additional studies and none were identified.
Review of Studies That Present New Clinical Data of CFTR Modulator Effect in Rhinosinusitis.
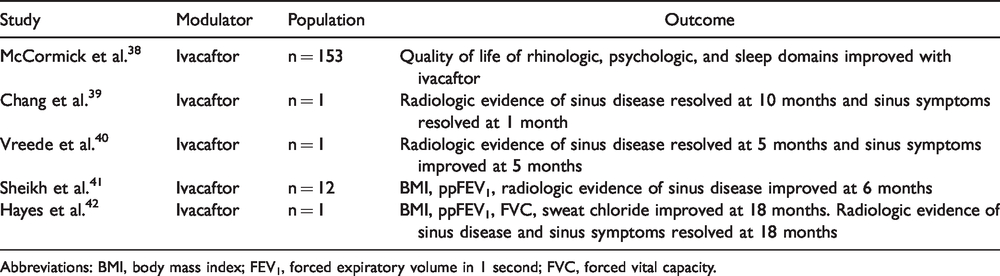
Abbreviations: BMI, body mass index; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity.
CRS is nearly universal in patients with CF and contributes to the morbidity of the disease. As in the lower airways, the CFTR protein is critical in the sinonasal mucosa for normal mucociliary clearance. Decreased mucociliary clearance in the sinonasal tract results in chronic sinus infections, hypoplastic sinuses, and potential for bacterial seeding of the lungs. Because of this, CF patient frequently has an otolaryngologist as part of the multidisciplinary care team among pulmonologists, endocrinologists, gastroenterologists, respiratory therapists, and pharmacists.
The effect of CFTR potentiators and correctors on CRS is poorly understood. One case describes reversal of CRS after initiation of ivacaftor. 39 Sinus symptoms resolved by 1 month of treatment initiation and radiologic improvement was noted at 10 months. 39 In the case report by Chang et al., a biopsy of nasal epithelium showed improvement in potentiation of transepithelial current with ivacaftor, similarly to that of the bronchial epithelia described during drug development. 39
A recent study utilized the Sino-Nasal Outcome Test (SNOT-20) to evaluate CRS symptoms after initiation of ivacaftor. 38 The SNOT-20 is a validated, disease-specific patient-reported outcome measure for rhinosinusitis that include rhinologic, psychologic, sleep, and ear/facial quality-of-life subdomains. 43 SNOT-20 scores were minimally but statistically significantly improved at 1, 3, and 6 months after therapy initiation, with notable improvement in rhinologic, psychologic, and sleep domains. Unfortunately, there were no data regarding changes in nasal endoscopy findings or to microbiome.
CFTR Modulator Challenges
Benefits achieved with CFTR modulator therapy are not sustained after treatment is stopped. For example, improvement in ventilation seen on magnetic resonance imaging after initiating ivacaftor therapy are lost if treatment with ivacaftor is discontinued. 44 In fact, clinical deterioration and symptom exacerbation can be seen after ivacaftor treatment cessation. This phenomenon is termed “ivacaftor withdrawal syndrome.” 45
There are several reasons why eligible patients may not be taking a modulator. Available modulators are metabolized through cytochrome P-450 enzymes. Ivacaftor and tezacaftor are substrates of CYP3A4. Use of CYP3A induces such as rifampin, phenobarbital, carbamazepine, phenytoin, and St. John’s wort will significantly decrease tezacaftor and ivacaftor levels; use of modulators in this setting is not recommended. Concomitant use of CYP3A4 inhibitors will lead to increased concentrations of ivacaftor and tezacaftor. Modulator dosing regimen can be adjusted if moderate or strong CYP3A4 inhibitors, such as triazole antifungals and macrolides (excluding azithromycin), are required. Ivacaftor is a weak inhibitor of CYP3A4 and P-glycoprotein (P-gp), which can increase drug levels of other enzyme substrates such as digoxin, cyclosporine, or tacrolimus. Since the latter 2 drugs are often used in solid organ transplant and many of these patients are also prescribed triazoles, CF patients who have undergone solid organ transplant are typically not prescribed CFTR modulators. The British National Formulary has listed organ transplant as a contraindication to modulation therapy, although this is not a listed contraindication on the product summary. 46
Additionally, there is limited efficacy of available monotherapies for some mutant alleles designated as class I, class II, or class III/IV. This could be explained by pleiotropic molecular defects caused by a single mutation with effects overlapping several classes. Furthermore, not all class III (gating mutations) respond the same to gating potentiators. 47
Cost of CFTR modulators is also a consideration, particularly for Medicare patients. Approximately 2700 adult CF patients in the Cystic Fibrosis Foundation Patient Registry have Medicare coverage. A portion of these patients will have Part D prescription drug coverage. While Part D will cover CF-specific medications, these specialty medications are Tier 5 and require patient cost sharing of 25% to 33% of total drug cost. The average annual cost without insurance is $306 600 for ivacaftor, $272 000 for ivacaftor/lumacaftor, $292 000 for ivacaftor/tezacaftor, and $311 000 for ivacaftor/tezacaftor/elexacaftor. Due to the antikickback statute, patients with government insurance (Medicare, Medicaid, Tricare) are not eligible for pharmacy assistance programs to lower the copay cost. Some Medicare Part D patients are simply unable to afford CFTR modulator therapy. 48 Additionally, in other countries, single-payer health systems are not covering CFTR modulators because of the high cost. There is limited data currently available on what percentage of CFTR modulator candidate are actually receiving treatment, but in a commercial insurance database study of 15 million members, only 54% of mutation-eligible patients were receiving modulators. 49 However, in cost-effectiveness analyses, quality-adjusted life year (QALY) gained with CFTR modulator use ranges from $840 600 to $974 300, far exceeding the commonly accepted thresholds of $100 000 to $150 000 per QALY. 50
Currently CFTR modulators are only approved for CF lung disease. Active research is being conducted utilizing ivacaftor coated stents to help reduce biofilm formation in preclinical, non-CF, animal models.51,52 In the future, CFTR modulators may have indications for CF sinus disease or for CRS in general.
Conclusions
CFTR modulators are a revolutionary CF treatment that have allowed for tailored, targeted therapies for specific gene mutations. These have improved objective and subjective measures in CF and are becoming increasingly available and accessible to patients. With the recent FDA approval of the novel triple therapy, treatment for CF patients has improved and is now accessible to many more patients. From a rhinologic standpoint, there are limited studies assessing the effect of these medications for CRS in CF patients. Further studies of the effect of CFTR modulators in CRS are warranted as development of these therapies continues.
Footnotes
Authors’ Note
This study was presented at American Rhinologic Society Annual Meeting, September 2019, New Orleans, Louisiana.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.