
Abstract
Late-onset Pompe disease is a rare autosomal recessive lysosomal storage disorder caused by acid α-glucosidase deficiency, resulting in progressive skeletal muscle weakness and respiratory failure. We present the case of a 43-year-old African American woman who was admitted to the intensive care unit with acute-on-chronic hypoxemic and hypercarbic respiratory failure, alteration of consciousness, and progressive weakness. Her recent medical history included respiratory distress and aspiration pneumonia, which had not fully resolved despite supplemental oxygen therapy. On admission, initial evaluations including imaging and laboratory tests did not reveal a diagnosis. Muscle biopsy showed a vacuolar myopathy with excess glycogen suggestive of glycogen storage disease. Enzyme testing was obtained through the dried blood spot testing and was low. Molecular genetic testing identified two pathogenic variants in the GAA gene, confirming the diagnosis of late-onset Pompe disease. This diagnosis enabled the prompt initiation of enzyme replacement therapy (ERT) with alglucosidase alpha. The early initiation of ERT in this patient was pivotal in managing her condition, given the progressive nature of late-onset Pompe disease and the potential for improved outcome when treatment is started early. This case highlights the importance of considering late-onset Pompe disease in adults presenting with unexplained progressive respiratory and neuromuscular symptoms. It also demonstrates the critical role of biochemical and molecular genetic testing, as early intervention can significantly impact treatment outcomes and quality of life.
Keywords
Background
Pompe disease (OMIM #232300), also known as glycogen storage disease type II (GSD II) or acid α-glucosidase deficiency, is a rare autosomal recessive lysosomal storage disease (LSD). This enzyme defect leads to the accumulation of glycogen within the lysosomes, 1 primarily in muscle tissue. Pompe disease is classified into two forms: infantile-onset and late-onset. 2 The infantile-onset form is more severe, typically presenting within the first few months of life with rapidly progressive hypotonia, cardiomyopathy, feeding difficulties, and respiratory failure. Without treatment, most patients die from cardiorespiratory failure or aspiration pneumonia before the age of 1 year. 3 In contrast, the late-onset form can present at varying ages, from the first to seventh decades of life. It is characterized by progressive skeletal muscle weakness and respiratory insufficiency, with a slower progression compared to the infantile-onset form. Unlike the infantile-onset form, cardiac involvement is rare in the late-onset form.
Case Presentation
A 43-year-old black woman was admitted to the intensive care unit (ICU) with acute-on-chronic hypoxemic and hypercarbic respiratory failure due to progressive neuromuscular weakness.
Nine months prior to this admission, she had respiratory distress and aspiration pneumonia, requiring a 2-week hospitalization. Subsequently she experienced persistent shortness of breath and was started on supplemental home oxygen therapy at 2 liters per minute as needed. The day prior to admission she needed this oxygen and then developed garbled speech and confusion. By the following morning, her condition worsened; she became increasingly somnolent and confused, prompting her presentation to the emergency department.
In the emergency department, she was alert but appeared ill. She was hypothermic to 94.5 F (34.7 C), and had a heart rate of 102 beats per minute. Her respiratory rate and oxygen saturation were normal on 4 liters per minute of oxygen via nasal cannula. Respiratory examination revealed bilateral chest rise with clear lung fields bilaterally, and no abnormal breath sounds. She was nonverbal and unable to follow commands. She had anti-gravity strength in both upper extremities and minimal movement in both lower extremities. Venous blood gas indicative of severe respiratory acidosis with a pH of less than 7.0, pCO2 greater than 125, and incalculable bicarbonate levels. Creatinine phosphokinase (CPK) was normal. A CT angiogram of the chest revealed findings consistent with aspiration in the left lower lobe but no evidence of pulmonary embolism. She was intubated for hypoxemic and hypercarbic respiratory failure.
On review of her medical history, she was born at term following an uncomplicated pregnancy and delivery. She achieved early developmental millstones appropriately. Her past medical history was notable for hypertension. She was not taking any prescribed medications. A review of her family history revealed no known instances of respiratory insufficiency, muscle weakness, or genetic conditions.
Electromyography (EMG) was conducted on HD11 and suggested potentially inflammatory myopathy and a muscle biopsy of right biceps was obtained on the same day. Following the biopsy, she was treated with intravenous methylprednisolone at 1 gram daily and intravenous immunoglobulin at 2 gram/kilogram for 5 days, but showed minimal improvement. The preliminary muscle biopsy report described a myopathic process with fibers showing centrally reduced NADH staining and basophilic aggregates, without inflammatory features, raising for the possibility of a myofibrillar myopathy. Given this tentative diagnosis, steroid therapy was discontinued on HD19. She subsequently developed respiratory failure and was re-intubated on HD26, with a tracheostomy performed on HD33.
Muscle tissue was sent to an external institution for expert and ultrastructural analysis, and the final report was returned on HD43. The biopsy report described a chronic vacuolar myopathy with excess glycogen (see Figure 1). The findings were suggestive of glycogen storage disease (GSD) or lysosomal storage disorder (LSD) with excess glycogen. Due to concerns of a metabolic disorder, genetics was consulted, and dried blood spot testing along with GSD and LSD gene panel testing was ordered. Her dried blood testing result showed that acid α-glucosidase enzyme activity was 0.488%. Five days later, the gene panels results were returned, revealing two pathogenic variants, c.1979G>A (p.Arg660His) and c.853 C>T (p.Pro285Ser), in the GAA gene (NM_000152.3). These results confirmed a diagnosis of late-onset Pompe disease, prompting the plan for enzyme replacement therapy (ERT) with avalglucosidase alfa-ngpt in an outpatient setting. (A). H&E-stained sections showed frequent fibers with central rimmed vacuoles (arrows), as well as occasional basophilic fibers taken up entirely by vacuolization (arrowhead). (B). Acid phosphatase showed extensive vacuolar positivity within the centers of many myofibers. (C) Granular PAS positivity was present throughout all myofibers. (D). Electron microscopy showed membrane-bound vacuoles containing glycogen (arrows). Panels (A)-(C): 200X magnification, panel (D): 15,000X.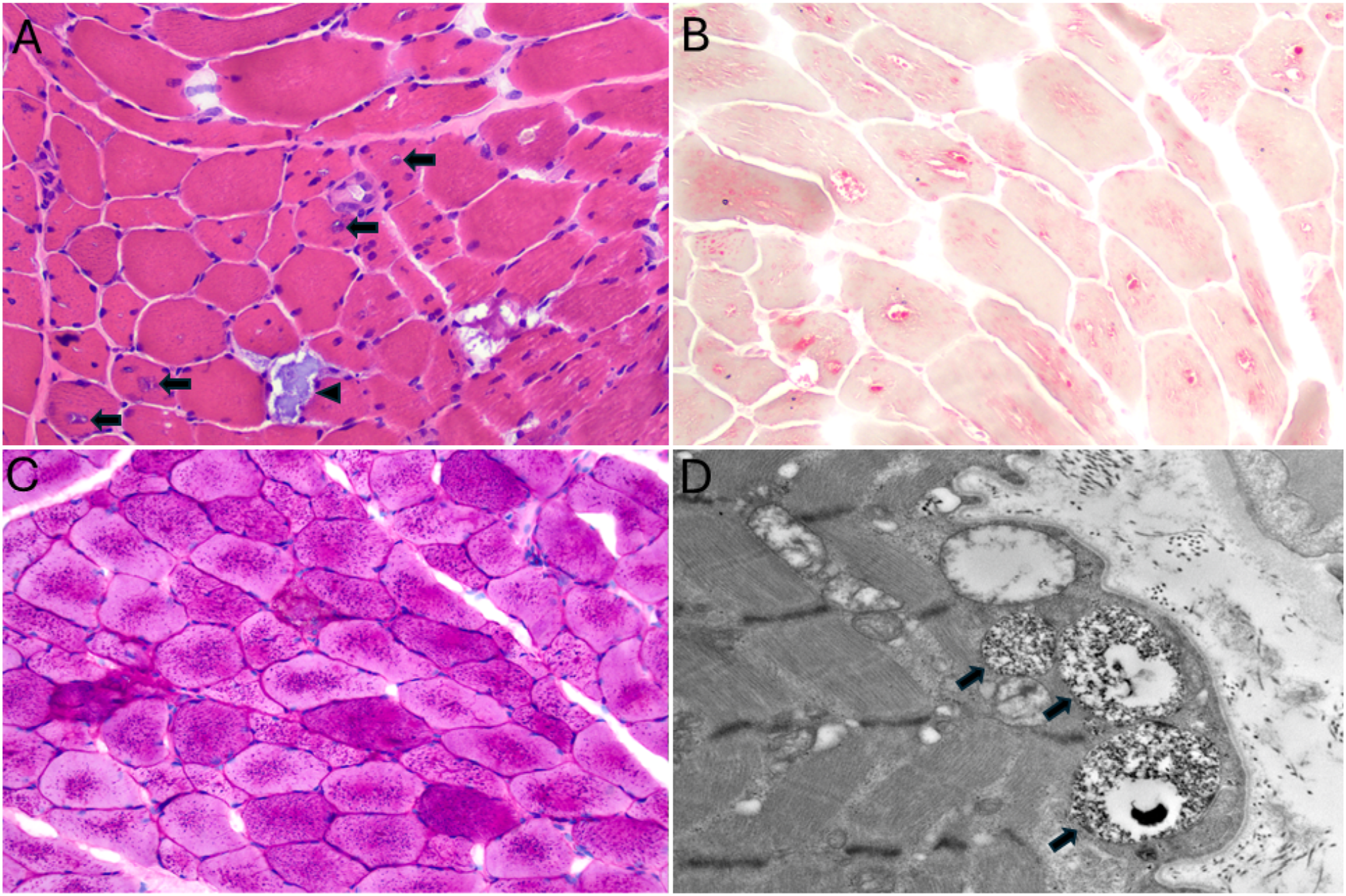
Discussion
Here we describe a 43-year-old black woman with late-onset Pompe disease leading to acute-on-chronic hypoxemic and hypercarbic respiratory failure. Her diagnosis was made through enzyme testing and molecular genetics testing, leading to the initiation of ERT.
Index of Suspicion and Differential Diagnosis
The diagnosis of late-onset Pompe disease often presents challenges because of its rarity, insidious progression, and nonspecific phenotypic features.4,5 The incidence of Pompe disease is approximately 1 in 40,000 in the United States and varies depending on ethnicity and geographic region. 6 Specifically, it is reported to occur more frequently in the African American population, at a rate of 1 in 14,000, compared to 1:100,000 in individuals of European descent.6,7 This higher incidence may lead to increased consideration of the diagnosis in this population, like our patient.
The majority of patients with late-onset Pompe disease present with progressive proximal muscle weakness (95%). 8 In addition to the limb-girdle muscles, diaphragm involvement is common, leading to acute respiratory failure. The diagnosis of late-onset Pompe disease often presents challenges because of its rarity, insidious progression, and nonspecific phenotypic features4,5. Therefore, the presence of unexplained respiratory insufficiency in patients with progressive proximal muscle weakness serves as a major index of suspicion for late-onset Pompe disease. It is important to distinguish it from other neuromuscular disorders–such as myasthenia gravis, Lambert-Eaton myasthenic syndrome, and botulism–which also present with weakness and subsequent respiratory insufficiency. Becker muscular dystrophy is an X-linked recessive disorder caused by variants in DMD gene, characterized by progressive proximal muscle weakness and respiratory impairment. Danon disease (OMIM #300257) is a rare X-linked disorder caused by a deficiency of lysosomal-associated membrane protein 2, encoded by the LAMP2 gene. 9 This condition typically presents after the first decade with muscle weakness, cardiomyopathy, and intellectual disability.
Diagnosis
To confirm a diagnosis of Pompe disease, it is crucial to either demonstrate a deficiency in acid α-glucosidase enzyme activity or identify biallelic pathogenic variants in the GAA through molecular genetics testing. Enzyme assays can detect acid α-glucosidase deficiency, typically using dried blood spot testing preferred as the first-line screening method over muscle tissue enzyme. In 2015, Pompe disease was added to the RUSP for NBS in the United States. 10 However, the screening is designed to detect the infantile-onset form, not the late-onset form. Therefore, it remains unclear to what extent NBS impacts the detection of the late-onset form, although NBS has significantly contributed to the early detection of the infantile-onset form.
Muscle biopsy is another valuable diagnostic tool, as vacuolar myopathy with glycogen accumulation in the lysosomes of muscle cells may be observed using periodic acid-Schiff (PAS) staining. While vacuolar myopathy is present in approximately two-thirds of adults with this condition, the biopsy in the remaining one-third of cases may appear normal or nonspecific due to partial enzyme activity, which can potentially result in misdiagnosis. 11 Due to the possibility of extended muscle biopsy turnaround times in the setting of in-depth analyses including electron microscopy, molecular testing should be considered as soon as clinical suspicion for Pompe disease arises. Given the turnaround time, non-specificity, and invasiveness of muscle biopsies, DNA sequencing-based gene panel testing can serve as a viable non-invasive alternative for diagnosing metabolic myopathies, including Pompe disease.
In addition, an increased T1 signal in the tongue on brain MRI–referred to as the ‘bright tongue sign’ – has been reported in patients with late-onset Pompe disease. 12 Since this finding is not typically seen in other neuromuscular diseases, it might serve as a useful diagnostic clue in identifying late-onset Pompe disease.
Treatment
ERT with infusion of algucosidase, a recombinant form of acid α-glucosidase, is a key treatment for Pompe disease. 13 Lumizyme® (algucosidase α) is currently used for the Late-onset Pompe disease and was approved by the Food and Drug Administration (FDA) in 2010. While experience with ERT in the late-onset form is more limited compared to the infantile-onset form, there is sufficient evidence demonstrating the short-term benefits of ERT for the respiratory function, muscular strength and function, and survival in late-onset Pompe disease.5,14,15 Although long-term outcomes are not yet well-established, ERT has been showed to positively affect the quality of life in adult patients with the late-onset form. 16 Subsequently, as a next-generation ERT, Nexviazyme® (avalglucosidase alfa-ngpt) received FDA approval in 2021 based on the efficacy and safety outcomes of the Phase 3 COMET trial (NCT02782741), in which avalglucosidase alfa-ngpt was compared with algucosidase alpha. 17 This study demonstrated that avalglucosidase alfa-ngpt provided meaningful clinical benefits over algucosidase alpha, particularly in mobility, daily functioning, respiratory function, pain, fatigue, and general health-related quality of life.17,18 To establish this therapy as the standard ERT in late-onset Pompe disease, its long-term efficacy and safety are currently being further investigated.
Lessons for the Clinician
• It is important to consider late-onset Pompe disease in adults presenting with unexplained respiratory insufficiency and progressive muscle weakness. • The more insidious nature of the progression in the late-onset form, compared to the infantile-onset form, may contribute to the delay in diagnosis. • In one-third of late-onset Pompe disease cases, muscle biopsy may appear normal or non-specific, necessitating confirmation through enzyme activity or molecular genetic testing. • Biochemical and molecular genetic testing is crucial for diagnosing late-onset Pompe disease, as initiating enzyme replacement therapy early is key to improving outcomes and quality of life.
Footnotes
Acknowledgements
We thank the Department of Pathology, Division of Neuropathology at Duke University Medical Center for their assistance with the diagnosis.
Author Contributions
YF drafted the article. NSA, ARG, RJT, BCM, KAJ, and TAC helped to draft the article. BCM and KAJ helped to create a figure. All authors read, corrected and approved the final article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Statement
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.