
Abstract
Introduction
Cercidophyllum japonicum Siebold et Zucc 1 (Cercidiphyllaceae, Japanese name: katsura) is found in the forests of East Asia at altitudes between 600 and 2700 meters. The long-lived tree has dull green colored leaves and reaches heights up to 30 meters with trunk diameters of over 2 meters (Figure 1). In autumn, the leaves change color to salmon pink, later to golden yellow and shortly before foliage fall they smell like freshly baked cake. Plant materials of C japonicum have long been used in Asian traditional medicine, owing to their antiinflammatory, antioxidative, antifungal, antimicrobial, and hair growth promoting activities, as well as arresting convulsion. 2 In light of these usages, numerous pharmacologically interesting ingredients have been identified in various above-ground parts of C japonicum. In 1986, the first publication reports on the isolation of a biphenyl phytoalexin from cortical tissue of C japonicum twigs. 3 More than 30 years later, a galloylflavonol glycoside together with anomeric tannins and flavonoids, isolated from the same plant part, were published. 4 In addition, maltol and phenolic glycosides were detected in the leaves,5,6 bark,7-9 flowers, 2 and fruits. 10 Furthermore, the whole methanol extract of the heartwood of C japonicum has been shown to stimulate proliferation of mouse hair epithelial cells. 11

Cercidophyllum japonicum, Whose Plant Materials are Used in Asian Traditional Medicine. (A) Whole Tree, (B) Trunk and Branches, and (C) Leaves of C japonicum. Shown is Europe's Tallest Specimen with a Height of Over 20 Meters, Located in the Botanical Garden of the Technical University of Darmstadt, Germany. Photos Were Taken by the Author in July 2020.
In 1991, a compound which possessed antimicrobial activity against Bacillus subtilis and Escherichia coli was isolated by Tada and Sakurai
12
using methanol extraction of fresh leaves of C japonicum. Structural analysis by high resolution mass spectrometry and 1H and 13C NMR spectroscopy revealed that the natural product, which they named cercidin
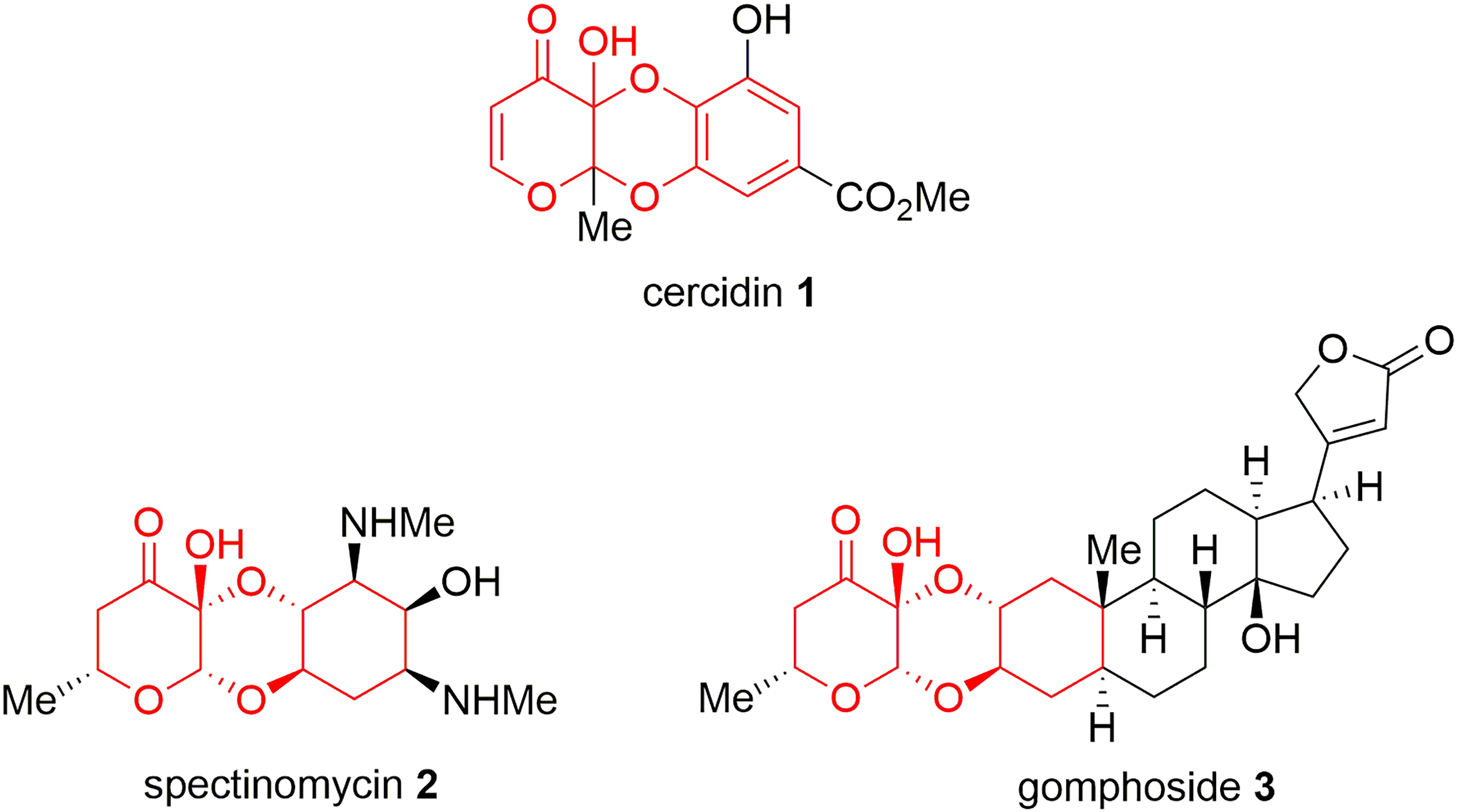
The Natural Products Cercidin
To date, no total synthesis of cercidin
Methods
Instruments
Thin-layer chromatography (TLC) was performed on POLYGRAM SILG/UV254 (Macherey Nagel & Co.). Preparative chromatographic separations were carried out on columns with Merck silica gel 60 (15-40 µm) and Merck precoated silica gel plates 60 F254, 20 × 20 cm, 0.25 mm. Melting points were determined on a Bock-Monoskop VS or on a Büchi SMP-20 and are uncorrected. Specific optical rotations were determined on a Perkin-Elmer Polarimeter 241 in 1 dm cuvettes at a wavelength of 589 nm. NMR spectra were measured on a (a) Bruker WM 300 spectrometer, (b) Bruker DRX 500 spectrometer, (c) Bruker 300 MHz spectrometer with Avance-II console and BBO probe, (d) Bruker 500 MHz spectrometer with DPX console and BBFO probe, and (e) Bruker Avance III HD 700 MHz spectrometer with a QCI cryo probe at 300 K using trimethylsilane as internal reference or by calibration with the shift of the solvent of the sample. Multiplicities of the shifts are abbreviated as s for singlet, d for doublet, t for triplet, q for quartet, sxt for sextet, oct for octet, m for multiplet, br for broad, and p for pseudo. Mass spectra were run on a Varian MAT 311 and Bruker Impact II spectrometer. Elemental analyses were performed on a Perkin Elmer 240 Elementar Analyser. NMR shifts with an asterisk * can be reversed.
Syntheses
The synthesis of the title compounds
The 700 and 500 MHz NMR spectra (1H, 13C, DEPT, HSQC, HMBC, COSY, and NOESY) for compounds
(2R,3R,4S,4aS,10aS)-2-((benzoyloxy)methyl)-4a,9-dihydroxy-7-(methoxycarbonyl)-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-3,4-diyl dibenzoate 13
With triethylamine in dichloromethane: A mixture of methyl gallate
C35H28O13 (656.6): calcd. C 64.02, H 4.30; found C 63.88, H 4.19.
MS (FD, 0-20 mA): m/z = 656 (100%, M+).
1H NMR (700 MHz, DMSO-d6) δ 3.84 (s, 3H, CO2CH3),), 4.34 (dd, 1H, 2'-HA), 4.42 (dd, 1H, 2'-HB), 4.62 (m, 1H, 2-H), 5.61 (pt, 1H, 3-H), 5.81 (s, 1H, 10a-H), 5.93 (d, 1H, 4-H), 7.13 (d, 1H, 8-H), 7.15 (d, 1H, 6-H), 7.42, 7.45, 7.49 (each 2H-pt, 6 meta-C6H5CO), 7.61 (m, 3H, 3 para-C6H5CO), 7.79 (pt, 4H, 4 ortho-C6H5CO), 7.92 (d, 2H, 2 ortho-C6H5CO), 8.02 (1H, 4a-OH), 9.96 (1H, 9-OH); J2,2'A = 3.98, J2’,2'B = 2.80, J2'gem = 12.48, J2,3 = 9.90, J3,4 = 9.68, J6,8 = 2.05 Hz.
NOE between 10a-H (5.81) and 2-H (4.62), 4-H (5.93), as well as 4a-OH (8.02).
13C NMR (175 MHz, DMSO-d6) δ 52.04 (CO2CH3), 62.27 (C-2’), 68.34 (C-3), 71.04 (C-2), 73.97 (C-4), 91.21 (C-4a), 91.76 (C-10a), 109.23 (C-6), 110.28 (C-8), 122.80 (C-7), 128.51, 3 × 128.57, 128.61, 128.72 (each m-C6H5CO), 2 × 129.07, 129.19, 129.21, 129.27, 129.46 (each o-C6H5CO), 132.74 (C-9a), 133.42, 133.46, 134.77 (each p-C6H5CO), 140,51 (C-5a), 146.26 (C-9), 164.82 (3-C6H5CO), 165.07 (2’-C6H5CO), 165.19 (4-C6H5CO), 165.68 (CO2CH3); 1JC10a,H10a = 175.7 Hz.
The synthesis of
Synthesis of the compounds
(2R,3R,4S,4aS,10aS)-2-((benzoyloxy)methyl)-4a,6-dihydroxy-8-(methoxycarbonyl)-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-3,4-diyl dibenzoate 14
The next fraction of the synthesis of compound
C35H28O13 (656.6): calcd. C 64.02, H 4.30; found C 64.02, H 4.13.
MS (FD, 0-20 mA): m/z = 656 (100%, M+).
1H NMR (500 MHz, CDCl3) δ 3.85 (s, 3H, CO2CH3), 4.29 (ddd, 1H, 2-H),), 4.43 (dd, 1H, 2'-HA), 4.58 (dd, 1H, 2'-HB), 5.45 (s, 1H, 10a-H), 5.60 (d, 1H, 4-H), 5.91 (pt, 1H, 3-H), 6.02 (broad m, 1H, 4a-OH), 7.32 (d, 1H, 9-H), 7.34 (d, 1H, 7-H), 7.25–8.05 (m, 15H, 3 C6H5CO); J2,2'A = 5.14, J2’,2'B = 3.15, J2'gem = 12.27, J2,3 = 9.95, J3,4 = 9.61, J7,9 = 1.99 Hz.
Weak NOE between 10a-H (5.45) and 2-H (4.29).
13C NMR (125 MHz, CDCl3) δ 52.22 (CO2CH3), 62.86 (C-2’), 68.07 (C-3), 72.63 (C-2), 77.20 (C-4), 91.86 (C-4a), 93.54 (C-10a), 111.07 (C-9), 111.45 (C-7), 124.38 (C-8), 128,35, 128,57, 128,67 (each m-C6H5CO), 129.30, 129.67, 129.85 (each o-C6H5CO), 131.52 (C-5a), 133.35, 133.93, 134.44 (each p-C6H5CO), 139.65 (C-9a), 144.72 (C-6), 165,50 (3-C6H5CO), 166.22 (2’-C6H5CO), 166.49 (CO2CH3), 168.13 (4-C6H5CO); 1JC10a,H10a = 172.6 Hz.
(2R,3R,4S,4aR,10aR)-2-((benzoyloxy)methyl)-4a,6-dihydroxy-8-(methoxycarbonyl)-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-3,4-diyl dibenzoate 15
The combined fractions of the synthesis of compound
C35H28O13 (656.6): calcd. C 64.02, H 4.30; found C 64.16, H 4.29.
MS (FD, 0-20 mA): m/z = 656 (100%, M+).
1H NMR (500 MHz, CDCl3) δ 3.85 (s, 3H, CO2CH3), 4.52 (dd, 1H, 2'-HA), 4.69 (m, 2H, 2-H, 2'-HB), 5.40 (s, 1H, 10a-H), 5.73 (d, 1H, 4-H), 5.98 (pt, 1H, 3-H), 7.15 (d, 1H, 9-H), 7.34 (d, 1H, 7-H), 7.20–8.20 (m, 15H, 3 C6H5CO); J2,2'A = 5.31, J2,2'B covered, J2'gem = 13.25, J2,3 = 9.95, J3,4 = 9.62, J6,8 = 1.82 Hz.
As expected no NOE between 10a-H (5.51) and 2-H (4.80).
13C NMR (125 MHz, CDCl3) δ 52.43 (CO2CH3), 62.72 (C-2’), 68.48 (C-3), 70.04 (C-4), 70.93 (C-2), 92.99 (C-4a), 94.72 (C-10a), 111.11 (C-7, C-9), 124.95 (C-8), 128,53, 128,58, 128,60 (each m-C6H5CO), 129.96, 129.98 130.12 (each o-C6H5CO), 132.63 (C-5a), 133.39, 133,78, (each p-C6H5CO), 140.14 (C-9a), 145.04 (C-6), 165,14 (CO2CH3), 165.38 (2’-C6H5CO), 166.47 (3-C6H5CO), 166.60 (4-C6H5CO); 1JC10a,H10a = 180.2 Hz.
(2R,3R,4S,4aS,10aS)-2-((benzoyloxy)methyl)-4a-hydroxy-9-methoxy-7-(methoxycarbonyl)-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-3,4-diyl dibenzoate 18
Methyl 3-O-methylgallate
C36H30O13 (670.6): calcd. C 64.47, H 4.51; found C 64.12, H 4.44.
MS (FD, 0-20 mA): m/z = 670 (100%, M+).
1H NMR (500 MHz, CDCl3) δ), 3.84 (s, 3H, CO2CH3), 3.90 (s, 3H, (aromat-OCH3), 4.32 (ddd, 1H, 2-H),), 4.47 (dd, 1H, 2'-HA), 4.59 (dd, 1H, 2'-HB), 5.54 (s, 1H, 10a-H), 5.58 (d, 1H, 4-H), 5.73 (broad s, 1H, 4a-OH), 5.96 (pt, 1H, 3-H), 7.16 (d, 1H, 8-H), 7.49 (d, 1H, 6-H), 7.28–8.03 (m, 15H, 3 C6H5CO); J2,2'A = 5.56, J2,2'B = 2.99, J2'gem = 12.28, J2,3 = 9.79, J3,4 = 9.54, J6,8 = 1.90 Hz.
Weak NOE between 10a-H (5.54) and 2-H (4.32), as well as 8-H (7.16) and aromat-OCH3 (3.90).
13C NMR (125 MHz, CDCl3) δ 52.34 (CO2CH3), 56.43 (aromat-OCH3), 63.26 (C-2’), 68.30 (C-3), 72.73 (C-2), 77.26 (C-4), 91.18 (C-4a), 93.48 (C-10a), 106.81 (C-8), 112.78 (C-6), 123.68 (C-7), 128,40, 2 × 128,62, (each m-C6H5CO), 129,88, 129,95, 130,42 (each o-C6H5CO), 133.21, 133,76, 134.21, (each p-C6H5CO), 133.47 (C-9a), 139.78 (C-5a), 148,54 (C-9), 165,16 (3-C6H5CO), 166.22 (2’-C6H5CO), 166.66 (CO2CH3), 167.95 (4-C6H5CO); 1JC10a,H10a = 171.8 Hz.
(2R,3R,4S,4aS,10aS)-2-((benzoyloxy)methyl)-4a-hydroxy-9-isopropoxy-7-(methoxycarbonyl)-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-3,4-diyl dibenzoate 19
A solution of methyl 3-O-isopropylgallate
C38H34O13 (698.7): calcd. C 65.32, H 4.90; found C 65.31, H 4.91.
MS (FD, 0-20 mA): m/z = 698 (30%, M+), 697 (100%, M + -1).
1H NMR (500 MHz, CDCl3) δ 1.38 and 1.39 (each 3H-d, 2 isopropyl-CH3), 3.84 (s, 3H, CO2CH3), 4.29 (ddd, 1H, 2-H),), 4.47 (dd, 1H, 2'-HA), 4.58 (m, 1H, isopropyl-CH), 4.60 (dd, 1H, 2'-HB), 5.53 (s, 1H, 10a-H), 5.60 (d, 1H, 4-H), 5.61 (broad s, 1H, 4a-OH), 5.96 (pt, 1H, 3-H), 7.18 (d, 1H, 8-H), 7.45 (d, 1H, 6-H), 7.32–8.20 (m, 15H, 3 C6H5CO); Jisoprpyl−Me,CH = 6.06, J2,2'A = 5.33, J2,2'B = 3.20, J2'gem = 12.16, J2,3 = 9.80, J3,4 = 9.55, J6,8 = 1.87 Hz.
Weak NOE between 10a-H (5.55) and 2-H (4.31).
13C NMR (125 MHz, CDCl3) δ 22.18, 22.21 (each isopropyl-CH3), 52.26 (CH3CO), 63.23 (C-2’), 68.36 (C-3), 72.21 (isopropyl-CH), 72.69 (C-2), 77.25 (C-4), 91.08 (C-4a), 93.39 (C-10a), 110.24 (C-8), 112.52 (C-6), 123.64 (C-7), 128,17, 128,20, 128,39 (each m-C6H5CO), 129.87, 129.95, 130.46 (each o-C6H5CO), 132.18, 133.68, 134.11, (each p-C6H5CO), 134.52 (C-9a), 139.92 (C-5a), 147.03 (C-9), 165.16 (3-C6H5CO), 166.22 (2′-C6H5CO), 166.70 (CO2CH3), 167.95 (4-C6H5CO); 1JC10a,H10a = 171.6 Hz.
Methyl 3-O-(2′S,6′S)-4′-benzoyloxy-6′-benzoyloxymethyl-3′-oxo-3′,6′-dihydro-2H-pyran-2-yl)oxy)-4,5-isopropylidenoxybenzoate 22
A solution of methyl gallate acetonide
MS: HR-ESI 575.1548 [M + 1]+ (calcd. for C31H26O11 + 1, 575.1552).
1H NMR (300 MHz, CDCl3) δ 1.67 and 1.69 (each 3H-s, 2 isopropyl-CH3), 3.80 (s, 3H, CO2CH3), 4.66 (pd, 2H, 6'-CH2OBz), 5.23 (sxt, 1H, 6'-H), 5.99 (s, 1H, 2'-H), 7.04 (d, 1H, 5'-H), 7.11* (d, 1H, 6-H), 7.49* (d, 1H, 2-H), 7.30–8.20 (m, 10H, 2 C6H5CO); J5’,6'=3.6, J2,6 = 1.0 Hz.
13C NMR (75 MHz, CDCl3) δ 25.78 and 25.86 (each isopropyl-CH3), 52.07 (CO2CH3), 65.98 (6'-CH2OBz), 71.69 (C-6’), 96.65 (C-2’), 105.25* (C-6), 113.33* (C-2), 120.61 (isopropyl-CMe2), 123.71 (C-1), 132.81 (C-5’), 138.87, 140.38, 142.06, 148.98 (C-4’, C-3, C-4, C-5), 163.7, 165.8, 166.0 (CO2CH3, 2 C6H5CO), 180.38 (C-3’); 1JC2’,H2’ = 174.1 Hz.
Methyl (2S,4aS,10aS)-4a-(benzoyloxy)-2-((benzoyloxy)methyl)-6-hydroxy-4-oxo-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-8-carboxylate 24
The solution of open-chain glycoside
MS (FD, 15 mA): m/z = 534 (100%, M+).
1H NMR (300 MHz, CDCl3) δ 2.88 and 3.17 (each 1H-dd, 3-He and 3-Ha), 3.87 (s, 3H, CO2CH3), 4.43 (m, 1H, 2-H), 4.47 (m, 2H, 2'-HA and 2'-HB), 5.92 (s, 1H, 10a-H), 6.23 (broad s, 1H 6-OH ; exchangeable with D2O), 7.33* and 7.39* (each 1H-d, 7-H and 9-H), 7.20–8.10 (m, 10H, 2 C6H5CO); J2,3a = 11.1, J2,3e = 2.3, J3gem = 15.1 Hz.
13C NMR (75 MHz, CDCl3) δ 41.10 (C-3), 52.20 (CO2CH3), 64.94 (C-2′), 70.27 (C-2), 92.60 (C-10a), 94.31 (C-4a), 111.72* (C-7), 110.86* (C-9), 125.32 (C-8), 128.4–130.6 (C-5a, C6H5CO), 139.03 (C-9a), 144.82 (C-6), 164.40, 2 × 165.94 (CO2CH3, 2 C6H5CO), 192.64 (C-4).
Methyl (2S,4aR,10aS)-2-((benzoyloxy)methyl)-4a,9-dihydroxy-4-oxo-3,4,4a,10a-tetrahydro-2H-benzo[b]pyrano[2,3-e][1,4]dioxine-7-carboxylate 28
Tetrabutylammonium acetate (687 mg, 2.28 mmol) was added to a stirred solution of
MS (FD, 0-20 mA): m/z = 430 (100%, M+).
MS: HR-ESI 431.0973 [M + 1]+ (calcd. for C21 H18 O10 + 1, 431.0973).
1H-NMR (300 MHz, CDCl3) δ 2.69 (dd, 1H, 3-He), 3.10 (dd, 1H, 3-Ha), 3.83 (s, 3H, CO2CH3), 4.19 (m, 1H, 2-H), 4.39 and 4.46 (each 1H-dd, 2'-HA and 2'-HB), 5.26 (s, 1H, 10a-H), 7.17 (d, 1H, 8-H), 7.30 (d, 1H, 6-H), 7.20–8.10 (m, 5H, C6H5CO); J2,2'A = 5.3, J2,2'B = 3.8, J2'gem = 12.0, J2,3a = 12.2, J2,3e = 0, J3gem = 14.5, J6,8 = 1.9 Hz.
13C-NMR (75 MHz, CDCl3) δ 39.73 (C-3), 52.45 (CO2CH3), 65.30 (C-2’), 70.12 (C-2), 90.36 (C-4a), 94.26 (C-10a), 111.63* (C-6), 111.12* (C-8), 124.27 (C-7), 131.94 (C-9a), 139.59 (C-5a), 145.04 (C-9), 166.38, 166.63 (CO2CH3, C6H5CO), 198.32 (C-4); 1JC10a,H10a = 174.0 Hz.
Results
Based on our previous work on the annulation of 2-ketosugars to glycol,
17
the synthesis of linear fused pyran-dioxan-cxclohexan tricycles,
18
of spectinomycin
Reactions of Ulosyl Bromides with Methyl Gallates
Glycosyl bromides are the most widely used glycosyl halides in one-step aromatic O-glycosylations with general yields of less than 60%. But their main advantage is their simplicity of generation as the thermodynamically favored α-anomers. Glycosylation generally results in the inversion of the stereochemistry and yields the β-products. 21
Thus, 2-keto glycosyl bromides, so-called ulosyl bromides, are easily synthetically accessible as well.15,16 The key reaction to construct cercidin analogues was the installation of the benzene moiety in the desired tricycle by aromatic O-glycosylation.21-25 Due to the lower nucleophilicity of phenols, phenolates of methyl gallate
With this knowledge, we first analyzed the conversion of methyl gallate
The same glycosylation reaction of
We also studied the reaction of methyl gallate
The para-selectivity was remarkably increased by blocking one of the two symmetric meta-hydroxyl groups of methyl gallate
By blocking the vicinal hydroxyl groups of methyl gallate
Keto Group Generation in the Pyran Ring
A pronounced structural feature of cercidin
The same reaction sequence already took place in part during the synthesis of compound
All synthesized tricyclic phenol glycosides
Structural Elucidations
The successful conversion of ulosyl bromides
In a first step, the structure of the main product
The 700 MHz 1H NMR spectrum of compound

700 MHz 1H NMR Spectrum of Compound
The group of shifts around 4.5 ppm (see also enlarged section of Figure 3) are part of the exocyclic methylene group 2′-CH2 and of pyran ring proton 2-H. As expected, the 2 geminal protons 2′-HA and 2′-HB are characterized by 2 separate double doublets with large geminal coupling constants (12.48 Hz) and smaller ones of 3.98 and 2.80 Hz, respectively. The latter 2 coupling constants are also present in the multiplet of 2-H, due to its further coupling with 3-H. The shift of 3-H, a pseudo triplet, is located in the middle of the spectrum (5.61 ppm) together with the sharp singlet of anomeric proton 10a-H (5.81 ppm) and the doublet of 4-H (5.93 ppm).
At the lower field of the spectrum, the phenolic part is represented by 2 very closely adjacent doublets of 8-H (7.13 ppm) and 6-H (7.15 ppm), both with the small meta-coupling constant of 2.05 Hz, respectively. At the lowest field of the spectrum, 2 singlets for the aliphatic hydroxyl proton 4a-OH (8.02 ppm) and 9-OH (9.96 ppm) are present, while the aromatic shifts of the 3 benzoyl groups are found as a characteristic broad multiplet between 7.4 to 7.9 ppm.
The assignment of the hydrogen-bearing carbon atoms of the pyran moiety (C-2, C-2′, C-3, C-4, and C-10a) could be easily confirmed via HSQC spectrum.
The 2 very closely adjacent doublets of 8-H (7.13 ppm) and 6-H (7.15 ppm,) could be assigned beyond any doubt by 1H-13C HMBC spectrum analysis (see Figure 4). The hydroxyl proton of 9-OH correlates with C-8 and therefore also determines C-6 and the shifts of the protons H-6 and H-8. Their HMBC correlations, 3JC8,H6 and 3JC6,H8, confirm these findings.

700 MHz 1H-13C HMBC Spectrum of Compound
Because of the sharp singlet shift of anomeric proton 10a-H, the quaternary carbon atom C-9a could be easily assigned on its 3JC9a,H10a correlation as well with its further correlations with 6-H, 8-H and 9-OH) (3JC9a,H6, 3JC9a,8a, 3JC9a,9OH,). Carbon C-4a could be determined in a similar way by two-bond correlation (2JC4a,H10a, 2JC4a,4OH).
The correlations for the 3 quaternary carbon atoms C-5a, C-7 and C-9 were not very pronounced in the HMBC-spectrum, therefore, a 700 MHz 1,1-ADEQUATE spectrum31,32 of compound

Section of the 700 MHz 1H-1,1-ADEQUATE Spectrum of Compound
Compound

Configuration and Conformation of Glycoside

Conversion of Methyl Gallate

Base-Induced Glycosylation of Ulosyl Bromides

Glycosylation of Ulosyl Bromide

Basic Elimination of (a) Tricycle
The magnitude of the anomeric coupling constant 1JC10a,H10a with 172.6 Hz also confirms the β-configuration of compound
As all other compounds were analyzed by NMR spectroscopy in CDCl3, we also performed an additional set of 700 MHz NMR spectra of compound
The structural determination of the derivatives
As expected, the open-chain glycoside
The structure of the derived product of
As expected in the 13C NMR spectra, the shifts of the 4-keto groups of the cercidin analogues
Since the phenol moiety remained unchanged in the synthesis of
In summation: Cercidin analogues
Discussion
Here, the aromatic O-glycosylation of multivalent hydroxyl phenols on α-bromo-2-keto pyranoses is reported for the first time, which yielded 10 different structural analogues of cercidin
Under basic reaction conditions, methyl gallate
Yields of up to 33% were obtained after recrystallization, which could be doubled by blocking one of the phenolic hydroxyl group in meta-position of aglycon
Exclusive phenolic meta-glycosylation could readily be achieved by the reaction of methyl gallate acetonide
The main product
Melting Points, Rotations (CHCl3) and NMR Data (CDCl3) of the Central 1,4-dioxan Ring of Cercidin Analogues

700 MHz, b500 MHz, c300 MHz, dvalue of the precursor 22; chemical shifts in ppm; coupling constants in Hz. In the literature, the only NMR shifts reported for cerdidin
All compounds have well defined melting points, negative rotations and β-configuration. Only glycosides
Consistently, a sharp singlet is found for the anomeric proton 10a-H and the chemical shifts of the quaternary carbons C-4a, C-5a, C-9a, and C-10a are very close. For cercidin analogues
All glycosides described above are novel and structurally related to cercidin
Conclusion
This study reports the total synthesis of the tricyclic pyran-dioxan-benzene ring framework of cercidin
Supplemental Material
sj-docx-1-npx-10.1177_1934578X251346329 - Supplemental material for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides
Supplemental material, sj-docx-1-npx-10.1177_1934578X251346329 for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides by Eckehard Cuny in Natural Product Communications
Supplemental Material
sj-docx-2-npx-10.1177_1934578X251346329 - Supplemental material for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides
Supplemental material, sj-docx-2-npx-10.1177_1934578X251346329 for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides by Eckehard Cuny in Natural Product Communications
Supplemental Material
sj-docx-3-npx-10.1177_1934578X251346329 - Supplemental material for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides
Supplemental material, sj-docx-3-npx-10.1177_1934578X251346329 for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides by Eckehard Cuny in Natural Product Communications
Supplemental Material
sj-docx-4-npx-10.1177_1934578X251346329 - Supplemental material for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides
Supplemental material, sj-docx-4-npx-10.1177_1934578X251346329 for Synthesis of Analogues of Cercidin, an Antimicrobial Compound of Cercidiphyllum japonicum: Glycosylation of Methyl Gallate with Ulosyl Bromides by Eckehard Cuny in Natural Product Communications
Footnotes
Acknowledgments
The author thanks Prof. Dr Michael Reggelin for the opportunity to work in his group, and Dr Schmidts and Dr Fohrer for the measurements of the 700 and 500 MHz NMR spectra and for helpful discussions.
Ethical Considerations
Ethical approval is not applicable for this article because it does not contain any studies with human subjects or animals.
Consent for Publication
There are no human subjects in this article and informed consent is not applicable.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of Human and Animal Rights
Not applicable because this article does not contain any studies with human subjects or animals.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.