
Abstract
During development and regeneration, bone is formed by endochondral ossification (EO) through the remodeling of a cartilage template. This complex process involves multiple cell types and interactions that cannot currently be modeled in vitro. This study aimed to develop a novel tissue-engineered human in vitro model of certain aspects of the early stages of EO by integrating cartilage which undergoes mineralization, self-assembled vascular networks, and osteoclasts into a single system. We first studied the dynamics of osteoclastogenesis and vascularization in an in vivo model of stromal cell-mediated EO, to inform our in vitro system. Next, we aimed to develop a fully human cell-based three-dimensional model of EO by combining pediatric bone marrow stromal cells differentiating into chondrocytes, osteoclasts derived from human CD14+ monocytes, and human umbilical vein endothelial cells and adipose-derived stromal cells as vessel-forming cells. We investigated how mineralizing cartilage affects osteoclast and vessel formation in vitro through separate cartilage-osteoclasts and cartilage-vessels cocultures. Finally, we combined these elements and established a complex in vitro model that supports the functionality of all these cell types and recapitulates chondrogenesis, cartilage mineralization, vessel formation and osteoclastogenesis. This integrated approach reaches unprecedented complexity and will enable new tissue engineering strategies to model skeletal diseases or cancer metastasis to the bone.
Impact Statement
Current in vitro models to study bone formation via endochondral ossification are relatively simple and generally do not include the multiple cell types involved in the process. In this study we added a new level of complexity to existing in vitro models by combining several cell types crucial for bone formation into a single system. In particular, mineralizing cartilage, human osteoclasts (cells that remodel cartilage into bone) and blood vessels were all incorporated into a single platform. This comprehensive model raises new opportunities for the in vitro study of bone formation and disease modeling applications, including bone metastasis.
Keywords
Introduction
Endochondral ossification (EO) is the natural process of bone formation which occurs in the majority of the bones in the body. 1 Several groups including ours have succeeded in recapitulating endochondral bone formation in vivo through a tissue engineering-based approach, allowing the investigation of cellular and molecular mechanisms or applications in bone regenerative medicine.2–4 In these studies, a template of hypertrophic cartilage is generally formed from human mesenchymal stromal cells (hMSCs) by inducing chondrogenic differentiation in vitro. Upon implantation in animals, the tissue-engineered cartilage undergoes EO and remodels into mature bone which includes all the elements of a functional bone niche, including blood vessels, clastic cells, bone marrow, and osteoblasts.5–13 Recapitulating EO in vitro would be highly desirable to establish a highly controlled and completely humanized system to recapitulate bone formation, while addressing ethical concerns related to using animals. Such a complex three-dimensional (3D) platform would allow for the in vitro investigation of diseases that affect bone formation or remodeling, including cancer metastasis to the bone.
However, the generation of useful in vitro bone models requires the incorporation of many of the aforementioned cellular elements. Many existing in vitro tissue-engineered bone models aim to recreate the mature bone microenvironment by the direct combination of osteoblasts, osteoclasts, and other cell types including osteocytes or endothelial cells. This generally mimics the process of intramembranous ossification. Here we aimed to recreate the stepwise bone formation process via endochondral ossification, involving a cartilage template that occurs during long bone development or repair. Current existing EO models are mainly limited to the recapitulation of separate processes, such as the mineralization of hypertrophic cartilage 14 or the combination of mineralized cartilage only with naïve vessels 15 or osteoclasts. 16 One major challenge is still the difficulty in maintaining the functionality of several cell types and recreating their intricate interplay in a single complex in vitro system.
During EO, hypertrophic chondrocytes produce a mineralized cartilage matrix and secrete factors that promote vascular invasion and the recruitment of remodeling osteoclasts. 17 Osteoclasts are important for bone vascularization and protect blood vessels against senescence. 18 The resorption of the cartilage template and the remodeling of immature bone structures are essential for the formation of a mature bone matrix which can host vasculature and marrow niche. While resorption of unmineralized cartilage is mainly associated with septoclasts19,20 and endothelial cells,20,21 chondroclasts/osteoclasts are thought to be responsible for the resorption of mineralized cartilage and bone. 22 Many studies have shown that when the function of osteoclasts is undermined via knockout of osteoclast-specific genes such as RANK or MMP9,23–26 the EO process in mice is disrupted, impeding normal bone development and bone remodeling. Recapitulating functional osteoclast activity is therefore essential for the development of a robust in vitro EO bone model. To date, there have been in vitro models demonstrating osteoclast–vasculature interactions18,27–29 but none yet in the context of modeling EO in vitro combined with a 3D vasculature and a self-assembled cartilage template.
During EO, vessel network formation is essential to transport nutrients as well as immune and progenitor cells, such as hematopoietic stem cells and osteoprogenitors, to the ossification site. Monocytes migrate through the vessel network and, once they reach the mineralization front, they differentiate into mature clastic cells. Besides providing access to the bone, blood vessels directly contribute to the regulation of signaling pathways involved in osteochondroprogenitor differentiation and cartilage remodeling during EO.30–32 In vitro studies with endothelial cells have also shown their stimulatory effect on MSC osteogenic differentiation.33,34 This highlights the importance of incorporating vascular components into in vitro bone models. Recent efforts have focused on diverse solutions to combine bone cells and vasculature in vitro, such as 3D-printed scaffolds with hollow channels, 35 self-assembly of vessel-forming cells 36 and microfluidic devices. 37 However, these studies mainly focus on intramembranous ossification and the interaction between remodeling cartilage and vasculature in vitro is still largely unexplored. We recently performed indirect coculture studies showing how in vitro mineralization affects the proangiogenic properties of tissue-engineered cartilage, providing some initial directions for combining these processes in vitro. 38 However, replicating the highly dynamic nature of cartilage mineralization and remodeling, which simultaneously involves vascularization and osteoclast activity, remains an arduous challenge.
We reasoned that the development of a comprehensive in vitro model of EO should start with the incorporation of mineralized cartilage, osteoclasts, and blood vessels in a single system, due to their intimate crosstalk and codependence. As a first step, the dynamics of osteoclastogenesis and vascularization were studied in an in vivo model of tissue-engineered endochondral bone. This provided temporal information for the recapitulation of these processes in vitro. Next, we established direct coculture systems of cartilage undergoing mineralization and osteoclasts or vascular networks. Finally, we developed a single complex in vitro model of EO that recapitulates certain aspects of this process, namely chondrogenesis, cartilage mineralization, osteoclastogenesis, and vascular network formation.
Materials and Methods
Cell isolation and culture
Human pediatric bone marrow stromal cells (pMSCs) (N = 5 donors in total) were isolated from leftover iliac crest bone chips of patients (ages 9–12) undergoing alveolar reconstruction surgery and expanded, as previously described. 39 Passage 3–4 cells at a confluency of 85–90% were used for the experiments. Material collection was performed with the approval of the Erasmus Medical Center Ethics Committee (MEC-2014-106 and MEC-2022-0163).
Human CD14+ monocytes were acquired from buffy coats provided by Sanquin blood bank (Amsterdam, The Netherlands; contract number: NVT0053.01) after receiving ethical approval (N = 5 adult male donors). Monocytes were isolated and stored in liquid nitrogen as previously described.16,40
Adipose-derived stromal cells (ASCs) were isolated from human adipose tissue obtained from a healthy patient undergoing plastic surgery, following informed consent and approval from the Ethics Committee of the Basel University Hospital (Ethikkommission beider Basel [EKKB], Ref. 78/07). The isolation procedure involved tissue mincing, collagenase digestion, centrifugation, and filtration steps as previously described. 38 Isolated cells were cultured in alpha minimum essential medium (αMEM; ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% v/v heat inactivated fetal bovine serum (FBS; Sigma Aldrich, Saint Louis, MO, USA, lot #BCCD0778), 50 µg/mL gentamicin (ThermoFisher Scientific), 1.5 µg/mL Amphotericin B (ThermoFisher Scientific), 10 mM HEPES (ThermoFisher Scientific), 2 mM glutamine (ThermoFisher Scientific), 1 mM sodium pyruvate (ThermoFisher Scientific), and 5 ng/mL fibroblast growth factor 2 (R&D Systems, Abingdon, UK). Passage 2 cells at a confluency of 85–90% were used for the experiments.
Human umbilical vein endothelial cells (HUVECs) were purchased from Promocell (Heidelberg, Germany; C-12203; N = 3 pooled donors) and expanded with a seeding density of 3300 cells/cm2, as previously described. 38 Endothelial cell growth medium-2 (Lonza, Basel, Switzerland) was used for HUVEC expansion. Cells were subcultured at 85–90% confluence, until Passage 4.
In this context, ASCs play the role of pericyte-like cells in this model, supporting the formation of microvascular structures by endothelial cells as occurs in vivo.
Chondrogenic differentiation and mineralization of pMSC pellets
In total, 200,000 pMSCs were transferred to individual 15 mL polypropylene tubes containing 500 µL of complete chondrogenic medium consisting of Dulbecco's modified Eagle medium high glucose (DMEM HG, ThermoFisher Scientific) supplemented with 50 µg/mL gentamicin, 1.5 µg/mL Amphotericin B, 1 mM sodium pyruvate, 40 µg/mL
Coculture of pMSC pellets, osteoclasts, and vascular networks
pMSC pellets were cultured for 11 days with chondrogenic medium, then TGF-β and dexamethasone were removed from day 11 to day 14. At day 14, cells were washed with PBS to remove any traces of TGF-β and dexamethasone. Next, CD14+ monocytes were seeded on and cocultured with the pellets from day 14 to day 28 with osteoclast medium (αMEM, 50 µg/mL gentamicin, 1.5 µg/mL Amphotericin B, 5% v/v FBS, 25 µg/mL ascorbic acid) supplemented with 25 ng/mL macrophage colony-stimulating factor (M-CSF; R&D Systems) (from day 14) and 30 ng/mL receptor activator of nuclear factor kappa-β ligand (RANKL; Peprotech, Cranbury, NJ, USA) (from day 17). On day 28, 300,000 HUVECs and 300,000 ASCs were resuspended in 50 µL of 20 mg/mL fibrinogen in 0.9% w/v NaCl (plasminogen, vWF, and fibronectin-depleted human fibrinogen; MILAN Analytica AG). For the enzyme solution, human thrombin (Sigma-Aldrich) and Factor XIII (CSL Behring, King of Prussia, PA, USA) were added at a concentration of 6 U/mL to 50 µL of 40 mM CaCl2 solution. Then, the cell suspension, enzymes and three chondrogenic pellets were mixed to generate 100 µL of fibrin hydrogels in an Ibidi 12-well chamber (Gräfelfing, Germany) and incubated for 15 min at 37°C to allow crosslinking. The hydrogels were cultured in 270 µL of vessel consensus medium (DMEM HG, 50 µg/mL gentamicin, 1.5 µg/mL Amphotericin B, 1 mM sodium pyruvate, 40 µg/mL
Histological analysis
Pellets and fibrin hydrogels were fixed with 4% formalin overnight followed by dehydration and paraffin-wax embedding. Then, paraffin blocks were sectioned at 6 µm thickness and deparaffinized. Von Kossa/thionine and hematoxylin and eosin (HE) staining were performed as previously described.38,41 For TRAP staining, slides were incubated with freshly prepared acetate-tartaric acid buffer (0.2 M sodium acetate [Sigma Aldrich] and 100 mM
Endomucin staining on paraffin sections (immunofluorescence)
Vascularization of implanted pellets was assessed by immunofluorescent staining of paraffin sections for endomucin. Before staining, antigen retrieval was performed with a microwave tissue processor at 95° for 20 min in a Tris-EDTA buffer solution (pH 9.0). Tissue sections were then incubated overnight with a primary rat anti-Endomucin antibody (Santa Cruz Biotechnology, sc-65495, 1:100). A fluorescently goat anti-rat labeled secondary antibody was used (Thermo Fisher Scientific, 1:200). To remove red blood cell autofluorescence, tissue sections were treated with Vector TrueVIEW quenching kit (Vector Laboratories, sp-8400) following manufacturer’s instructions. Images were acquired with a Nikon Ti2 Eclipse microscope (Nikon, Tokyo, Japan). Details of the lectin/laminin staining on paraffin sections can be found in the Supplementary Data S2.
Calcium uptake assay
Calcium content in culture medium supernatants was calculated using a standard curve of 0–3.0 mM CaCl2 (Sigma-Aldrich) in calcium-free DMEM (ThermoFisher). Then, 100 μL of reagent [1:1 of reagent 1 (1 M ethanolamine pH 10.5; Sigma-Aldrich) and reagent 2 (0.35 mM o-cresolphthalein complexone; Sigma-Aldrich 19.8 mM 8-hydroxyquinoline; Sigma-Aldrich, and 0.6 M hydrochloric acid)] was mixed with 10 μL medium or standard. Samples were measured at 570 nm on a Versamax (Molecular Devices, Wokingham, UK) spectrophotometer. Medium only (no cells) samples were taken during culture and used for data normalization.
Gene expression analysis
Gene expression analysis was performed as previously described. 38 Samples were manually homogenized with a pestle and lysed with 400 µL RNA STAT-60 (Tel-Test Inc., Friendswood, Texas, USA). Gene expression analysis was normalized to a bestkeeper index value generated from averaging gene expression values of three different housekeepers (B2M, UBC and GAPDH). The analysed genes and their sequences can be found in Table 1.
RNA sequencing data analysis
The RNA sequencing dataset analyzed in this study was previously generated by culture of pMSC pellets for 7 days with TGF-β (7-day-primed pellets) or without TGF-β (7-day-unprimed pellets) (N = 4 pMSC donors). Differential expression analysis was performed using DESeq2. A prefiltering heuristic was used prior to running DESeq2 to remove single strong outliers in either one of the classes. Among these, genes encoding for secreted factors related to vascularization and osteoclastogenesis were identified based on the database of Uniprot and Gene Ontology (GO) and GO Annotations.
Whole-mount laminin staining, confocal imaging and vessel length density quantification
For whole-mount laminin staining, fibrin hydrogels were first fixed with 1% w/v paraformaldehyde overnight at 4°C. To block nonspecific binding, 3% w/v BSA and 5% v/v donkey serum (Sigma-Aldrich) in PBS were added overnight. The gels were subsequently incubated with 7 µg/mL antihuman laminin antibody (Abcam, Cambridge, UK) in PBS with 3% w/v BSA and 5% v/v donkey serum overnight. Samples were then incubated with 10 µg/mL Alexa Fluor 647 secondary antibody (ThermoFisher Scientific) and DAPI (1 µg/mL) at 4°C, overnight. The hydrogels were imaged using Leica Stellaris 5 low-incidence angle upright microscope or Leica TCS SP5 microscope (Manheim, Germany), with an excitation wavelength of 638 nm and an 650–750 band-pass emission filter for laminin signal. The gels were optically scanned using a 6 µm stepsize in Z-direction (see Supplementary Video) and images processed using FIJI. 42 Vessel lengths were measured by overlaying captured microscopic images with a square grid (field size = 200,000 μm2). Squares were randomly chosen and the length of each vessel (if any) in the selected squares was measured and summed up. For each sample, 10 fields of the whole image for vessel length measurements were obtained, with triplicate samples for each experimental group.
Statistical analysis
Data representation was performed using GraphPad Prism (software version 8.0), and statistical analyses were performed with SPSS 24 (IBM). Subsequently, for all multiple comparisons, a linear mixed model with Bonferroni correction was used; the different conditions were considered as a fixed parameter and the donor as a random factor. Statistical significance was evaluated between conditions within the same time-point and defined as p < 0.05. All results are presented as mean ± standard deviation (SD).
Results
pMSC pellets chondrogenically primed as early as 7 days instruct vascularization and osteoclastogenesis
We initially investigated the dynamics of vascularization and osteoclastogenesis in vivo in the context of a tissue engineering model of pMSC-mediated endochondral bone formation. Our aim was to assess how the timing of these processes is affected by the duration of chondrogenic differentiation and evaluate whether a briefer priming protocol than our standard 21-day protocol is still able to preserve the dynamics of bone formation in vivo. Our interest in briefer priming protocols lies on the fact that it would facilitate the development of a complex coculture system in vitro. Of note, their potential to form bone was already observed in a previous study performed in our group. 41 Sections of available samples from previous in vivo experiments of endochondral bone formation performed with 2 pMSC donors were stained to visualize osteoclasts and blood vessels. For these studies, pMSC pellets had been chondrogenically primed in vitro for 7 (brief priming) or 21 days (standard priming), implanted subcutaneously in mice, and retrieved at several time-points up to 84 days postimplantation. Of note, no unprimed condition was considered in this work since previous studies with this setup showed that unprimed conditions could only be found up to 2 weeks postimplantation and without clear signs of vessel and osteoclast infiltration. Endomucin staining of the implanted pellets showed that at the early stages after implantation (3–7 days) vessels were present close to the surface of the pellets for both priming conditions (Fig. 1A). Between 14- and 28-days postimplantation, vessel infiltration was more evident for 7-day primed pellets, while only superficial for 21-day primed pellets (Fig. 1A). At the later stages of bone formation (days 56 and 84), large vessel structures were present in the newly formed bone marrow stroma for both 7-day and 21-day primed pellets (Fig. 1A).
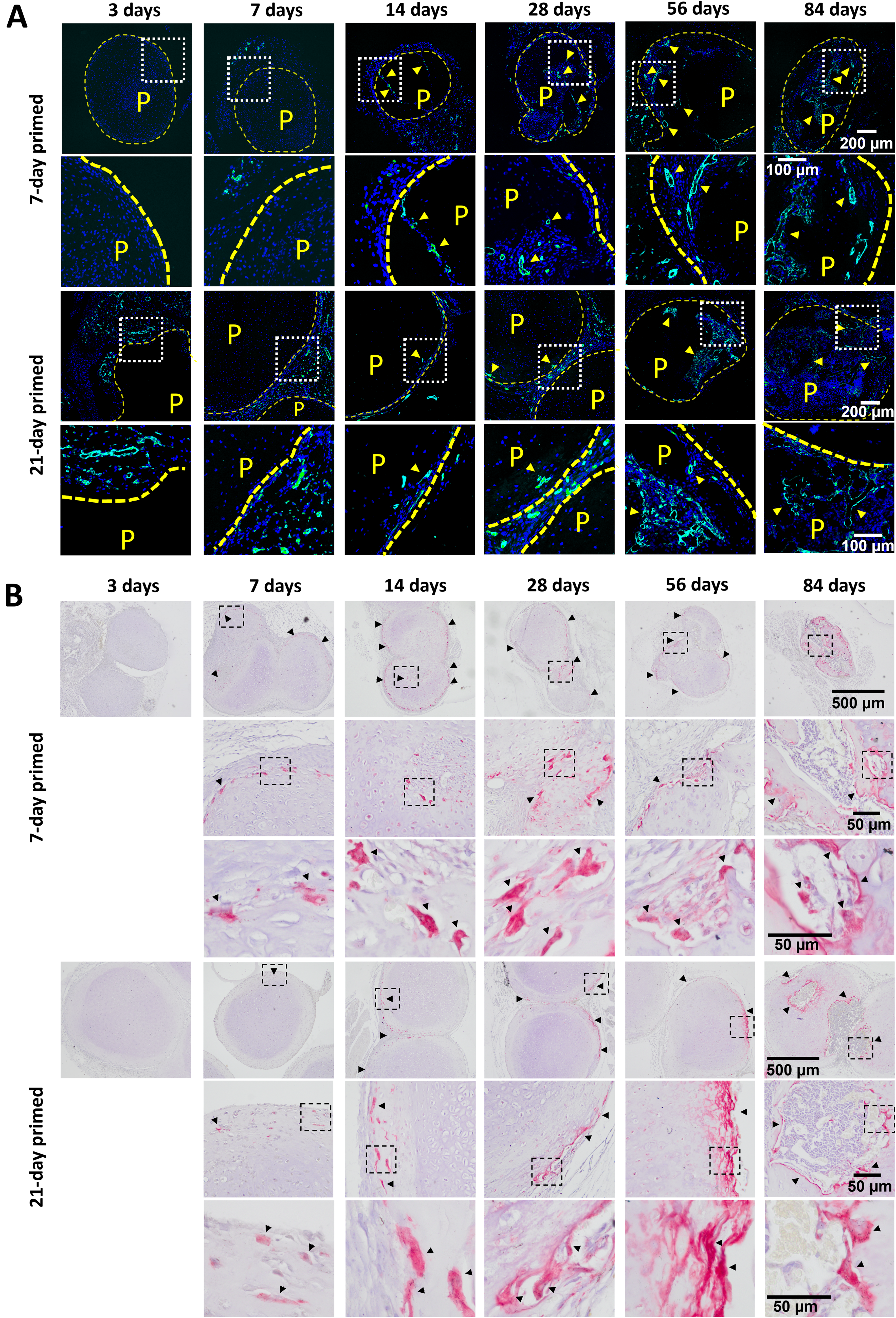
Vessel and osteoclast recruitment by chondrogenically primed pediatric bone marrow stromal cell (pMSC) pellets in vivo.
Osteoclast recruitment was evidenced by TRAP staining around the pellets as early as 7–14 days postimplantation for both 7-day and 21-day primed pellets (Fig. 1B). While osteoclasts were mainly localized on the surface of the pellets at these early time-points, in the late stages of bone formation (day 84) osteoclasts were observed within the bone marrow at the resorption front of the unresorbed cartilage matrix, supporting their active role in bone remodeling (Fig. 1B). Thus, the evaluation of vessel and osteoclast infiltration into in vitro pMSC pellets with different chondrogenic priming times (7 or 21 days) showed that priming pellets as brief as for 7 days has potential to instruct vessel and osteoclast infiltration.
In order to further characterize the proangiogenic and pro-osteoclastogenic properties of 7-day primed pellets, their full transcriptome was analyzed by bulk RNA-seq and compared with unprimed pellets (cultured without TGF-β for 7 days; N = 4 pMSC donors) (Fig. 2A). Differential gene expression analysis showed that 3920 genes were significantly changed (log2 fold change > 1) when comparing 7-day primed and 7-day unprimed pMSC pellets, of which 1969 were upregulated and 1951 downregulated (Fig. 2B). Next, we used human secretome data to identify differentially regulated genes that encode for secreted factors. 43 Interestingly, the significantly upregulated genes included several secreted factors known to modulate vascularization (Fig. 2C) and osteoclastogenesis (Fig. 2D). Altogether these data suggest that pMSC pellets subjected to brief chondrogenic priming may represent a suitable system to support vessel formation and osteoclast differentiation in vitro.
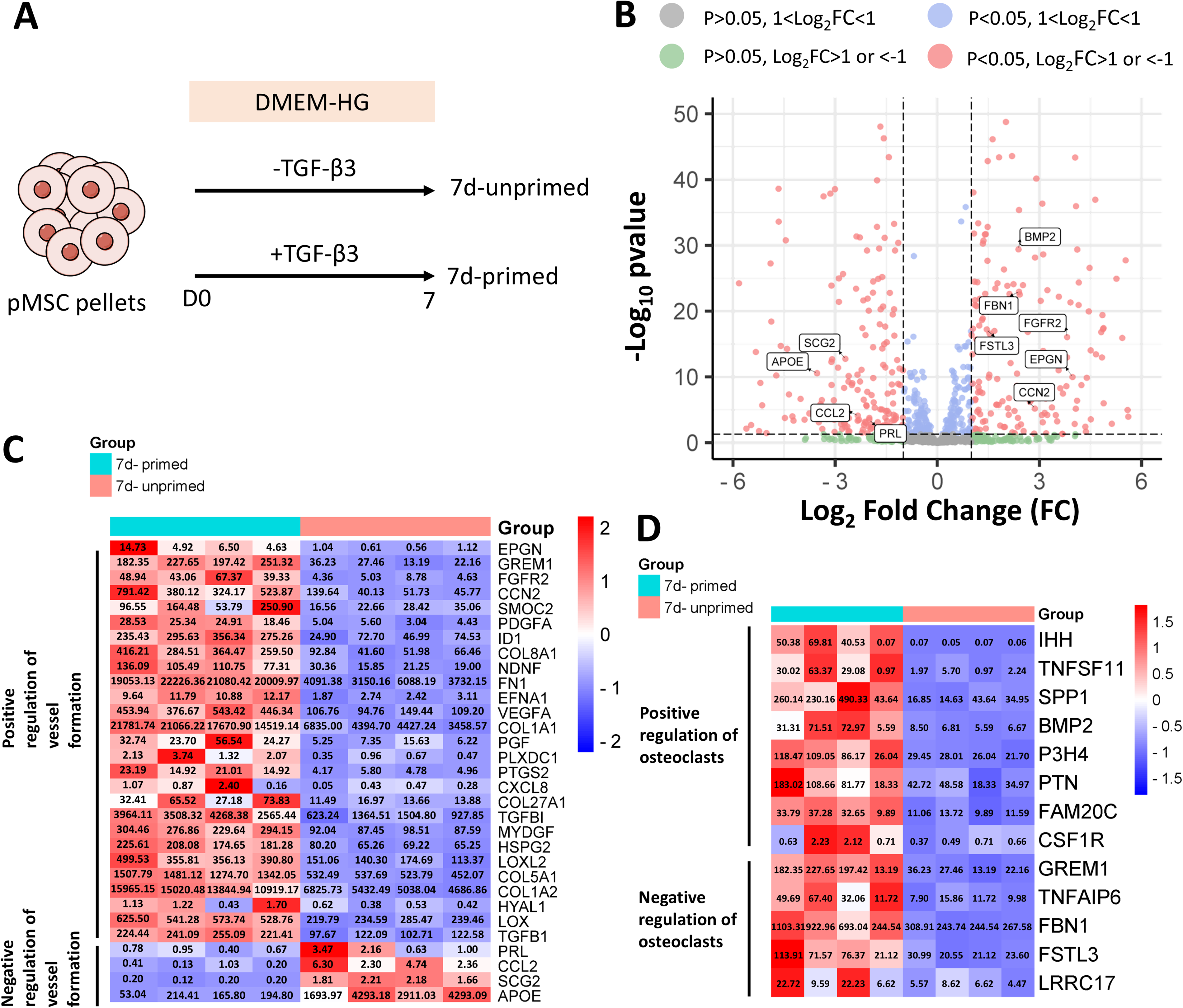
Bulk RNA-seq analysis of 7-day primed vs 7-day-unprimed pellets.
Osteoclasts successfully form on chondrogenic pellets at different stages of mineralization
To develop an in vitro model with chondrogenic pellets undergoing mineralization and osteoclasts, pMSC pellets were exposed to BGP after 7 days of chondrogenic priming and subjected to different mineralization periods (7 or 14 days), prior to monocyte seeding and induction of osteoclast formation for further 14 days. The pellets were therefore cultured for 28 or 35 days in total, of which the last 14 days in coculture with monocytes/osteoclasts (Fig. 3A). Chondrogenic pellets cultured without BGP and seeded with monocytes were included as nonmineralized control. Longitudinal measurements of calcium concentration in the medium revealed significant calcium uptake by mineralizing pMSC pellets starting from day 14 (Fig. 3B). Von Kossa staining confirmed the successful formation of mineral deposits within the pellets, which could be maintained in pellets seeded with monocytes (Fig. 3C). Interestingly, the addition of monocytes to pellets at day 14 and harvesting of pellets at day 28 caused a delay in mineralization in 2 out of 4 donors analyzed, as observed by the absence of Von Kossa positivity in those groups with monocyte seeding (Supplementary Fig. S1A). However, the groups without monocyte seeding all eventually mineralized (Supplementary Fig. S1A). In addition, the seeding of monocytes to pellets at day 21 and harvesting of pellets at day 35 led to mineralization in all four donors (Fig. 3C, Supplementary Fig. S1B). Seeding of chondrogenic pellets with monocytes did also not cause changes in calcium concentrations in the medium (Supplementary Fig. S1C). Expression levels of the chondrogenic marker COL2A1 and the hypertrophic marker COL10A1 were similar for all conditions and not significantly affected by monocyte seeding (Fig. 3D). Osteoclastogenesis was assessed by HE and TRAP staining, revealing the presence of TRAP+ multinucleated cells on the surface of all mineralized pellets seeded with monocytes (Fig. 3E, Supplementary Fig. S2). A certain degree of variability was observed between pMSC donors: three out of four donors clearly evidenced osteoclasts on the surface of day 28 mineralized pellets, while the size of the osteoclasts formed on the surface of chondrogenic pellets also varied between donors. Induction of the expression of the markers of osteoclast fusion DC-STAMP and OC-STAMP and the markers of osteoclast activity TRAP and CTSK was found for all mineralized pellets seeded with monocytes in comparison with mineralized pellets not seeded with monocytes where some genes were nondetectable (ND) (Fig. 3F). This phenomenon was also observed in the chondrogenic condition without monocytes in our previous work, which, similarly to mineralized pellets without monocytes, showed very low to undetectable levels of osteoclast markers. 16 Based on these results, we conclude that osteoclasts can be formed and cocultured with chondrogenic pellets at different stages of in vitro mineralization.
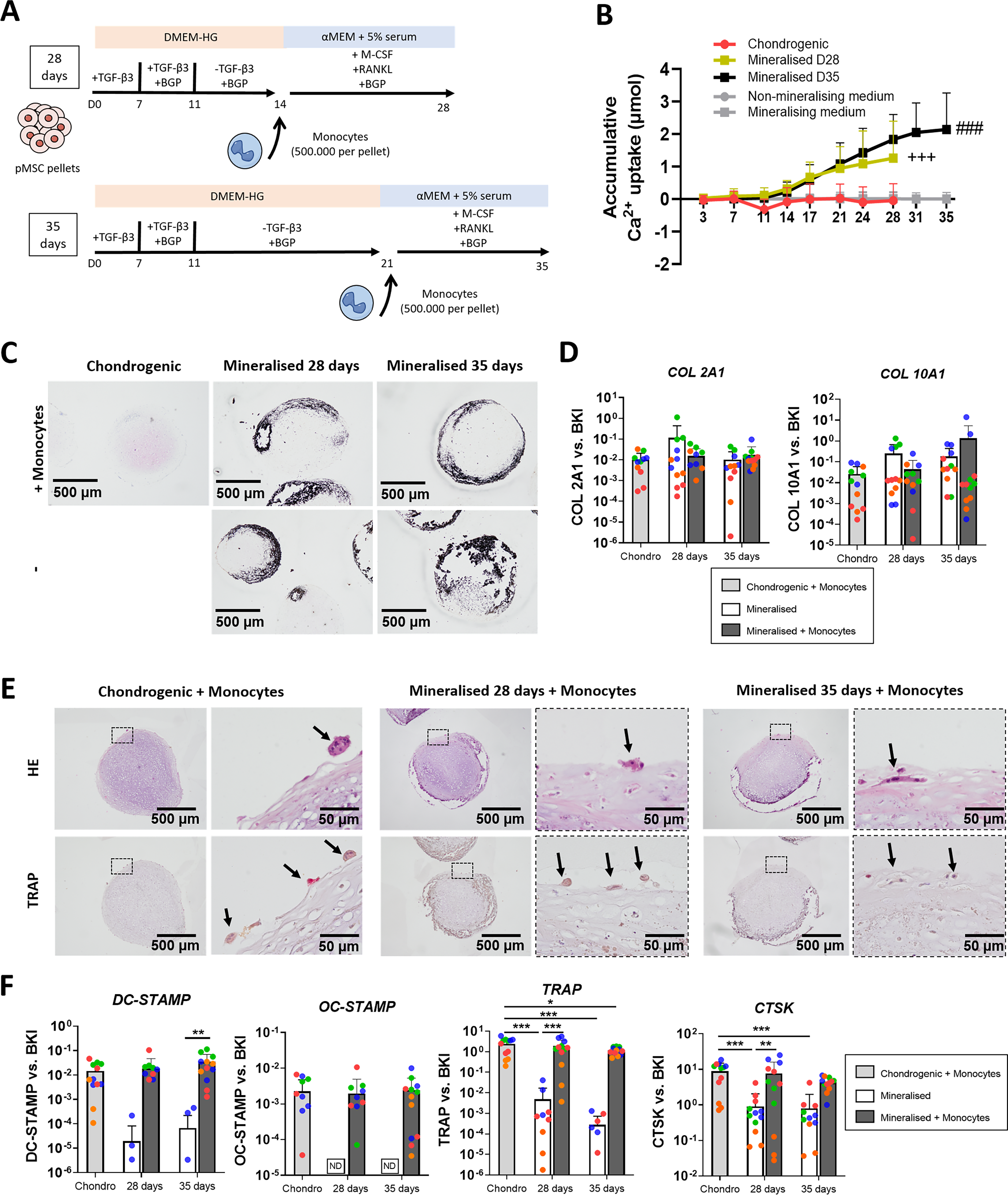
Coculture of pediatric bone marrow stromal cell (pMSC) pellets undergoing mineralization with osteoclasts.
Microvascular networks can be formed and cocultured with chondrogenic pellets undergoing mineralization
To establish an in vitro model which combines cartilage undergoing mineralization and vascular networks, we next directly cocultured pMSC pellets with microvessel-forming HUVECs and ASCs as endothelial- and pericyte-like cell sources, respectively, in fibrin hydrogels. Since we previously showed that pellets subjected to prolonged mineralization have reduced proangiogenic properties, 38 BGP was added to the medium only at the start of the coculture (day 21). During initial studies, we also found that the exposure of pellets to TGF-β until the start of the coculture decreased the efficiency of vascular network formation, likely due to a detrimental effect of TGF-β retained by the pellets (Supplementary Fig. S3). Based on these observations, we set up a coculture scheme where pMSC pellets were chondrogenically primed for 14 days with TGF-β, then further cultured without TGF-β for 7 days. The pellets where then embedded in fibrin hydrogels with ASCs and HUVECs and cultured until day 31 with or without BGP (Fig. 4A). 44 Calcium measurements in the medium demonstrated progressive Ca2+ uptake for pellets exposed to BGP, suggesting that mineralization of the cartilage matrix can occur during coculture (Fig. 4B). This was further confirmed by Von Kossa and thionine staining of the pellets (Fig. 4C). HE staining of the hydrogels evidenced the presence of vessel lumina, mainly in the proximity of chondrogenic pellets (Fig. 4C, yellow arrows). To assess vascular network formation, the hydrogels were subjected to whole-mount staining for the endothelial marker laminin (Fig. 4D, F) and paraffin section immunofluorescence staining for both lectin and laminin (Fig. 4F). Vessel length densities (VLDs) within 200 μm distance from the pellets (zone 1) or further (zone 2) were also quantified (Fig. 4E). The results indicated that vascular networks could form around chondrogenic or mineralized pellets. In the case of mineralized pellets, vessel formation in the area close to the pellets (zone 1) was significantly decreased in comparison with zone 2. This effect was due to pellet mineralization and not uniquely related to BGP addition (Supplementary Fig. S4). No significant differences were found in zone 2 between chondrogenic and mineralized pellets.
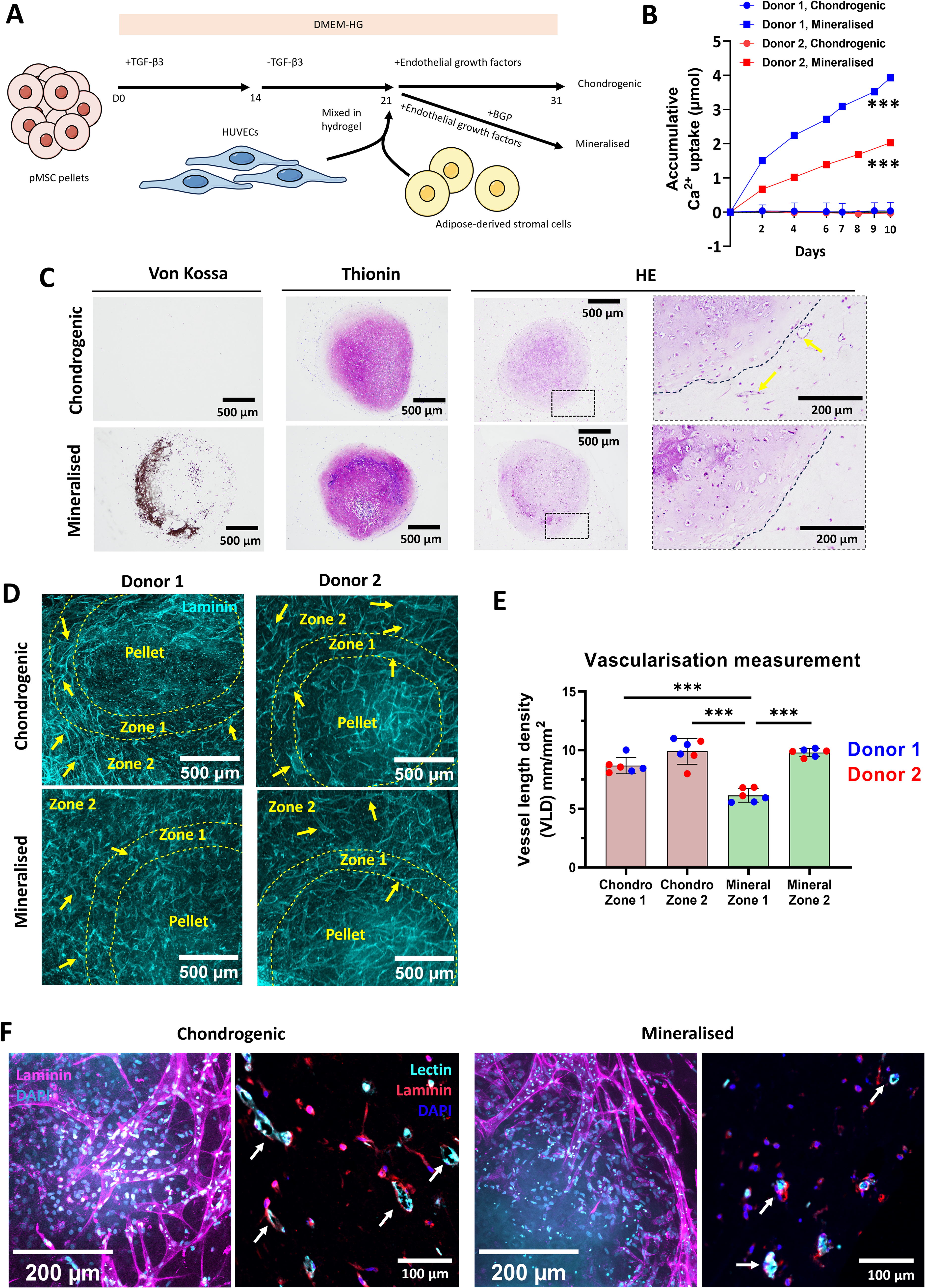
Coculture of pediatric bone marrow stromal cell (pMSC) pellets undergoing mineralization with vascular networks.
In conclusion, we showed that microvascular networks can be formed and cocultured with chondrogenic pellets undergoing mineralization. Under in vitro conditions, cartilage mineralization can locally reduce microvascular assembly.
Integration of chondrogenesis, mineralization, vascular network formation and osteoclastogenesis into a single 3D coculture system
We finally aimed to setup a coculture system which captures cartilage mineralization, vascular network formation and osteoclastogenesis (Fig. 5A): pMSC pellets were chondrogenically primed with TGF-β for 11 days. At day 14, they were seeded with monocytes and osteoclastogenesis was induced via M-CSF and RANKL addition until day 28. Finally, pellet-osteoclast cocultures were combined with vessel-forming cells (ASCs and HUVECs) in fibrin hydrogels up to day 38. For the last coculture stage where all cell types are present, we tested consensus media consisting of either DMEM-HG or αMEM supplemented with osteoclastogenic and angiogenic factors. The use of both media generally supported microvascular network formation and osteoclastogenesis under coculture conditions. A variable but overall similar number of osteoclasts was observed for both conditions at days 35 and 38 (Fig. 5B, D, black arrows). Overall, while VLDs of the DMEM-HG group showed similar values to the αMEM group (Supplementary Fig. S5), more vessel branching was observed for DMEM-HG based consensus medium as opposed to αMEM (Fig. 5C, D, black stars). Interestingly, some of the histological sections evidenced loose fragments of pellets in close contact with osteoclasts, which may suggest a resorptive effect of these cells (Fig. 5E, black arrows). This phenomenon was observed for both medium types. Based on this study, the DMEM-HG based consensus medium was chosen for further cocultures.
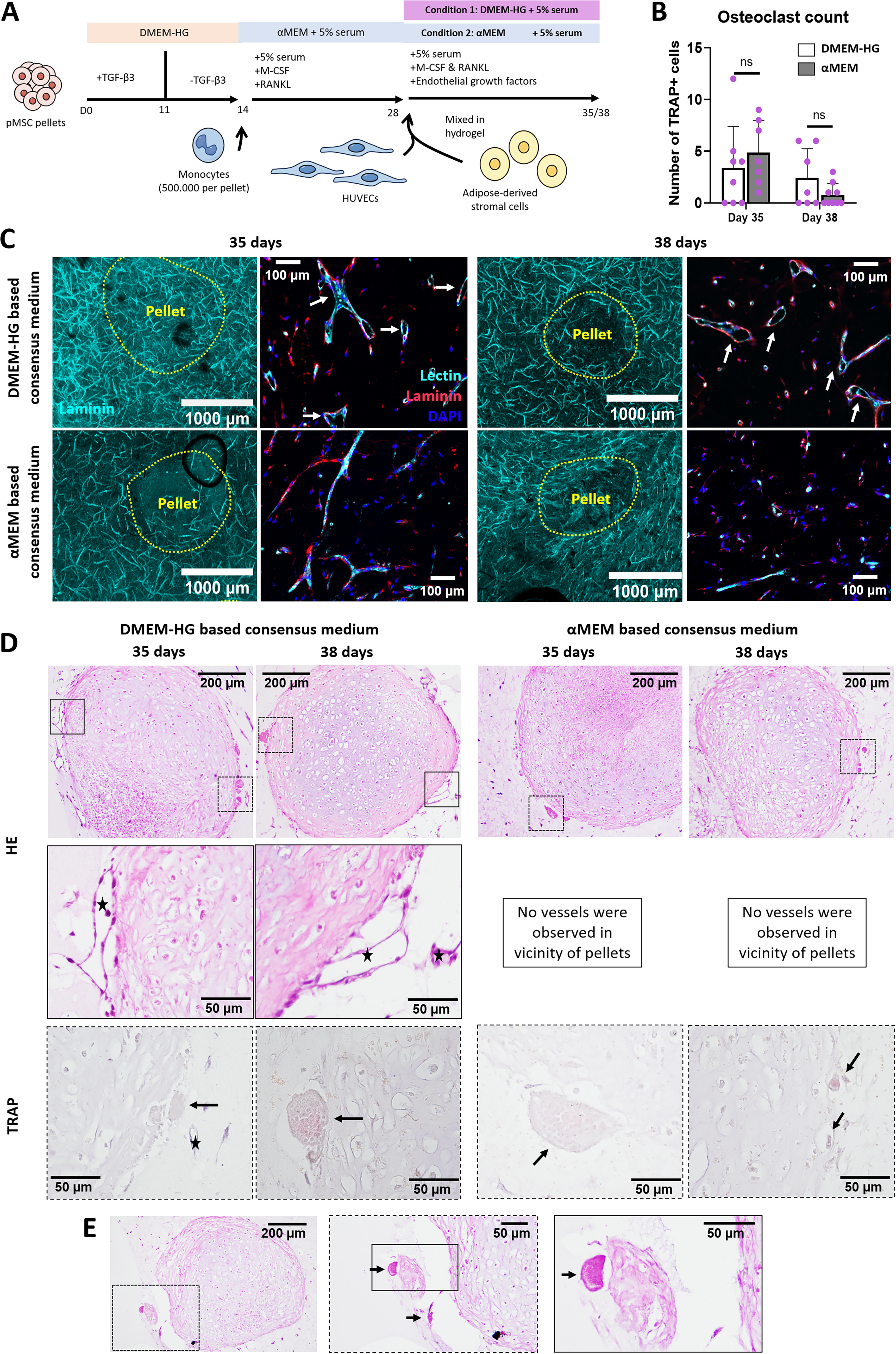
Coculture of chondrogenic pediatric bone marrow stromal cell (pMSC) pellets, vascular networks, and osteoclasts with different consensus media.
As a last step, the induction of cartilage mineralization was introduced in the coculture system to add a further level of complexity to the model. This was achieved by the addition of BGP from day 28 onwards, while the coculture was continued until day 35 or day 42 to allow sufficient time for mineralization (Fig. 6A). Mineralization of chondrogenic pellets was confirmed via Von Kossa/Thionine staining at both time-points of analysis (Fig. 6B).
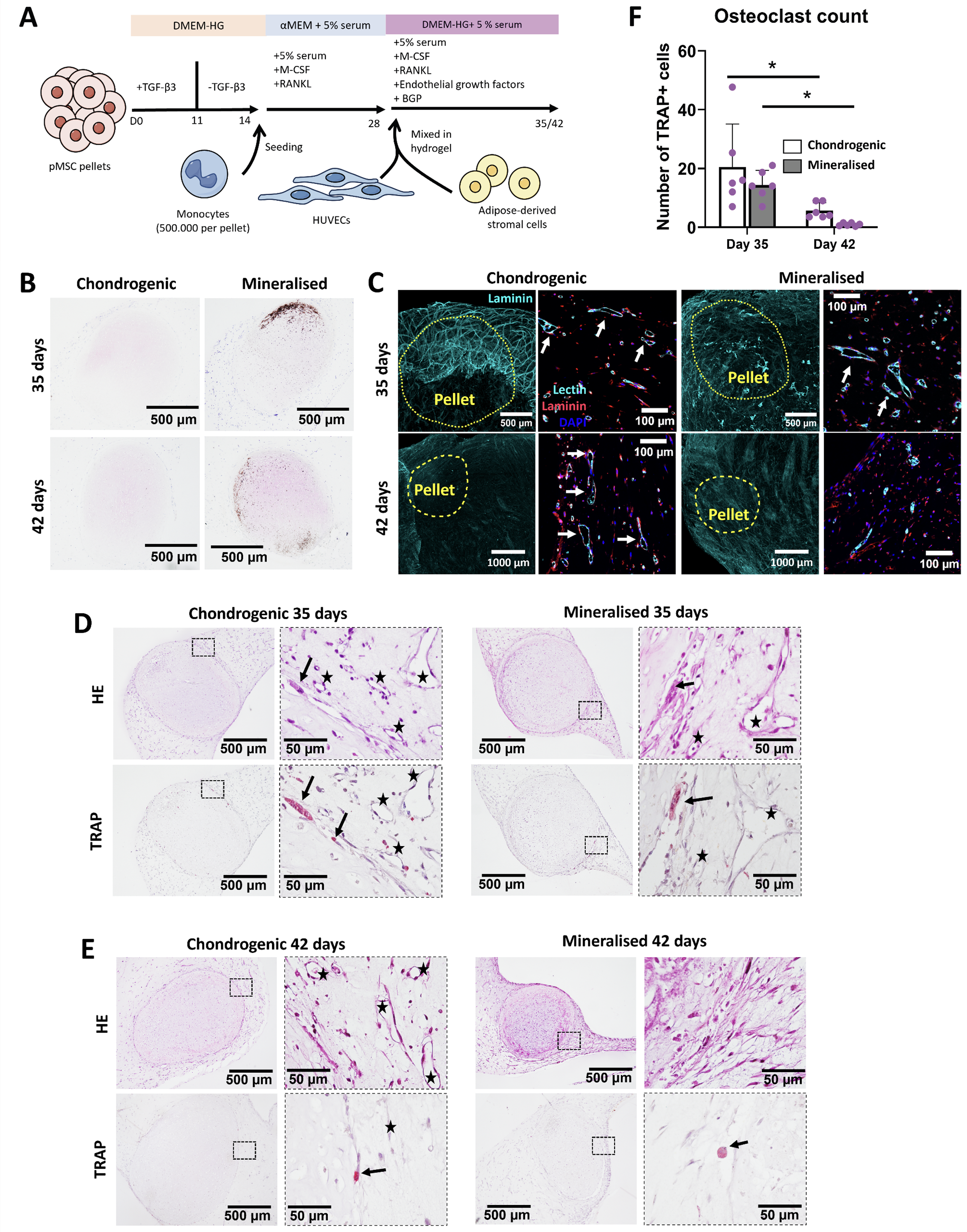
Coculture of chondrogenic pediatric bone marrow stromal cell (pMSC) pellets undergoing mineralization, vascular networks and osteoclasts.
List of Genes of Interest Used for RT-qPCR Analysis in This Study

At day 35 we observed the characteristic vessel structures around both chondrogenic and mineralized pellets (Fig. 6C). Prolonging the culture time to 42 days had a detrimental effect on the vascular networks as evidenced by the faint laminin signal and reduced vessel lumen structures by lectin staining (Fig. 6C). This was particularly noticeable for the mineralized group where no vessels could be observed by HE staining, as opposed to the chondrogenic group (Fig. 6E, black stars). In terms of osteoclast formation, HE and TRAP staining of adjacent sections of the hydrogels showed successful formation of multinucleated osteoclasts and their presence in close contact with the surface of the pellets and close to the vessels (Fig. 6D–E, black arrows). No significant differences in terms of number of formed osteoclasts were observed between chondrogenic and mineralized pellets for 35-day cultures (Fig. 6F). Prolonging the culture until day 42 led to significantly reduced osteoclast numbers, further highlighting the need to select proper culture periods for such a complex system.
Discussion
In this study, we successfully established a complex in vitro model of certain aspects of EO where the functionality of multiple cell types is supported. This model provides a controlled environment that mimics some of the early physiological steps of EO, including the formation and calcification of the cartilage template, the recruitment of monocytes and their differentiation into osteoclasts, and the formation of microvascular networks. We thus provide a platform to study the interplay of these processes in physiology or bone-related diseases. Such model can not only enhance our fundamental understanding of bone biology but also serve as a valuable tool for testing potential therapeutic interventions, drug screening, and exploring the pathogenesis of skeletal disorders.
To design an in vitro model of EO with physiological relevance, we initially investigated how vessels and osteoclasts are recruited in vivo after the implantation of chondrogenically primed pellets. After implantation of pMSC-derived cartilage in an animal, mineralization starts already within 7 days postimplantation and continues henceforth. Here we found that during these early stages of the bone formation process, osteoclasts are rapidly recruited to the surface of the pellets and vessels start to form in close proximity. This led us to reason that a relevant in vitro model of the early stages of EO should necessarily integrate cartilage mineralization, osteoclast formation and activity, and angiogenesis. The in vivo data showed that these processes occur simultaneously and capturing the cross-talks between these cell types will likely affect the response of these cells to stimuli or drugs. In the context of these in vivo analyses, we also took into account the effect of the duration of the chondrogenic priming on the dynamics of bone formation. The chondrogenic differentiation phase leads to the formation of a mature cartilage template for implantation, and it is generally achieved by culturing pMSCs in the presence of chondrogenic growth factors for a period of 3–6 weeks. Such an extensive period is obviously not practical as a starting point to develop a model where multiple cell types need to be added and further differentiation stages are required. Furthermore, there are indications that a prolonged chondrogenic priming (5–6 weeks), which leads to the formation of a mature and very dense cartilaginous matrix, may potentially reduce in vivo remodeling and be overall detrimental for bone formation. 41 We found that 7 days of chondrogenic priming are sufficient to induce osteoclast recruitment and vessel invasion after implantation, with similar dynamics in comparison to 21 days of priming. In the case of 7 days of priming, vessel infiltration reached further depth at the earlier stages of bone formation, possibly due to smaller size of the construct and presence of less (dense) cartilage matrix. Based on this evidence, we concluded that in the setting up of the in vitro model a minimum of 7 days priming with TGF-β is necessary prior to the stimulation of other processes and the addition of other cell types. We then corroborated this evidence by showing that 7 days of in vitro chondrogenic priming are sufficient to induce the expression of a number of genes related to the modulation of angiogenesis and osteoclastogenesis. These include well-known proangiogenic genes such as VEGF and PDGFA, which are significantly increased with 7 days of chondrogenic priming, as well as genes relevant to osteoclastic activity such as, TNFSF11 (RANKL) and SPP1. Overall, this suggested that chondrogenically primed pellets may contribute to the establishment of a supportive environment for the coexistence of vasculature, osteoclasts, and mineralized cartilage in an in vitro model.
Setting up a coculture of several human primary cell types and guaranteeing their functionality is a complex task. Of note, culture media to induce cartilage formation, vascular network formation and osteoclastogenesis all include a number of factors which may affect the viability and activity of the other cell types. We thus decided to establish first separate cocultures of (mineralized) cartilage with osteoclasts or vascular networks, as these would provide relevant information on how all these processes can be combined in a later stage. Monocyte seeding on mineralized pellets and culture with M-CSF and RANKL led to the successful formation of TRAP+ osteoclasts at the pellet surface. Osteoclasts in this context were also observed to be CTSK+ in our previous work. 16 This is a very similar pattern to those observed for in vivo implanted pellets where osteoclasts are mainly localized on the edge of the pellets at the early stages of bone formation. Interestingly, smaller and less multinucleated osteoclasts formed on the surface of mineralized pellets in comparison to chondrogenic pellets. This is not necessarily a detrimental phenomenon for our model as very large osteoclasts may represent culture artifact. 45 However, an inhibitory effect might also be happening due to the increase on the levels of local calcium in the mineralization front of pellets. Previous studies have indeed shown that high extra and intracellular levels of calcium promote osteoclast apoptosis as well as inhibition of osteoclast maturation.46–49 In order to provide a more controlled environment in vitro, it is therefore necessary to further understand the exact interactions of osteoclasts with mineralized and unmineralized cartilage and how this regulates their formation and resorption activity. Future studies could employ the model developed in this study to that specific purpose.
By coculturing self-assembled vessel structures with lumen and mineralized cartilage in vitro, we next established a model that allows recapitulation of the intricate interplay between vascularization and cartilage mineralization in vitro. Of note, this process is normally studied in vivo as in vitro models of tissue vascularization still face many challenges. By applying our in vitro coculture system of cartilage and vascular networks, we demonstrated that cartilage mineralization leads to a local decrease in vessel formation. This is in line with our previous work showing that the transition from hypertrophic to mineralized cartilage involves a certain level of chondrocyte cell death with a decrease in the expression and production of proangiogenic factors such as VEGF, which could explain the reduced vessel formation. 38 Furthermore, increased concentrations of calcium and phosphate have been shown to increase oxidative stress and apoptosis in endothelial cells. 50 Future mechanistic studies could make use of our newly developed model to investigate the exact cellular and molecular mechanisms by which cartilage mineralization can negatively impact angiogenesis in the context of the early stages of EO. These may be highly relevant for the tissue engineering field, where the ability to better control cartilage vascularization and remodeling after implantation could lead to more effective or more rapid bone formation in the context of a large bone defect.
In the last step of the work, we succeeded in incorporating all of the selected processes into a single in vitro model of EO. Cocultures of chondrogenic/mineralized pellets, monocytes and vessel-forming cells led to the formation of TRAP+ osteoclasts and vessels in close proximity and in contact with the pellets. To the best of our knowledge, this represents unprecedented complexity for an in vitro model of bone formation via EO. This model thus opens up a new range of possibilities to investigate a complex cross-talk between chondrocytes, osteoclasts, and vascular cells in a controlled environment. Performing single-cell RNA-sequencing experiments in future studies could allow for the investigation of such cross-talks. Osteoclasts have, for example, been previously described to promote angiogenesis18,29 and osteogenesis,51,52 while blood vessels have shown pro-osteogenic effects.33,34 Similarly, macrophages have also been previously shown to have capacity to promote angiogenesis. 53 Given the usual presence of nonfused CD14+ monocytes in our models, it would be interesting to investigate their differentiation stage and their possible effect on the system. Of note, a longer culture period led to a reduced osteoclast and vessel network formation, particularly for mineralized pellets. These findings highlight the complexity of recapitulating a bone-like environment in vitro, where a delicate balance between mineral deposition and osteoclast and vascular network formation needs to be achieved. It should be considered that the current absence of the renewal of existing osteoclasts and vessels in vitro by fresh cells sets a time limit to the survival of these cells. We expect that further developments of the in vitro model, which could include dynamic fluidic system with periodic cell renewal, will allow to guarantee cell functionality for more extended periods of time, ultimately allowing the recapitulation of later stages of the bone formation process.
Within this study we mainly focused on establishing a complex coculture which could capture some of the early processes which normally occur after implantation of tissue-engineered cartilage templates and guarantee the functionality of the cell types involved. Our choice of recapitulating in vitro bone formation following EO rather than intramembranous ossification relied on the relevance of such a model for the study of the dysregulation, therapeutic rescue or manipulation of the various stages of bone formation, and their use for disease modeling or tissue engineering approaches whenever endochondral formation is involved. Future steps could focus on the subsequent stages of EO where the cartilage is resorbed and osteoprogenitors home via the vasculature leading to osteoblast differentiation and generation of a bone matrix. Osteogenic cell emergence from the perichondrium as well as through their transdifferentiation from hypertrophic chondrocytes are aspects that should also be incorporated into the models, which could also be useful for the study of the effect of pro-osteogenic effect of osteoclasts in this transdifferentiation process. Given the possibility that osteogenic cells could emerge early on in EO, future iterations of the model could choose an appropriate timing of osteoprogenitor addition to the model. It is also important to consider that this study employed bone marrow MSCs for the creation of tissue engineered chondrogenic constructs, which might represent a limitation when trying to capture developmental aspects. Related applications should be thus carefully considered, or the model adapted to host an alternative progenitor cell type. It is also very promising that we already observed the formation of loose cartilage fragments in proximity to osteoclasts in our cocultures, which may be indicative of resorptive phenomenon. However, further analysis is necessary in order to evaluate whether this colocalization is not incidental and there is indeed a resorptive action of osteoclasts. Optimization of culture conditions to further stimulate remodeling may allow to create space for the vessels to invade the dense cartilage matrix, eventually providing a mean to deliver cells, factors and drugs to the forming bone niche. Importantly, this growing level of in vitro complexity can be of enormous value for research on the physiology of EO or bone formation-related diseases.
Potential research applications include, but are not limited to, the study of undesired cartilage hypertrophy and vascularization in osteoarthritis or disturbances of endochondral bone formation in growth and metabolic diseases. In vitro models of bone formation could also allow to recapitulate the migration of metastatic cancer cells to the bone under controlled conditions. Despite great research effort in this field, many questions still remain to be answered, e.g., related to the effect of the bone microenvironment on cancer cell migration and survival or the differential tropism of metastatic cancers toward bone. Given that two-dimensional models cannot recapitulate the complexity of bone, while animal models pose interspecies differences and lack of high-throughput options, in vitro models of higher complexity and relevance could be of vital importance for shedding new light on the metastatic process.
Conclusion
This study presents a novel in vitro model that integrates mineralized cartilage, vascular structures and osteoclasts to recapitulate the early stages of EO. Inspired by the in vivo dynamics of EO in the tissue-engineered setting, we adopted a systematic approach to develop cocultures of increasing complexity, finally combining all processes into a single in vitro platform. These models will contribute to our understanding of bone formation and remodeling, offering a valuable tool for future research into skeletal and oncological disorders and potential therapeutic strategies.
Authors’ Contributions
E.L. and A.G.U. designed the study, performed the experiments, analyzed and interpreted the data, and wrote the article. A.L. and E.F. designed the study, interpreted the data, and edited the article. J.W.-B. performed the experiments, interpreted the data, and edited the article. N.D., A.B., and G.-J.K. interpreted the data and edited the article. All authors contributed to the article and approved the submitted version.
Footnotes
Funding Information
This project was supported by the following funding: the Erasmus MC-Health∼Holland TKI-LSH Grant EMCLSH21004; the European Union Horizon 2020 Research and Innovation Program under grant agreement 801159; the Dutch Research Council (NWO)-XS Science grant OCENW.XS5.074; Encheng Ji is supported by the China Scholarship Council (CSC, NO. 202207720011) and Wenzhou Wangqiao Orthopedic Hospital.
Disclosure Statement
There are no conflicts to declare.
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.