
Abstract
Introduction
Macrophages are pivotal in native immunity by responding to pathogens or tissue damage and releasing mediators that recruit and activate immune cells. 1 The recruitment of immune cells to sites of inflammation is vital for host protection against invading pathogens and tissue damage, and disruptions in this procedure can lead to chronic inflammatory diseases, cancer, atherosclerosis, sepsis, and autoimmune disorders. 2 Consequently, regulating macrophage migration has emerged as a potential therapeutic approach for these conditions. Focal adhesion kinase (FAK), an intracellular tyrosine kinase, is involved in cellular functions including cell motility, migration, and survival. 3 The expression of FAK in circulating monocytes is low but rises in segregated macrophages. 4 Decreases in the expression of FAK results in diminution of macrophage migration. 5 FAK activation is induced by lipopolysaccharides (LPSs) and requires the presence of inducible nitric oxide synthase (iNOS). 2 Src, a member of the steroid receptor coactivator family, phosphorylates FAK and disturbs the complex with paxillin and vinculin, thereby activating downstream pathways (eg, mitogen-activated protein [MAP] kinase pathways) that regulate cellular morphology and migration.6,7 The interaction between FAK and paxillin is controlled by vinculin through the extracellular signal-regulated kinase (ERK) pathway. 8 Inhibiting FAK/paxillin interaction in macrophages holds promise for halting cancer progression and inflammatory diseases. 8 Src activation, stimulated by factors such as iNOS/nitric oxide (NO), facilitate FAK phosphorylation and enhances cell migration in LPS-exposed macrophages. 2 Thus, the involvement of Src/FAK signaling in macrophage migration provides insights into the molecular mechanisms underlying macrophage function for potential therapeutics in inflammatory diseases.
Macrophages respond to LPS by releasing inflammatory mediators and the expression of cytokine genes in macrophages is regulated by the activation of various MAPKs. 9 The Akt pathway not only regulates macrophage survival, migration, and proliferation, but also orchestrates the response to different metabolic and inflammatory signals in macrophages. 10 Numerous studies have reported the importance of Akt signaling in inflammation-mediated diseases, such as rheumatoid arthritis, psoriasis, and atherosclerosis.11,12 Furthermore, β-catenin plays a crucial role in cell migration. 13 In macrophages, LPS exposure has been shown to activate β-catenin and promote its nuclear accumulation. 14 An earlier study has shown that introduction to LPS results in the activation of Akt and nuclear translocation of β-catenin upon LPS stimulation, highlighting the significance of these molecules in LPS-induced signaling. 14 LPS induces the activation of downstream MAPKs and nuclear factor-κB (NF-κB) via TLR4, leading to the amplification of inflammatory mediators. 15 Therefore, the inhibition of Src/FAK, MAPK, NF-κB, and β-catenin pathways in macrophages may have potential therapeutic benefits in preventing the progression of inflammatory diseases.
Morin hydrate (MH), 2’3,4’5,7-pentahydroxyflavone (Figure 1A), is a yellow-colored flavonoid isolated from Moraceae species, 16 herbs, red wine, guava, sweet chestnut, and almond. 17 MH has been reported to have several biological activities, such as antioxidant, neuroprotective, anti-inflammatory, antihypertensive, and anticancer properties.16,17 Moreover, it has been shown that MH exhibits antioxidant and anti-inflammatory effects, which can lead to the inhibition of liver injury and fibrosis in animal models.18,19 While several studies have investigated the influence of LPS-induced macrophages on the production of proinflammatory mediators and the release of granular components, limited research has focused on the role of cell migration. Therefore, we speculated whether the Src/FAK, MAPK, and β-catenin signaling pathways may play a role on macrophage migration in response to LPS, which could be recovered by MH.

(A) The chemical structure of MH. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to detect the cell viability of (B) MH (10-200 µM) alone and (C) LPS-induced that had already been pretreated with MH RAW264.7 cells. (D) Optical microscopic view of the morphology of RAW264.7 cells treated with either LPS alone or with MH.
Results
Effects of MH on Macrophage Viability
MH concentrations (10-40 µM) did not affect the viability of macrophages by LPS. However, a 24 h treatment with the highest concentration of MH alone (200 µM) resulted in decreased macrophage viability (Figure 1B). After incubating LPS (1 µg/mL) with MH-pretreated cells for 24 h, they did not show any toxic effect against macrophages (Figure 1C), as evidenced by cell morphology images, as shown in Figure 1D. Therefore, the ideal concentrations of 10 and 20 µM MH were used in subsequent experiments.
MH Reduces the Generation of NO and Expression of iNOS, but has no Effect on COX-2
In Figure 2A and B, MH significantly decreased the NO production in LPS-treated macrophages. The protein and mRNA expression levels of iNOS and COX-2 in LPS-treated macrophages were also determined. RT-PCR was used to detect the effects of MH on the mRNA expression of iNOS and COX-2. As shown in Figure 2C and D, the mRNA expression of iNOS and COX-2 was markedly increased in LPS-induced macrophages. However, MH treatment resulted in a significant inhibition of iNOS mRNA expression, while COX-2 mRNA expression remained unaffected. These results indicate that MH reduces NO production in LPS-treated macrophages by downregulation of iNOS.
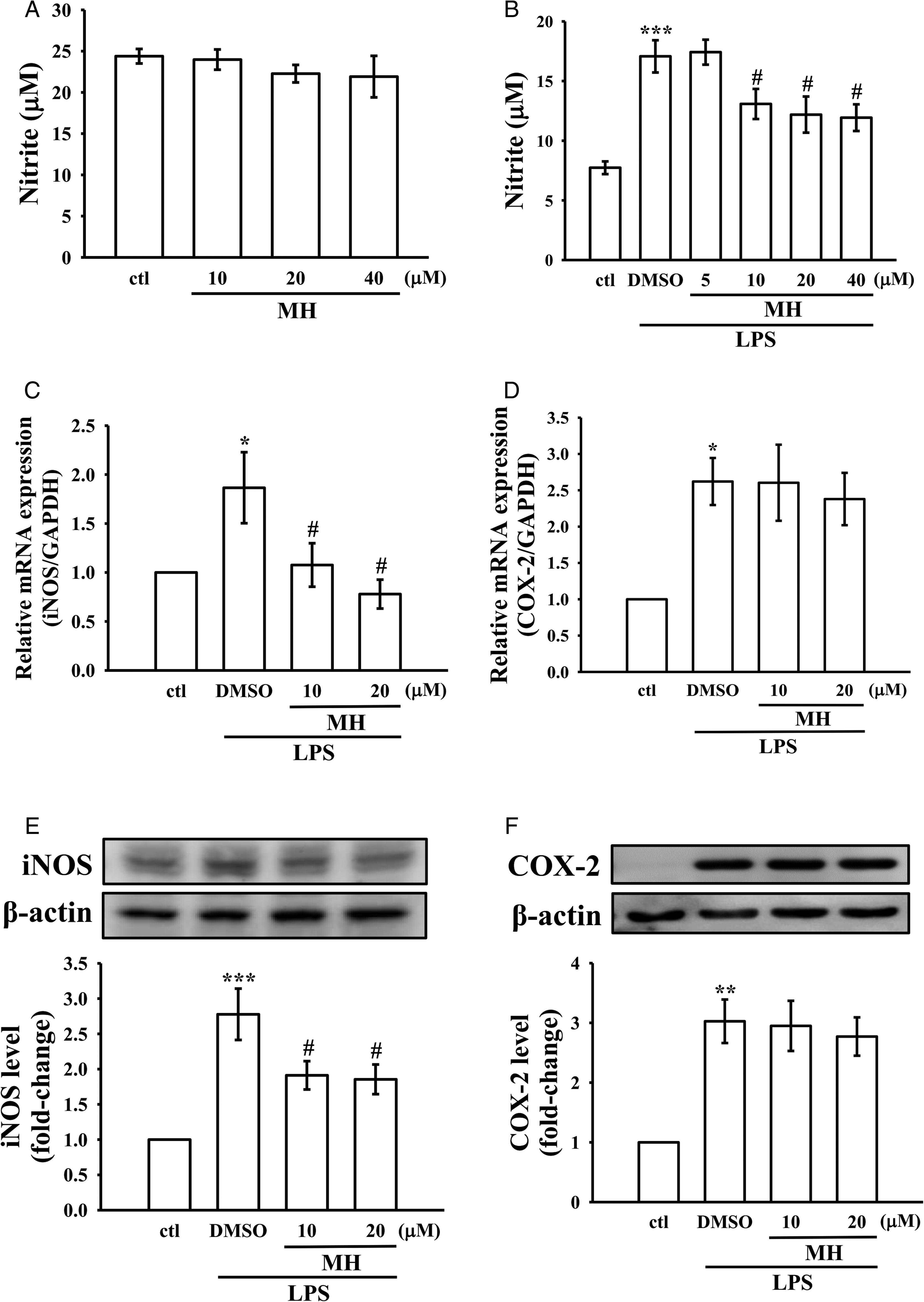
Effects of MH on NO production, and expression of mRNA and protein of iNOS, and COX-2 in LPS-stimulated RAW264.7 cells. (A-B) Cells were pretreated with either MH alone (10-40 µM) or 0.1% DMSO for 1 h and then stimulated by LPS (1 µg/mL) for 24 h. NO was measured using Griess reagent. (C-F) Cells were pretreated with MH (10 and 20 µM) for 1 h and then stimulated by LPS (1 µg/mL) for 24 h. The respective mRNA (C, E) and protein (D, F) levels of iNOS and COX-2 were detected as mentioned in the Materials and Methods section.
For detecting the protein expression levels of iNOS and COX-2, Western blot analysis was performed. Figure 2E and F demonstrates that LPS intensely induced the expression of both iNOS and COX-2 protein, and MH treatment effectively inhibited LPS-induced expression of iNOS protein in a concentration-dependent manner, but it did not affect the inhibition of COX-2 protein. These results are almost consistent with the effect MH has on iNOS and COX-2 mRNA expression.
The Phosphorylation of ERK1/2 and JNK in LPS-Induced RAW264.7 Cells is Inhibited by MH
To explore if MH's inhibitory effect on iNOS involves the MAPK pathway, the effects of MH on the phosphorylation of ERK1/2, p38, and JNK1/2 MAPKs induced by LPS in RAW264.7 cells were evaluated using Western blotting. The results presented in Figure 3A-C demonstrate that LPS stimulation strongly increased MAPK phosphorylation after 1 h, but MH treatment led to a significant reduction in LPS-induced phosphorylation of ERK1/2 and JNK1/2, whereas p38 phosphorylation remained unaffected. These findings suggest that the ability of MH to inhibit the phosphorylation of ERK1/2 and JNK1/2 could contribute to its anti-inflammatory effect in RAW264.7 cells.

MH inhibits LPS-induced phosphorylation of ERK, JNK, and NF-κB p65 in RAW264.7 cells. (A-D) Cells were treated with either 0.1% DMSO or MH (10 and 20 µM) for 1 h, followed by LPS (1 µg/mL) for another 1 h. The cell lysate was used to detect the phosphorylation of (A) ERK, (B) JNK, (C) p38, and (D) NF-κB p65 by immunoblotting assay, as defined in the Methods section. (E) Anti-p65 antibody and FITC-conjugated antirabbit IgG antibody (green) were used to detect the p65 protein using the immunofluorescence staining analysis. 4’,6-Diamidino-2-phenylindole (DAPI) was used to label the nuclei (blue). The confocal microscopy images were captured with the scale bar of 25 μm.
MH Inhibits LPS-Induced Phosphorylation and Nuclear Translocation of p65
This study further investigated whether NF-κB involves MH-mediated anti-inflammatory signaling molecules leading to cell migration. We examined the levels of p65 phosphorylation and its nuclear translocation. Exposure of RAW264.7 cells to LPS significantly increased (P < .001) p65 phosphorylation and its nuclear translocation, as shown in Figure 3D and E. NF-κB translocation into the nucleus in response to LPS was established by immunostaining the cells with p65 antibody in RAW264.7 cells. Moreover, both Western blotting and immunostaining methods showed that MH, particularly at 20 µM, potently inhibited p65 phosphorylation and its nuclear translocation, respectively. This result indicated that MH shows its anti-inflammatory activity mediated by the regulation of both NF-κB p65 and ERK/JNK signaling pathways.
The Activation of Src/FAK Induced by LPS Is Suppressed by MH in RAW264.7 Cells
Src is reported to be activated by inflammatory activators resulting in macrophage migration.20,21 Macrophage migration is significantly influenced by FAK, which plays a critical role as a substrate for Src. 5 Hence, the impact of MH on the activation of Src/FAK was investigated. Western blot analysis presented in Figure 4A and B revealed that RAW264.7 macrophages treated with LPS showed activation of Src and phosphorylation of FAK. Nevertheless, MH strongly inhibited this LPS-stimulated phosphorylation when cells were pretreated with 20 µM, but not effective at 10 µM. In addition, as demonstrated by the results represented in Figure 4C and D, an enhanced activation of Akt/β-catenin phosphorylation was observed in LPS-induced macrophage cells; however, MH only inhibited β-catenin phosphorylation, but not Akt phosphorylation. This indicates the role of β-catenin in MH's anti-inflammatory as well as antimigratory roles in RAW264.7 macrophages.
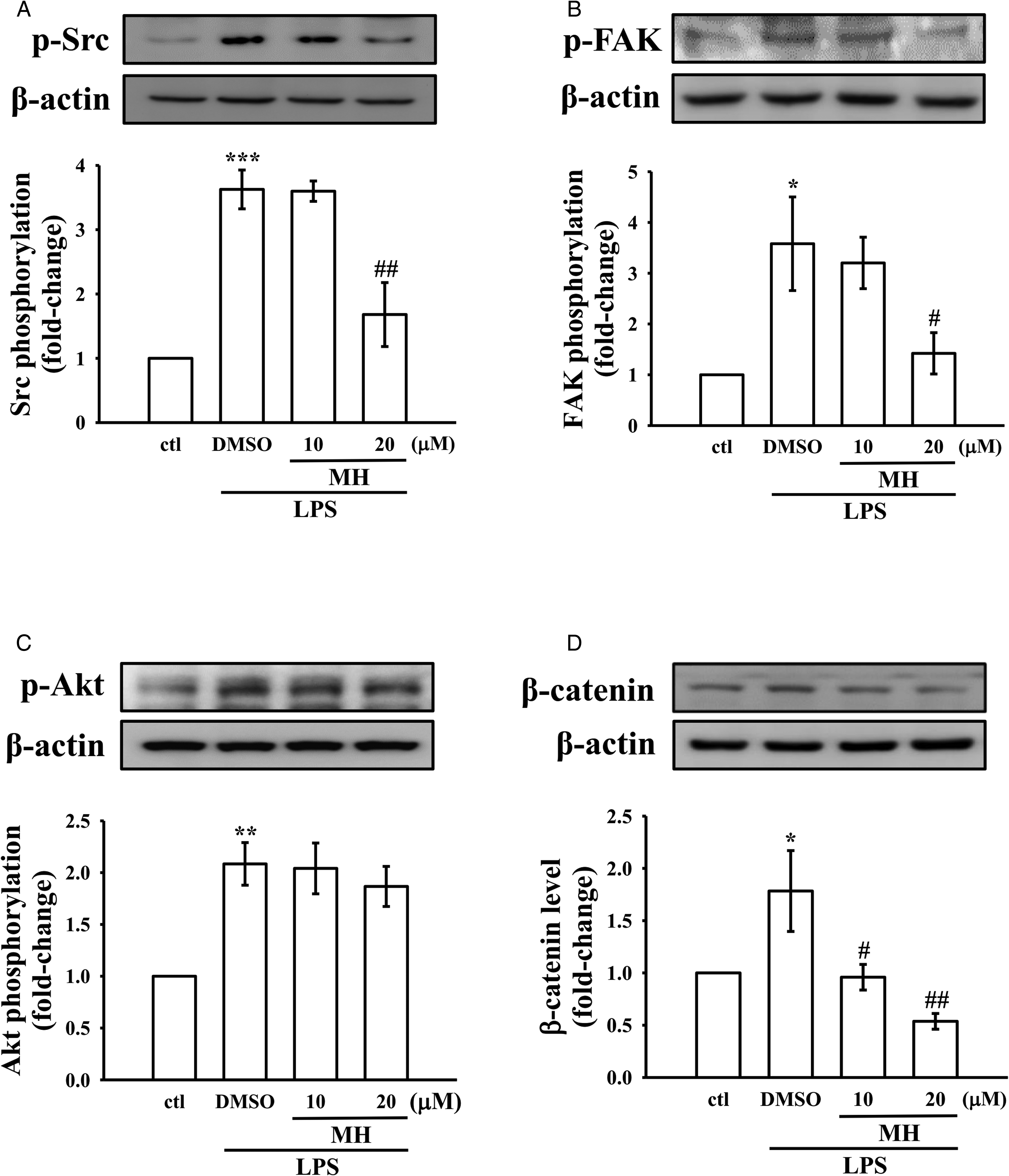
Effects of MH on the phosphorylation levels of Src/FAK and Akt, and on β-catenin expression in LPS-stimulated RAW264.7 cells. (A-D) Cells were treated with either 0.1% DMSO or MH (10 and 20 µM) for 1 h, followed by LPS (1 μg/mL) for 24 h. Cell migration of all the treatment groups was measured by wound healing assay. Phosphorylation of (a) Src, (B) FAK, and (C) Akt, as well as (D) β-catenin expression were detected by immunoblotting.
MH Inhibits LPS-Elicited Macrophage Migration
In macrophages, LPS is a well-recognized stimulator that can induce diverse cellular functions including cell migration. 22 Therefore, the effect of MH on the migratory ability of RAW264.7 cells was assessed before exposure to LPS. As shown in Figure 5, compared to control cells, a significant increase was detected in the migratory ability of LPS-stimulated RAW264.7 cells, and pretreatment with MH hindered the LPS-exerted migration. Based on these results, it can be inferred that the inhibition of Src/FAK-associated signaling pathways might be the mechanism by which MH suppresses macrophage migration.

MH suppresses LPS-activated migration in RAW264.7 cells. Cells were pretreated with either 0.1% DMSO or MH (10 and 20 µM) for 1 h, followed by LPS (1 µg/mL) for 24 h. Cell migration images were taken at 0 and 24 h time intervals.
Discussion
MH has been stated to have several beneficial characteristics, such as antioxidant, neuroprotective, anti-inflammatory, antihypertensive, and anticancer effects.16,17,23 In various animal models, MH has been shown to hinder liver inflammation, injury, and fibrosis through its antioxidant and anti-inflammatory mechanisms.18,19 However, the exact molecular mechanism of MH, particularly on the role of Src/FAK, NF-κB, and MAPK signaling in macrophage migration remains partially understood. Therefore, the objective of this study was to examine the modulatory impact of MH against inflammatory events induced by LPS, as well as to elucidate its underlying mechanisms. According to the findings, MH suppressed the generation of NO induced by LPS and also downregulated the expression of iNOS at both the protein and mRNA levels. MH did not inhibit the expression of COX-2 at either mRNA or protein levels induced by LPS. MH strongly inhibited ERK and JNK phosphorylation, but was not effective on p38. This compound suppressed phosphorylation and subsequent translocation of NF-κB p65 to the nucleus elicited by LPS in macrophage cells. MH exerts its inhibitory effects on macrophage migration through the modulation of iNOS/NO-mediated signaling pathways, specifically the activation of Src/FAK signaling, thereby attenuating the migratory potential of macrophages. Additionally, MH downregulates β-catenin signaling, which further contributes to the suppression of macrophage migration. These signaling pathway modulations collectively result in the prevention of macrophage migration and the control of inflammatory responses. Overall, this work may suggest that MH exerts its effect on the migration of RAW264.7 cells by suppressing Src/FAK and β-catenin, as well as ERK/JNK and p65 signaling pathways.
LPS is the most commonly used endotoxin to induce macrophage activation, and it is a component of the outer membrane of Gram-negative bacteria. 24 The LPS-stimulated RAW264.7 macrophages model has been widely utilized to investigate inflammatory responses, demonstrating the upregulation of inflammatory mediators and proinflammatory enzymes. 25 The results of this study showed that MH suppressed the production of NO and the expression of iNOS at both protein and mRNA levels, but had no significant impact on COX-2. Generally, inflammatory cells that are activated can produce excessive amounts of NO, which may result in inflammatory damage to the target tissue. 26 One of the significant factors contributing to the inflammatory process is the change in the release of NO, mediated by iNOS. 27 COX-2 is an enzyme that can be induced and is responsible for catalyzing the biosynthesis of PGE2. 28 This molecule is known to have a crucial role in the development of various inflammatory conditions, as well as processes such as edema, angiogenesis, invasion, and tumor growth. 29 Several studies have revealed the critical role of COX-2 in the initiation and perpetuation of inflammation. Macrophages can induce COX-2 expression in response to various stimuli, including cytokines and LPS, which can lead to the production of high levels of PGE2 at the site of inflammation. 30 Consequently, a substance that can prevent proinflammatory mediators released by inflammatory cells may have the potential to exhibit anti-inflammatory properties. A previous study demonstrated that taraxasterol, a pentacyclic triterpene, suppressed LPS-induced NO production in RAW264.7 macrophages 31 and significantly decreased upregulation of both iNOS and COX-2 at both mRNA and protein expressions. 32 Similarly, NO production suppressed by MH indicates that it may be a result of iNOS gene expression inhibition. Overall, while the present study demonstrates that MH did not have a significant impact on COX-2 expression, the observed suppression of NO production and iNOS expression suggests its potential anti-inflammatory properties by modulating specific pathways involved in macrophage activation and inflammation. Additionally, the lack of a dose-dependent relationship in the COX-2 expression suggests that MH may have differential effects on different inflammatory mediators.
ERK, p38, and JNK are MAPKs that regulate cells, are activated by unique pathways, and are vital in signaling. 33 These MAPKs play a critical role in regulating cell growth and differentiation, as well as mediating cellular responses to cytokines and stress stimuli. 34 MAPKs exert their effects by phosphorylating downstream targets, including transcription factors and other kinases, thereby regulating the functional responses of cells. 35 A previous study reported that MAPK signaling pathways are known to play a vital role in the biological activities of LPS and can have a positive impact on anti-inflammatory mechanisms. 36 The MAPK signaling pathways are known to regulate the expression of various genes, including iNOS and COX-2. 37 Specifically, both the p38 and ERK signaling pathways have been implicated in the regulation of iNOS production 38 and COX-2 expression in LPS-induced murine RAW264.7 cells, 39 while JNK is responsible for the up-regulation of COX-2 mRNA 40 Moreover, MAPKs have been shown to regulate the expression of iNOS in murine macrophages and human colon epithelial cells. 41 Specific MAPK inhibitors have also been found to suppress the expression of iNOS and COX-2 genes. 42 Furthermore, NF-κB is chiefly responsible for LPS-induced production of NO and PGE2. 43 Therefore, MAPKs and NF-κB are identified as significant targets for molecules with anti-inflammatory properties. In this study, it was detected that MH suppressed the phosphorylation of ERK1/2 and JNK1/2 and also p65 phosphorylation and nuclear translocation in LPS-induced RAW264.7 macrophages. These findings indicate that MH may regulate the expression of inflammatory mediators by modulating the signaling pathways of ERK/JNK and NF-κB.
The Src family kinases (SFKs) are important for inflammatory stimulus-induced macrophage migration, with Src being one of the key members.19,44 Nitrosylation promoted by NO can trigger the up-regulation of Src expression. A study indicated that Src is hardly detectable in normal cells, but LPS stimulation results in a significant increase in Src expression in the RAW264.7 cells. FAK, a substrate of Src, is essential for macrophage migration. 20 Hence, the impact of MH on the activation of Src/FAK was investigated. Consistent with these results, our study detected a significant phosphorylation of both Src and FAK in RAW264.7 cells induced with LPS. A recent study has identified, by using the experimental model of LPS-induced lung injury and macrophage cultures, that FAK is a crucial facilitator of inflammatory responses. 45 They also showed that LPS induces FAK phosphorylation in macrophages, leading to increased formation of the FAK-TAK1 complex and activation of NF-κB, and hence it was suggested that inhibition of FAK in macrophages leads to decreased LPS-induced MAPKs/NF-κB. 45 Miao et al 46 found that inhibitors of FAK and Src proficiently decreased the capability of macrophages to migrate induced by hydrogen sulfide. Another previous study reported that in LPS-stimulated macrophages, the induction of Src led to increased activation of FAK activation and enhanced cell migration. 2 The flavonoid quercetin inhibits macrophage migration by hampering the production of NO and obstructing the FAK pathway induced by LPS. 47 LPS has been reported to induce macrophage migration via the Src-FAK cascade, and this has been shown to be highly reliant on iNOS.2,48 Thus, in the present study, MH may have inhibited macrophage migration induced by LPS via Src/FAK pathways by suppressing the release of NO. This study's results align with those presented in a study conducted by Maa et al. 3 Their research showed that activating the Src/FAK pathways increased the movement of macrophages, whereas butyrate, a short-chain fatty acid, inhibited both the pathways and the migration of RAW264.7 and rat peritoneal macrophages induced by LPS. 3 The results of Gao et al 49 are highly in harmony with our study, as they found that sinomenine, an alkaloid, expressively inhibited macrophage mesenchymal migration by down-regulating the activation of Src/FAK by suppressing the expression of iNOS and production of NO.
β-Catenin has a critical role in regulating cell proliferation, differentiation, and migration. A study found that LPS triggers Akt and causes nuclear accumulation of β-catenin. 14 Therefore, Akt and β-catenin inhibition may be regarded as possible and potential targets for anti-inflammatory drugs. While p-Akt is known to positively regulate β-catenin expression, it is not the sole determinant of β-catenin levels. Other factors and signaling pathways can also influence β-catenin expression independently of Akt. In the present study, the observed decrease in β-catenin expression upon MH treatment suggests that MH may exert its inhibitory effect on β-catenin through alternative mechanisms rather than directly targeting p-Akt. It is possible that MH interferes with other regulatory mechanisms involved in β-catenin stability. Previous studies provided evidence supporting the notion that ERK enhances the signaling cascades involving β-catenin in macrophage cells exposed to LPS 50 and the inhibition of the JNK pathway blocks migration in mammalian cells. 51 This finding aligns with earlier observations in hepatocellular carcinoma, where the activation of ERK was shown to stimulate β-catenin signaling 52 and JNK binds to β-catenin and regulates cell migration. 53 Consistently, in this study, MH significantly inhibited LPS-induced β-catenin expression, indicating the anti-inflammatory role of MH via β-catenin inhibition.
The present study offers significant insights into the anti-inflammatory potential of MH and its impact on macrophage migration. Notably, the investigation reveals a complex interplay between MH, macrophage migration, and various regulatory molecules. However, the intricate relationship between MH and macrophage migration, along with the observed mechanism of cell death at higher concentrations of MH, warrants further in-depth investigations to unravel the involvement of additional related molecules and their interactions with MH and the underlying signaling pathways.
Conclusions
The NF-κB pathway activated by LPS in macrophages leads to increase expression of iNOS and production of NO, which in turn stimulates Src and promotes macrophage migration via the Src-FAK axis. Therefore, the iNOS/NO-mediated signaling of Src/FAK/NF-κB could be a potential target for preventing macrophage migration and controlling inflammatory responses. This study revealed that MH may have potential as an anti-inflammatory agent by targeting the iNOS/NO-mediated Src/FAK/NF-κB pathway (Figure 6) and offers a new approach for regulating inflammatory processes by controlling macrophage migration and Src/FAK/NF-κB signaling.

The schematic illustration shows the effects of MH on cell migration in vitro and subsequent pathways to exert its effect. Upon LPS stimulation, NF-κB becomes activated, which leads to increases in iNOS expression and NO production; NO promotes SFK activation, including Src/FAK. MH decreases the production of NO through NF-κB and ERK/β-catenin signaling cascades, ultimately decreasing macrophage migration via the Src/FAK pathway.
Materials and Methods
Chemicals and Reagents
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA) provided the Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and L-glutamine penicillin/streptomycin. Sigma-Aldrich (St. Louis, MO, USA) was the source of MH, LPS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO). Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) was the source of the purchased anti-iNOS polyclonal antibody (pAb) and β-catenin monoclonal antibody (mAb). COX-2 pAb was obtained from Novus Biologicals (Centennial, Colorado, USA). Antiphospho-c-JNK, antiphospho-Akt pAb, as well as antiphospho-p65, anti-p65, antiphospho-Src, and antiphospho-FAK mAbs were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Antiphospho-p44/p42 ERK and antiphospho-p38 MAPK pAbs were purchased from Affinity Biosciences (Cincinnati, OH, USA). Anti-β-actin mAb was obtained from Proteintech Group (Chicago, IL, USA). Horseradish peroxidase-conjugated donkey antirabbit immunoglobulin G and sheep antimouse IgG antibodies were supplied by Amersham, GE Healthcare Life Sciences (Chalfont, UK). The Hybond™-P polyvinylidene difluoride (PVDF) blotting membranes and enhanced chemiluminescence for Western blotting were provided by GE Healthcare Life Sciences provided.
RAW 264.7 Cell Cultivation
The murine macrophage cell line RAW264.7 (BCRC 60001) was procured from the Bioresource Collection and Research Center (BCRC) located in Hsinchu, Taiwan. The cells were grown using DMEM medium, which was enriched with 10% FBS, 100 U/mL penicillin G, and 100 mg/mL streptomycin. The cells were maintained in a controlled environment with a humidified atmosphere containing 5% CO2 and 95% air at a temperature of 37 °C.
Cell Viability Assay
The RAW264.7 cells were seeded in 24-well culture plates at a density of 5 × 104 cells per well and cultured in DMEM medium containing 10% FBS for 24 h. Upon reaching the required confluence, cells were treated with varying concentrations of MH (10-200 µM) or 0.1% DMSO for 1 h and then subjected to LPS stimulation (1 µg/mL) for 24 h at 37 °C. To assess cell viability, the MTT assay was performed, and the result was expressed as a percentage using the following formula: (absorbance of treated cells/absorbance of control cells) × 100%.
NO Assay
To evaluate the level of NO production, the nitrite/nitrate content, which is a stable end product of NO, was measured using a modified version of a previously described method. 54 RAW264.7 cells were cultured and treated with MH (10-40 μM) or 0.1% DMSO for 1 h, followed by stimulation with LPS (1 μg/mL) for 24 h. The culture supernatants were collected, and Griess reagent was added in equal volumes. The absorbance at 540 nm was read. A standard curve was generated using sodium nitrite.
RT-qPCR
Total cellular RNA was isolated using the NucleoSpin® RNA kit, and Fast SYBR®-Green Master mix was used for the analysis. To normalize the results of the targeted gene, GAPDH was used. The StepOne Real-Time PCR system was utilized for amplification, and the following cycling conditions were used: Hot-start activation at 95 °C for 20 s followed by 45 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 30 s. 55
The used primer details are iNOS, forward 5’-AGCCAAGCCCTCACCTACTT-3’and reverse 5’-GCCTCCAATCTCTGCCTATC-3’; COX-2, forward 5’-CCAGCAC-TTCACCCATCAGT-3’ and reverse 5’- GGGATACACCTCTCCACCAA-3’; and GAPDH, forward 5’-GAACATCATCCCTGCATCCA-3’ and reverse 5’-GCCAGTGAGCTTCCCGTTC-3’. The comparative CT method (2−ΔΔCt) was used for densitometry quantification. 56
Immunofluorescence Staining Assay
RAW264.7 cells were cultured over coverslips at a density of 5 × 104 cells per well. After treatment, the cells were fixed in PBS containing 4% paraformaldehyde for 10 min at room temperature. The fixed cells were then permeabilized with 0.1% Triton X-100 for 10 min and blocked with 5% BSA for 30 min. Primary and secondary antibodies were added and incubated overnight and for 1 h, respectively. After washing thrice with PBS, the cells were stained with 30 μM of 4,6-diamidino-2-phenylindole (DAPI) and mounted using a mounting buffer. The images of the target protein were captured using a Leica TCS SP5 confocal spectral microscope imaging system equipped with either an argon or krypton laser (Mannheim, Germany) for further analysis of the target proteins. This method was described previously and the description of the method partly reproduces their wording. 57
Western Blotting Assay
The experimental design involved pretreating cells with MH (10 and 20 μM) for 1 h, followed by stimulation with LPS (1 μg/mL). Following drug treatment, cellular proteins were extracted using a lysis buffer consisting of 50 mM HEPES, 5 mM EDTA, 50 mM NaCl, and 1% Triton X-100, as previously described. 56 Fifty micrograms of extracted proteins was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a 12% gel and then transferred onto 0.45 μm PVDF membranes by electrophoretic transfer. The membranes were treated with 5% skimmed milk in TBST buffer (10 mM Tris-base, 100 mM NaCl, and 0.01% Tween-20) for 30 min at room temperature. The membranes were then exposed to primary antibodies against various target proteins for 2 h at 4 °C and then incubated with a secondary antibody for 1 h at room temperature. The protein band was detected using a Biolight Windows Application, V2000.01 (Bio-Profil, VilberLourmat, France).
Macrophage Migration Assay
The wound was created over the 80% grown RAW264.7 cells by scratching with a 200 μL sterile pipette tip, as described previously. 57 The debris in the cells was removed with PBS. Subsequently, cells were incubated with 1 μg/mL LPS in the absence or presence of MH (10 and 20 μM) for 24 h. Photographs of the wound area were taken at 0 and 24 h using a light microscope (Nikon, Tokyo, Japan). Cell migration in the wound area was analyzed by quantifying the number of cells using ImageJ 1.47v software. Cell migration was calculated by comparing images of the initial wounded monolayers with corresponding images taken 24 h later.
Statistical Analysis
The data are presented as the means ± SEM. The individual experiment was performed at least 4 times. The statistical analysis was conducted through 1-way ANOVA, and significant differences between the groups were compared using the Newman–Keuls method. A P-value < .05 was considered statistically significant.
Footnotes
Acknowledgments
The authors thank the Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University for providing facilities and technical support.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Ministry of Science and Technology of Taiwan (MOST 110-2320-B-341-001-MY3 and MOST 111-2320-B-038-036-MY3), Taipei Medical University-Taipei Medical University Hospital (108TMU-TMUH-21), and Shin Kong Wu Ho-Su Memorial Hospital (2022SKHAND003 and 2023SKHAND006) financially supported this work.
Ethical Approval
Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.