Spirocarbocyclic natural products have been attracting considerable attention from synthetic organic chemists. This review focused on total syntheses of sesquiterpenoids involving spiro[4.5]decane and spiro[5.5]undecane scaffolds, compiling syntheses of colletoic acid, cubebol, axenol, vetispirene, hinesol, agarospirol, axenol, gleenol, exiguamide, exigurin, erythrodiene, spirojatamol, antroalbocin A, omphalic acid, dactylone, and aplydactonee since 2015.
Sesquiterpenoids are widely distributed in nature and have drawn long-standing attention of synthetic as well as medicinal chemists due to their diverse carbocyclic frameworks along with a variety of biological activities, in which polycyclic structures such as fused, bridged, and spiro carbocyclic rings in confined frameworks are characteristic. These aspects stimulated the interest of synthetic organic chemists as attractive synthetic targets in early days in the total synthesis of natural products.1 The synthetic studies of sesquiterpenoids at that time contributed especially to the development of how to construct polycyclic carbon frameworks, and at the same time to the understanding of the concepts of stereochemistry of carbocyclic structures such as conformational analysis and stereoelectronic effect. Thereafter, the attention of synthetic organic chemists was broadened to the syntheses of natural products having much more complex frameworks with larger molecular weight, which led to the flourishing in total syntheses of natural products in these days. Control of stereochemistry in acyclic compounds and syntheses of macrocyclic compounds were major outcomes. Synthetic efforts for sesquiterpenoids also have been continuing by introducing new concepts and methodologies in organic synthesis. Development in asymmetric synthesis and transformation of functional groups contributed to novel synthesis and improvement of synthetic design. The discovery of new sesquiterpenoids having new structures and biological activities have inspired the interest of synthetic organic chemists.2,3 Spirocarbocyclic sesquiterpenoids have especially attracted much attention due to their intriguing frameworks among a variety of carbocyclic sesquiterpenoids (Figure 1). In the synthesis of spirocyclic sesquiterpenoids, how to install quaternary carbon at the spirocyclic center is the key to synthetic approach. A quaternary carbon center is sterically congested due to the adjacent substituents, which gives difficulty to install especially in enantiomerically enriched form. A variety of methodologies have been reported to synthesize spirocyclic compounds, though mainly spiro-heterocyclic compounds.4–7 On the total syntheses of spirocarbocyclic natural products, Smith and Baxendale8 presented in 2015 a comprehensive review according to the key reaction to construct spirocyclic rings. Since then, much results have been accumulated in the total syntheses of spirocarbocyclic sesquiterpenoids, which exemplifies the lasting interest and importance of the synthetic study in the area. In this review, some selected works on the total syntheses of spirocarbocyclic sesquiterpenoids from the last eight years are compiled specifically focusing on sesquiterpenoids involving spiro[4.5]decane and spiro[5.5]undecane scaffolds.
Representative frameworks of spirocyclic sesquiterpenoids.
Syntheses of Sesquiterpenoids Involving spiro[4.5]decane Scaffolds
Colletoic acid (14), a member of the acorane family, was isolated from the endophytic fungus Colletotrichum gloeosporioides,9 and is a potent and selective 11β-hydroxysteroid dehydrogenase (11β-HSD1) inhibitor. Rivas et al disclosed the improved synthesis of colletoic acid (14) and its derivatives to evaluate the inhibitory activity of 11β-HSD1, which regulates intracellular cortisol levels (Figure 2).10 The construction of spiro[4.5]decane system began with β,g-unsaturated acid 6. Reaction with (R)-4-benzyl-2-oxazolidinone (7) led to Evans’ amide 8 via a mixed anhydride with pivaloyl chloride. Alkylation with iodide 9 proceeded in gram quantities with high enantioselectivity (98% ee) and diastereoselectivity (dr > 20:1). After removal of the benzyl-2-oxazolidinone moiety with NaBH4 and subsequent protection of resulting primary alcohol, vinyliodide 11 was subjected to intramolecular Heck-type cyclization to afford spirocyclic compound 12 as a single diastereomer. It is presumed that the exo-transition state is favored to avoid clashing interactions between the palladium complex and the isopropyl group in the transition state. The unsaturated ketone 13 has already been transformed to (+)-colletoic acid (14) by Rivas and co-authors.11 Over 60 derivatives were synthesized from the enone 13, and their 11β-HSD1 inhibitory activities were evaluated.
Formal synthesis of (+)-colletoic acid (14) by Rivas et al.10
Alternative formal synthesis of (+)-colletoic acid (14) by List et al is described in Figure 19.
Cubebol (24) is a main constituent of cubeba oil from berries of Piper cubeba. Its synthesis by Derksen et al has started from tetrahydrocarvone (15) employing tandem photocyclization/1,4-sigmatropic rearrangements to construct a cubebane-type carbon framework. Introduction of a cyano group to tetrahydrocarvone (15) followed by the Robinson annulation led to decalones 17 and 18 (Figure 3).12 Cross-conjugated dienone 19, a precursor for photocyclization, was obtained through dehydration of the major β-cyanodecalone 18 followed by Saegusa reaction. Irradiation of the dienone 19 by UV-C afforded tricyclic enones 20 and 21. Catalytic hydrogenation of the minor diastereomer 21 led to tricyclic ketone 22. After the protection of the carbonyl group as an acetal to avoid the cleavage of the cyclopropane ring, the cyano group was removed successfully by the reductive elimination with potassium in HMPA, maintaining the tricyclic carbon framework.13 Deprotection of the acetal followed by stereoselective addition of methyllithium provided cubebol (24).
(–)-Axenol (34), an enantiomer of natural product isolated from a New Zealand Sponge of the Genus Eurypon, was synthesized by Ozaki and Kobayashi, employing stereoselective allylic substitution to construct the quaternary carbon center, in which ring-closing olefin metathesis reaction (RCM) to close the spiroring was employed (Figure 4).14 (–)-Menthol (25) was led to ketone 27 via 4 known steps.15 Peterson olefination of the ketone 27 provided (Z)-olefin 28, which was transformed to picolinate 30 after a series of reactions, reduction with DIBAL-H, esterification, and protection. The SN2′ substitution of the copper ate complex 31, which was originally developed by Kobayashi et al,16 proceeded with perfect stereocontrol through an equatorial attack of the reagent 31 to afford bis-olefin 32. RCM reaction furnished (–)-axenol (34) in high yield.
Synthesis of (–)-axenol (34) by Ozaki and Kobayashi.14
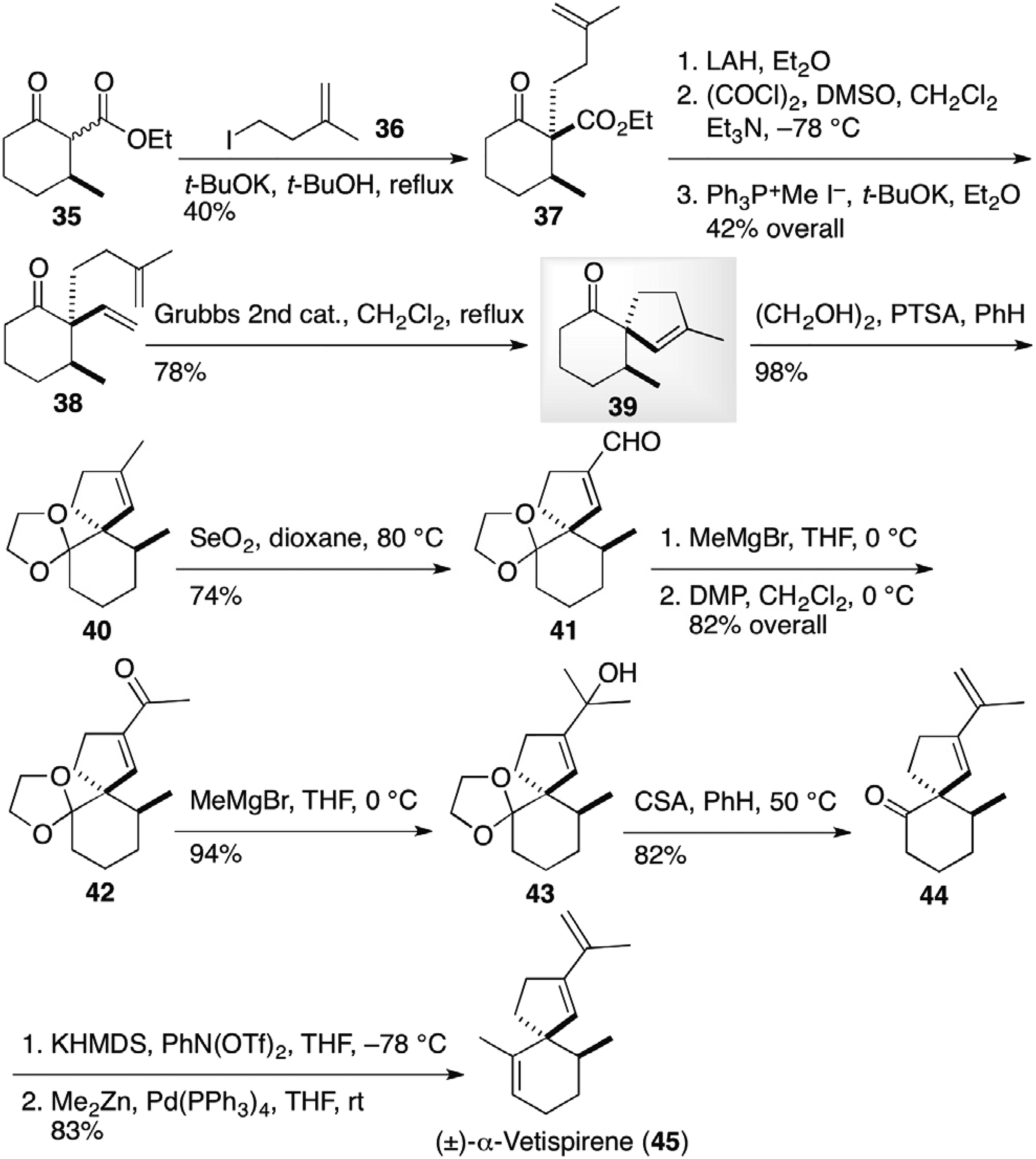
Mehta and co-authors17 delineated syntheses of a variety of spiro[4.5]decane-based sesquiterpenoids from common intermediate 43 derived via RCM reaction (Figures 5 to 7). A quaternary carbon center of the spirocyclic ring was introduced by alkylation of cyclic β-ketoester 35 with iodide 36 to provide stereoselectively alkylation product 37, in which the ester group was transformed to a vinyl group by LAH reduction, Swern oxidation and Wittig condensation to afford bis-olefin 38 (Figure 5). RCM reaction delivered spiro[4.5]decane 39. After the protection of the carbonyl group, allylic oxidation of a vinyl methyl group led to unsaturated aldehyde 41. The introduction of methyl groups sequentially led to tertiary alcohol 43, a key precursor for the further synthesis of spirocyclic sesquiterpenoids. Treatment of the alcohol 43 with CSA resulted in dehydration along with deprotection to furnish keto-diene 44. The olefinic methyl group was installed by the palladium-catalyzed coupling of enoltriflate with dimethylzinc to complete the synthesis of racemic α-vetispirene (45).
Syntheses of hinesol (53), agarospirol (54), axenol (34), and gleenol (56) by Mehta et al.17
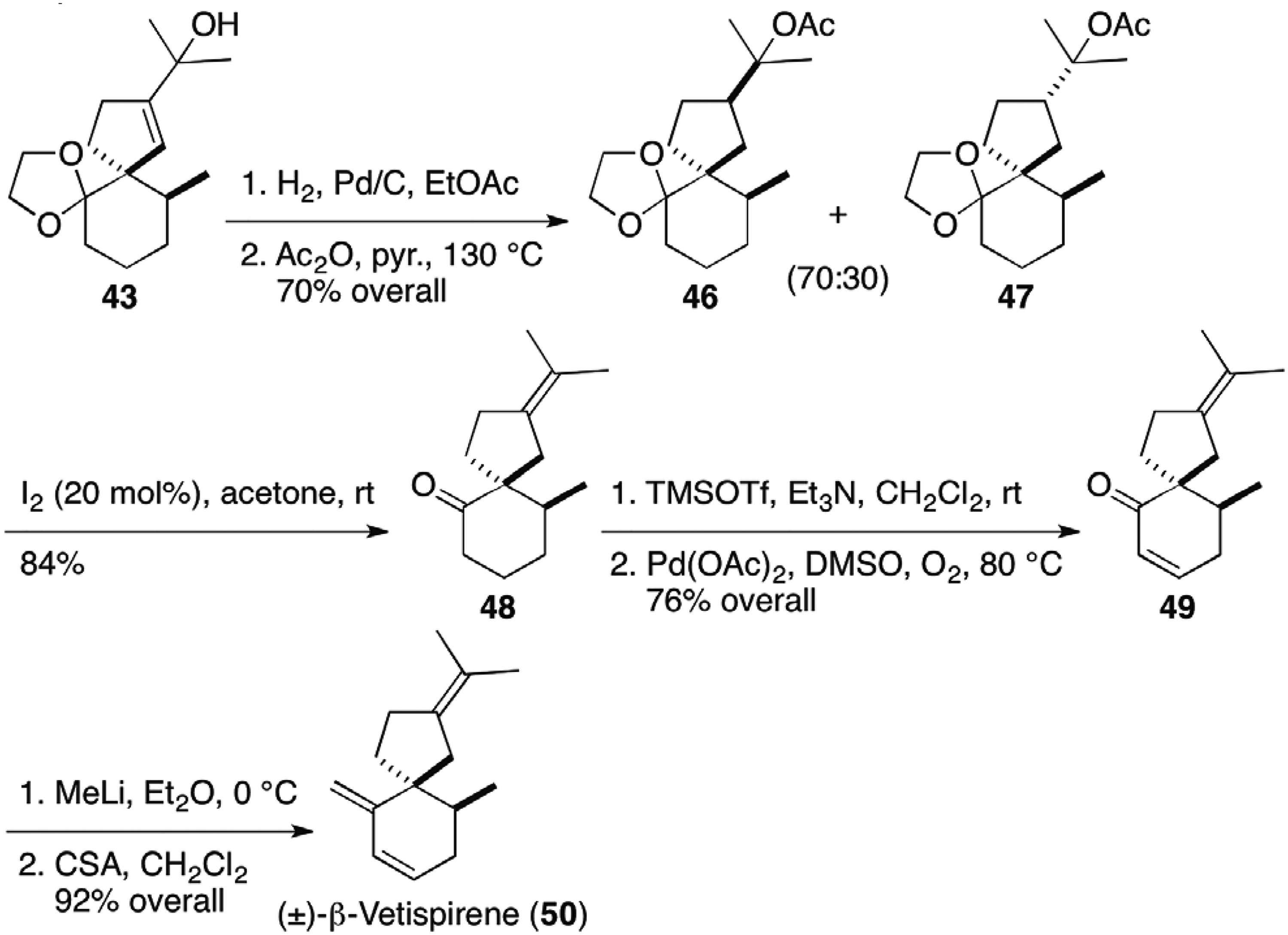
The α-vetispirene (45) synthesis in Figure 5 was extended to the synthesis of β-vetispirene (50) from the intermediate 43 (Figure 6).17 Catalytic hydrogenation followed by acetylation at high temperature led to diastereomeric tertiary acetate 46 and 47, which were treated with a catalytic amount of iodine at elevated temperature to afford isopropylidene-ketone 48. After Saegusa oxidation, resulting enone 49 was transformed to racemic β-vetispirene (50) by methyllithium addition and subsequent dehydration.
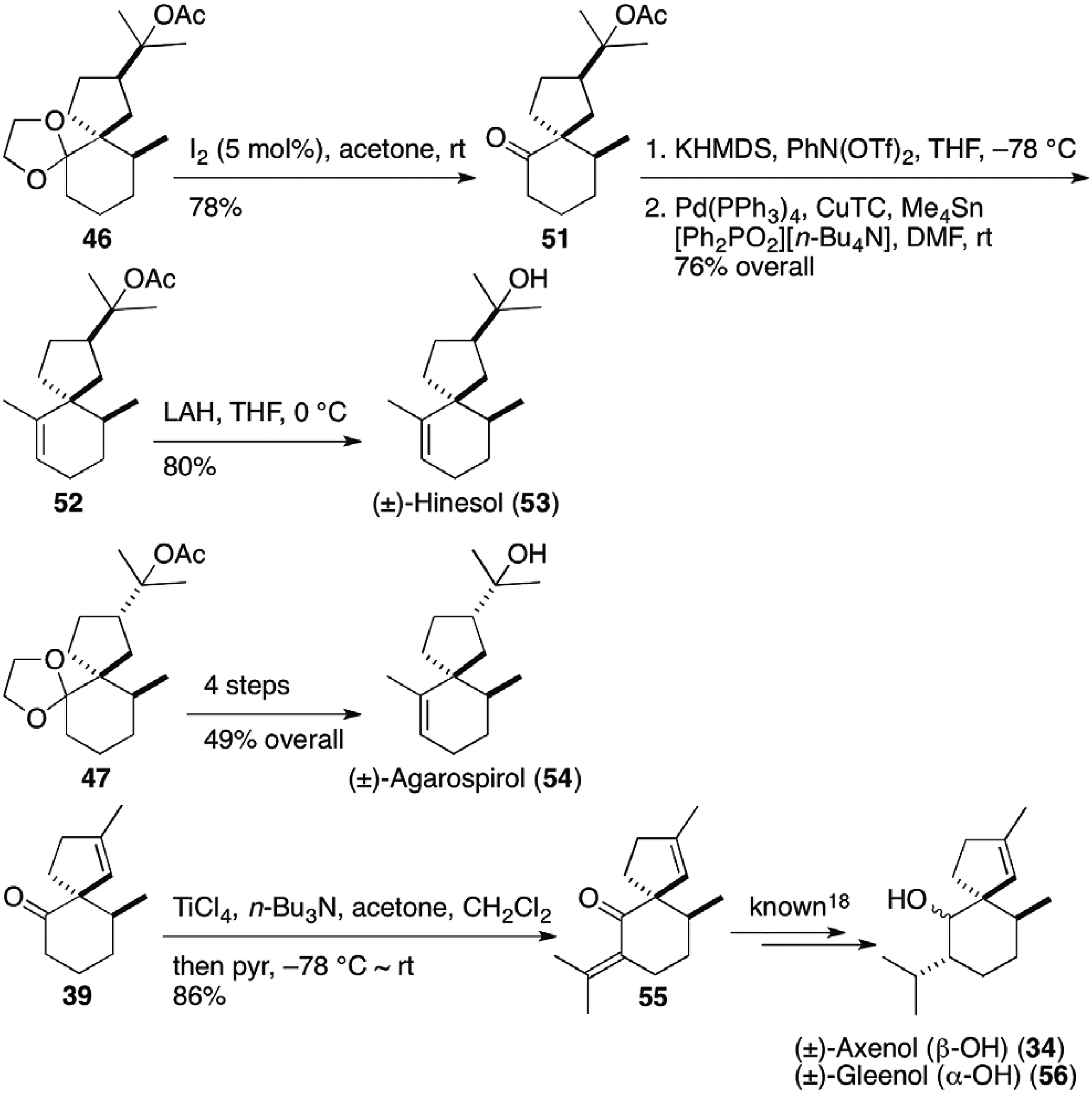
Furthermore, deprotection of the acetal group of the major diastereomer 46 in Figure 6 with a catalytic amount of iodine at room temperature provided keto-acetate 51, which was transformed to hinesol (53) via palladium-catalyzed coupling reaction of enol triflate to install an olefinic methyl group (Figure 7).17 Starting from the minor diastereomer 47, agarospirol (54) was furnished by the same sequence of reactions. In addition, Lewis acid promoted aldol condensation of the intermediate 39 in α-vetispirene (45) synthesis in Figure 5 gave isopropylidene-ketone 55, a precursor for the syntheses of axenol (34), and gleenol (56).18
Exiguamide (68), isolated from the marine sponge Geodia exigua, is a nitrogen-containing sesquiterpenoid and reported to exhibit the micromere formation of sea urchin embryos and subsequent spicule formation at a quite low concentration19 Takikawa et al completed the first total syntheses of both enantiomers of exiguamide (68) via stereoselective intramolecular cyclopropanation and stereoselective homoconjugate addition of an azide to cyclopropyl ketone as the key steps (Figure 8).20 Elongation of side chain was carried out by Heck reaction of enol triflate 58 derived from (+)-carvomenthone (57) to afford unsaturated ester 59, which was reduced with diimide regioselectively to provide γ,d-unsaturated ester 60. Condensation with the anion of methylphenlsulfone to β-ketosulfone 61 followed by a diazo-transfer reaction led to α-diazo-β-ketosulfone 62. The intramolecular cyclopropanation of the resulting diazoketosulfone 62 with the copper catalyst proceeded stereoselectively on the more open space to furnish cyclopropylketone 63 having cubebane scaffold. Homoconjugate addition of sodium azide to doubly activated cyclopropane ring proceeded with the aid of magnesium triflate to present spirocyclic β-ketosulfone 64. The olefinic methyl moiety was introduced by Negishi coupling to furnish vinylsulfone 66. Reduction of the vinylsulfone 66 was successful only with magnesium powder to result in removal of the sulfonyl group with concomitant reduction of the azide to amino group affording amine 67, which was led to (+)-exiguamide (68) after N-formylation. Synthesis of (–)-enantiomer was also achieved from (–)-carvomenthone.
Synthesis of (+)-exiguamide (68) by Takikawa et al.20
Exigurin (80) was isolated with (–)-10-epi-axisonitrile-3 (76) from the marine sponge Geodia exigua and involves terpene and amino acid units connected through an amide linkage.21 Shortly after the synthesis of exiguamide (68) by Takikawa et al, the first synthesis of exigurin (80) was presented by Hosokawa et al, in which a similar strategy via ring opening of cubebane scaffold in the synthesis of exiguamide (68) in Figure 8 was employed independently to construct cubebane scaffold (Figure 9).22 The amino acid moiety was installed by Ugi five-center four-component reaction.23 Introduction of a formyl group to (+)-menthone (69) accompanied epimerization of the isopropyl group to give hydroxymethylene ketone 71 as a major inseparable diastereomer. Hydride reduction gave a separable mixture of diastereomers 72 and 73. Authors proposed conjugate addition of hydride to deprotonated exo-cyclic olefin followed by β-elimination and subsequent 1,2-reduction of the carbonyl group. Elongation of the side chain by Johnson-Claisen rearrangement of the major allyl alcohol 73 led to γ,d-unsaturated ester 60, the same intermediate used for exiguamide (68) synthesis in Figure 8. After transformation to β-ketosulfone 61 followed by diazotization to diazosulfone 62, intramolecular cyclopropanation with the copper catalyst 75 effected cyclopropyl ketone 63. Opening of the cyclopropane ring of 63 with sodium azide was successful with the aid of magnesium perchlorate in the presence of a phase transfer catalyst in DMF at elevated temperature. After the conversion of the resulting ketosulfone 64 to vinyl triflate 65, the olefinic methyl group was introduced by palladium-catalyzed coupling with trimethylboroxine to furnish vinylsulfone 66. Removal of the sulfonyl group with samarium iodide proceeded with concomitant reduction of the azide to the amino group to afford amine 67, which was led to exiguamide (68) by N-formylation and subsequently to (–)-10-epi-axisonitrile-3 (76) by dehydration with phosgene. Finally, the Ugi reaction with sarcosin (77) and formaldehyde for the introduction of amino acid moiety was successful to complete the (+)-exigurin (80) synthesis. It was speculated that the discrepancy of the sign of specific rotations between synthetic and natural exigurin (80) was a consequence of inaccuracies in measuring specific rotations since the sign of rotation of (–)-10-epi-axisonitrile-3 (76) was the same with the natural one.
Synthesis of (+)-exigurin (80) by Hosokawa et al.22
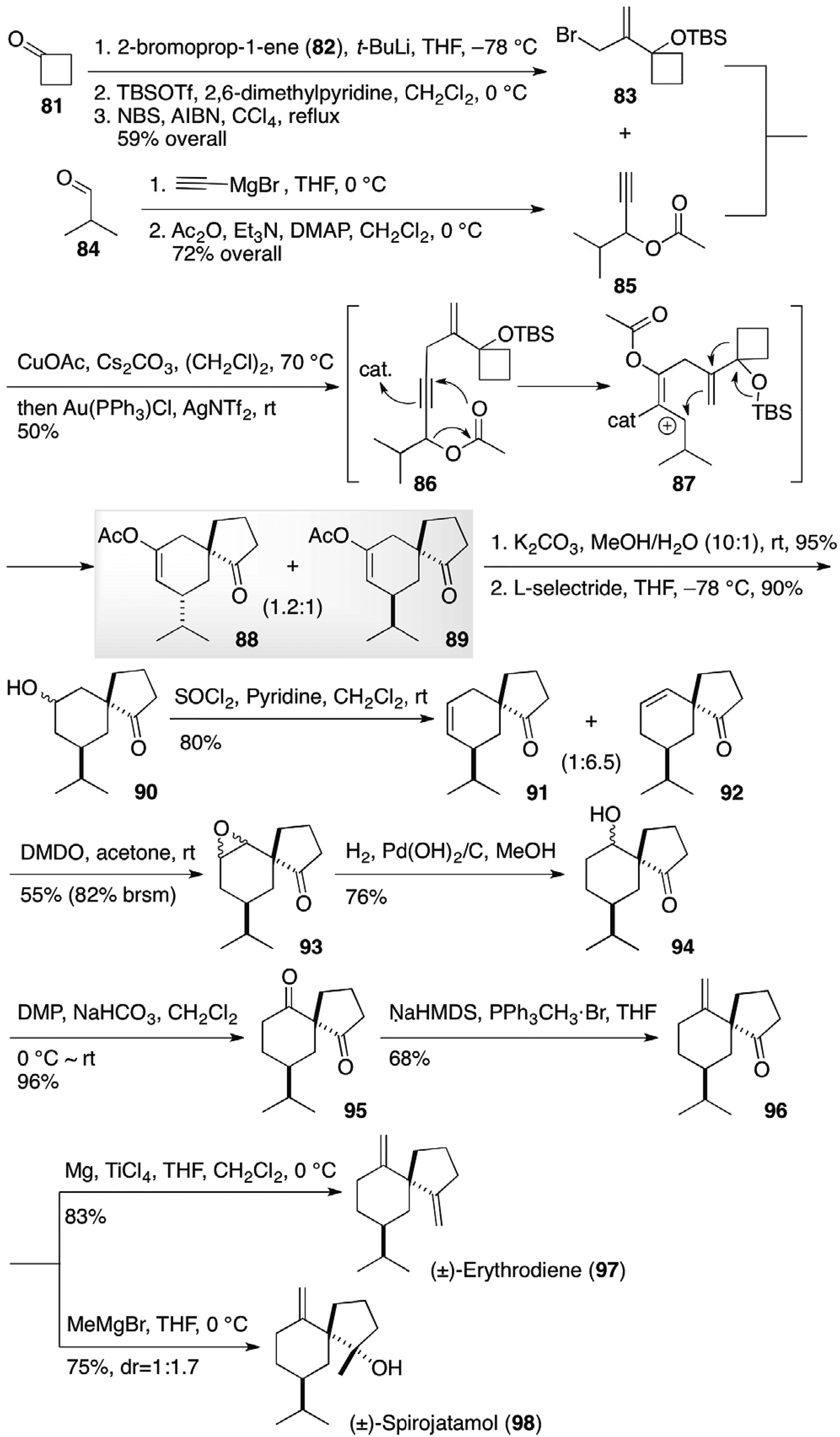
(–)-Erythrodiene (97) and (+)-spirojatamol (98) were isolated from the roots of the Caribbean soft coral Erythropodium caribaeorum and the Indian plant Nardostachys jatamansi, respectively, and have opposite absolute configurations for their quaternary carbon centers.24,25 Wang et al completed the total syntheses of racemic erythrodiene (97) and spirojatamol (98) by constructing the spiroyclic ring through a tandem Castro-Stephens coupling/1,3-acyloxy shift/cyclization/semipinacol rearrangement sequence26 (Figure 10).27 Addition of 2-lithioprop-1-ene to cyclobutanone 81 followed by protection of the tertiary alcohol and subsequent allylic bromination provided allylbromide 83. Propargyl acetate 85 was obtained by the addition of ethynylmagnesium bromide and subsequent acetylation. The Castro-Stephens coupling reaction of the allylbromide 83 with propargylic ester 85 in the presence of copper catalyst led to coupling product 86, which was transformed sequentially to spirocyclic compounds 88 and 89 via 1,3-acyloxy shift/cyclization/semipinacol rearrangement with the aid of gold and silver salts. Hydrolysis of the minor product 89 followed by hydride reduction afforded keto-alcohol 90, which was converted to regioisomeric keto-olefins 91 and 92. Epoxidation of the major keto-olefin 92 and subsequent epoxide opening by a catalytic hydrogenation led to a diastereomeric mixture of keto-alcohol 94, which was transformed to keto-olefin 96 after oxidation and Wittig condensation. Racemic erythrodiene (97) was obtained by methylenation,28 while spirojatamol (98) was obtained by methylation.
Syntheses of erythrodiene (97) and spirojatamol (98) by Wang et al.27
Antroalbocin A (107) isolated from the higher fungus Antrodiella albocinnamomea belongs to a cedrane family, in which bicyclo[4.5]decane scaffold is embedded. It exhibits antimicrobial activity against Staphylococcus aureus.29 Kalesse et al started synthesis from conjugate addition of known diketone 10030 with methyl vinyl ketone to provide triketone 101 as a single diastereomer (Figure 11).31 The enantioselective intramolecular aldol condensation of the triketone 101 in the presence of organocatalyst 10232,33 under sonication effected enone 103 in 99.6% ee after two recrystallizations. Reduction with sodium borohydride led to alcohol 104 regio- and stereoselectively. Construction of the cedrane framework was accomplished by irradiation with medium pressure mercury lamp in dichloromethane to afford unstable keto-alcohol 106 by photo-equilibrated deconjugation to β,g-unsaturated ketone 105 followed by 1,3-acyl shift. Deconjugated enone 105 was not observed. Since the resulting 1,3-shift product 106 soon equilibrated back after a few minutes by daylight to the starting enone 104, the reduction was carried out rapidly with DIBAL-H to furnish (−)-antroalbocin A (107).
Synthesis of (−)-antroalbocin A (107) by Kalesse et al.31
Omphalic acid (121) was isolated from the Colombian liverwort Omphalanthus filiformis,34 and involves spiro[4.5]decane scaffold. Zhang et al synthesized for the first time through a Diels-Alder reaction leading to spiro[5.5]undecane scaffold followed by ring contraction to spiro[4.5]decane scaffold (Figure 12).35 A Diels-Alder reaction is suitable to introduce a quaternary carbon center and to install a spirocyclic scaffold at the same time. Silyl enol ether 109 was obtained by conjugate addition to cyclohexenone 108 followed by trapping of resultant enolate. Mukaiyama aldol reaction led to hydroxyketone 110, which was transformed to exo-methylene ketone 111 by mesylation and subsequent β-elimination. Since the exo-methylene ketone 111 was active to dimerize by hetero-Diels-Alder reaction at room temperature, the Diels-Alder reaction was carried out with 3 equivalents of isoprene 112 in the presence of an equivalent amount of Me2AlCl at −78 °C to afford spirocyclic compound 113. The use of a catalytic amount of Me2AlCl resulted in homodimerization of the exo-methylene ketone 111. The double bond of the spirocyclic compound 113 was cleaved by dihydroxylation followed by Criegee oxidation to give diketo-aldehyde 115, while ozonolysis of 113 was not successful. The aldol reaction of the diketo-aldehyde 115 under the acidic condition led to spiro[4.5]enone 116, which was transformed to tricyclic enone 117 through catalytic hydrogenation and subsequent aldol reaction. Since the catalytic hydrogenation of the enone 117 gave undesired 10α-isomer, the double bond was deconjugated leading to ketal 118, which was reduced either by catalytic hydrogenation or Co-catalyzed HAT reduction36 to afford desired 10β-diastereomer 119. After the transformation of the ketone 119 to enol triflate 120, the total synthesis of omphalic acid (121) was accomplished by palladium-catalyzed carboxylation. Synthetic racemic omphalic acid (121) was resolved by derivatizing with (S)-1-(4-bromophenyl)ethan-1-ol and the absolute configuration of natural (+)-omphalic acid (121) was determined to be 5R,7R,10S.
Synthesis of (±)-omphalic acid (121) by Zhang et al.35
Alternative formal synthesis of omphalic acid (121) by List et al is described in Figure 19.
Syntheses of Sesquiterpenoids Involving spiro[5.5]undecane Scaffolds
The brominated chamigrene sesquiterpenes constitute a large subclass of bromocyclohexane-containing natural products. (–)-Dactylone (128) was isolated with aplydactone (144) from the sea hare Aplysia dactylomela37 and suppresses phenotype expression at noncytotoxic doses in human lung, colon, and skin tumor cell lines. Snyder et al achieved syntheses of dactylone (128), 10-epi-dactylone (131), and related bromochamigrenes 126 and 127 (Figure 13).38 Considering the difficulty to introduce a bromine atom at the neopentyl position at the late stage of the synthetic sequence, β-bromocyclohexanone 122 having a bromine atom at the neopentyl position was chosen as a starting material. The Diels-Alder reaction of isoprene 112 and α-methylenecyclohexanone 123, which was prepared in 3 steps from β-bromocyclohexanone 122, proceeded in the presence of the stoichiometric amount of dimethylaluminum chloride to afford chamigrene scaffolds 124 and 125 in 1:4.9 diastereoselectivity. Methylenation of the minor diastereomer 124 led to (±)-10β-bromochamigrene (126), which was transformed to (±)-10-bromo-3,4-dihydroxy-β-chamigrene (127) by dihydroxylation and subsequently to (±)-dactylone (128) by oxidation and dehydration. The same protocol was applied to the major diastereomer 125 to afford 10-epi-dactylone (131).
Syntheses of (±)-dactylone (128), (±)-epi-dactylone (131), and related bromochamigranes by Snyder et al.38
Aplydactone (144), which was isolated with dactylone (128), has a unique and characteristic polycyclic strained structure embedding [2]-ladderane scaffold in bromochamigrane framework.37 Although the [2]-ladderane structure was anticipated to be formed by intramolecular [2 + 2]-photocycloaddition of dactylone (128), the report on the stability of dactylone (128) on photoirradiation in the original paper led Meier and Trauner to introduce the ladder like cyclobutane scaffold at the initial stage of their synthetic project (Figure 14).39 Ozonolysis of olefinic ester 132 followed by allylation gave homoallyl alcohol 133, which was cleaved with ozone, reduced, and protected to provide γ-butyrolactone 134. Introduction of a methylene group to the lactone 134 followed by reduction afforded diol 135, which was protected as bis-benzylether and subsequently brominated to obtain bromide 136. Coupling of the bromide 136 and sulfone 137 followed by hydrolysis of the acetal and subsequent β-elimination led to cyclopentenone 138. The first cyclobutane ring of the [2]-ladderane scaffold was installed by photochemical [2 + 2]-cycloaddition at low temperature to give ketone 139. The second cyclobutane ring was installed by a ring contraction of the cyclopentanone ring through photochemical Wolff rearrangement. Diazoketone 141 was prepared via enaminone 140 by the treatment with 4-acetamidobenzenesulfonyl azide (p-ABSA) in one pot operation. Irradiation to the diazoketone 141 provided ladder-like compound 142 in a 3:1 diastereomeric ratio. The introduction of a methyl group proceeded from the less hindered α-face to give ester 143.
Syntheses of the intermediate for the total synthesis of (±)-aplydactone (144) and (±)-epi-aplydactone (153) by Meier and Trauner.39
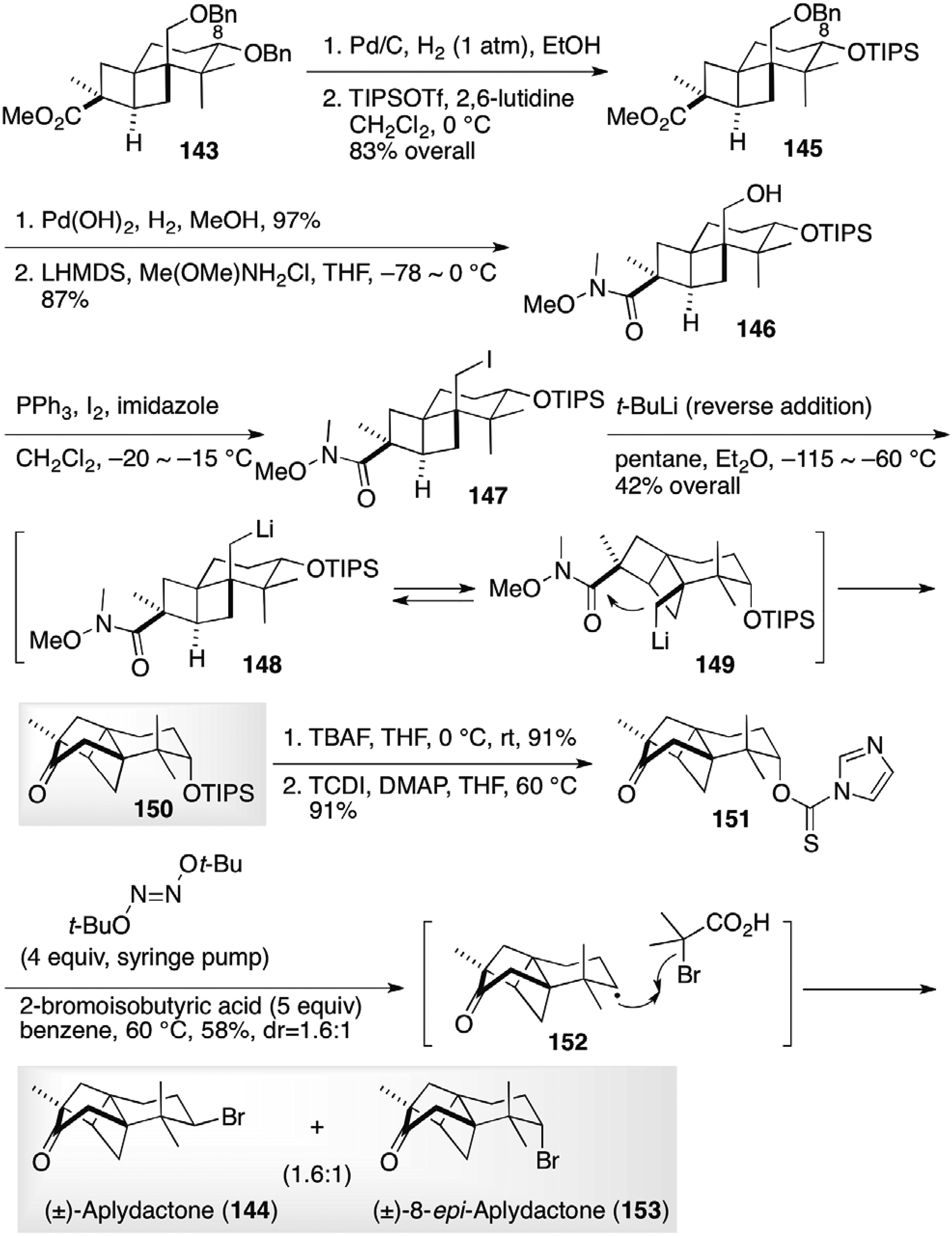
With the ladderane compound 143 in hand, the closure of the six-membered ring to the target aplydactone (144) was set forth (Figure 15).39 Selective exchange of the protecting group at C-8 gave ester 145. Deprotection of the benzyl ether and subsequent transformation to Weinreb amide afforded amide 146, which was converted to labile iodide 147. Ring closure to the full carbon skeleton of aplydactone (144) was carried out by treatment of the iodide 147 with t-BuLi. Metal-halogen exchange to 148 followed by ring flipping to 149 brought the nucleophilic site into proximity to the electrophilic Weinreb amide to close the cyclohexane ring to ketone 150. Substitution of the secondary hydroxyl group at C-8 with bromine was a formidable task due to the steric hindrance at the neopentyl position and a ring contraction by a Wagner-Meerwein rearrangement by a carbocation at C-8, which was effected by a radical protocol. After deprotection of the TIPS ether of the ketone 150, thiocarbonylimidzolide 151 was obtained. Combination of di-t-butylhyponitrile as a radical initiator and 2-bromoisobutyric acid as a bromine source was optimal for the successful introduction of the bromine atom at C-8 to complete the total synthesis of racemic aplydactone (144) and 8-epi-aplydactone (153). Di-t-butylhyponitrile was favorable for low-temperature radical initiation preventing the elimination of the thiocarbonylimidazoyl group, and 2-bromoisobutyric acid was used for better reactivity along with easy removal of residue from the reaction mixture.
Completion of syntheses of (±)-aplydactone (144) and (±)-epi-aplydactone (153) by Meier and Trauner.39
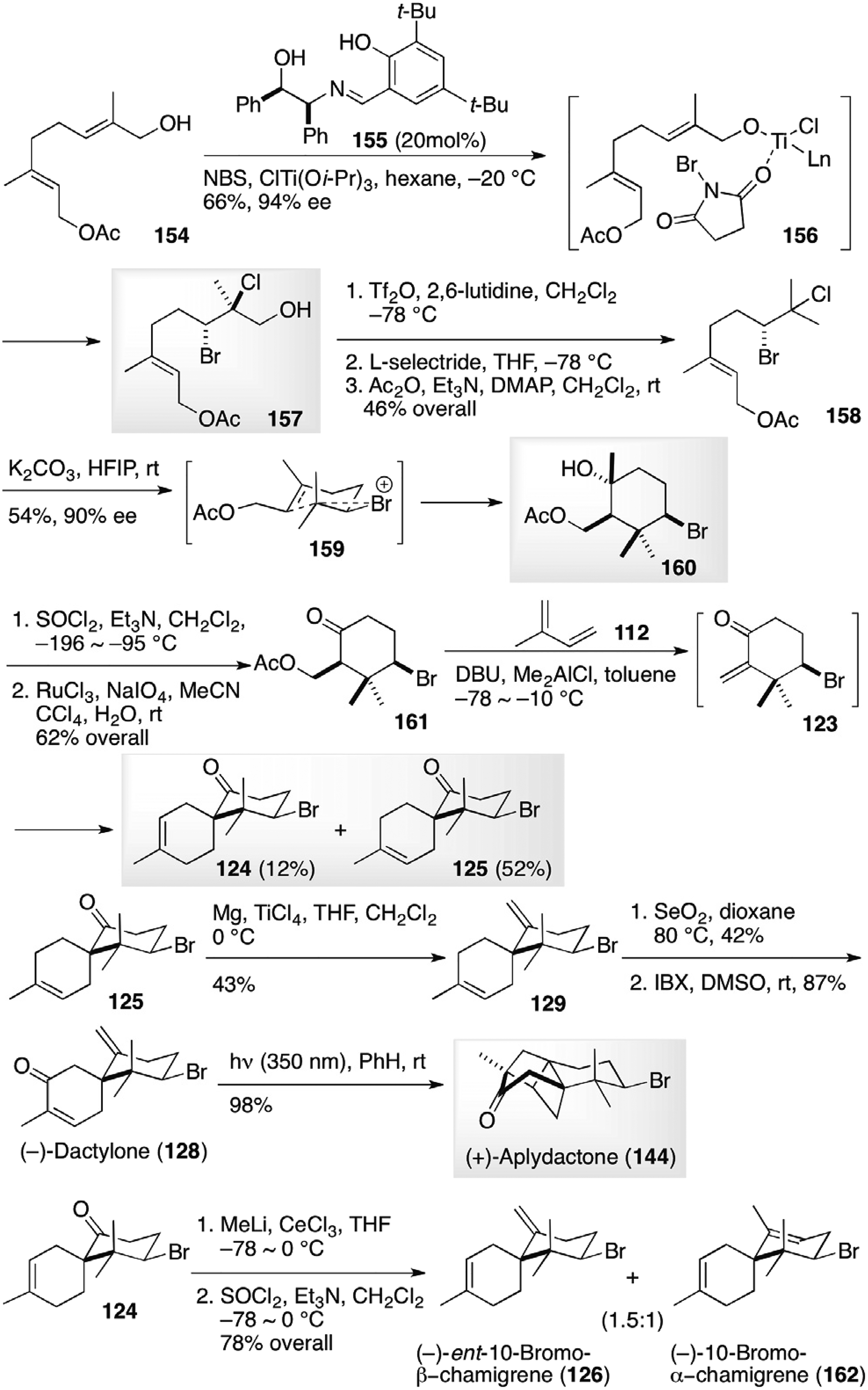
At the same time, Burns et al disclosed the syntheses of (–)-dactylone (128) and (+)-aplydactone (144) in 12 steps via enantioselecive cyclization and Diels-Alder strategy, along with the syntheses of (–)-10-bromo-α-chamigrene (162) and (–)-ent-10-bromo-β-chamigrene (126) (Figure 16).40 Authors challenged the photocycloaddition of dactylone (128) leading to aplydactone (144). The stereogenic centers at C-8 of the targets 128 and 144 were arranged at first because the difficulty was anticipated to install at the neopentyl position at the late stage in synthesis. An enantioselective bromochlorination of allylic alcohol 154 with the Schiff base catalyst 155 delivered bromochloride 157 highly enantioselectively.41 The primary hydroxyl group of 157 was removed by triflation and subsequent reduction to give acetate 158 after acetylation. The stereospecific solvolytic cyclization of the bromide 158 was successful via bromonium ion 159 in strongly ionizing and less nucleophilic medium, hexafluoroisopropanol (HFIP),42 to afford bromocycle 160 with high enantiospecificity in gram scale. The dibromoalcohol analogous to 157 was unstable under the deoxygenation conditions, and the cyclization under the ionizing conditions resulted in racemization. The bromocycle 160 was transformed to cyclohexanone 161 via dehydration and cleavage of the resulting exo-methylene group. The chamigrane framework was elaborated by the intermolecular Diels-Alder reaction of intermediary exo-cyclic enone 123 with excess isoprene 112 in the presence of excess dimethylaluminum chloride, in which the dienophile 123 was generated with DBU in situ at low temperature to prevent homodimerization, giving regioselectively spirocycle 124 and its double bond isomer 125. Methylenation of the hindered carbonyl group of the major spirocycle 125 to diene 129 was only successful with magnesium powder and titanium tetrachloride employing dichloromethane as a methylene source.28 Allylic hydroxylation of the diene 129 with selenium dioxide followed by 2-iodoxybenzoic acid (IBX) oxidation furnished (–)-dactylone (128). Irradiation of 350 nm light to dactylone (128) for 36 h successfully furnished (+)-aplydactone (144), in spite of the original report on the failure of intramolecular photo [2 + 2]-cycloaddition.37 The bromine-carbon bond at C-8 was stable under the irradiation condition. From the minor spirocycle 124, (–)-α-10-bromo-α-chamigrene (162) and (–)-ent-10-bromo-β-chamigrene (126) were prepared. Since methylenation with Mg/TiCl4/CH2Cl2 resulted in a lower yield, two-step methylenation was employed.
Syntheses of (+)-dactylone (128), (+)-aplydactone (144), bromochamigrenes 126, and 162 by Burns et al.40
Moreover, Meier et al finished an alternative more concise, and stereoselective synthesis of racemic aplydactone (144) in 11 steps via the Diels-Alder strategy (Figure 17).43 A copper-mediated conjugate addition of trimethylsilylmethylmagnesium chloride to enone 163 and subsequent trap of the enolate with trimethylsilyl chloride gave silylenol ether 164, which was led to exo-methylene ketone 165 by Eschenmoser's protocol. The key Diels-Alder reaction between the enone 165 and isoprene 112 was accomplished in the presence of zinc bromide under high pressure to afford spirodecanone 166. The presence of the trimethylsilyl group in the dienophile 165 was important for higher diastereoselectivity in the Diels-Alder reaction. Methylenation of the sterically hindered ketone was successful with Nysted/Utimoto reagent44 in the presence of titanium tetrachloride at 50 °C to give diene 167, in which the trimethylsilyl and the t-butyldimethylsilyl groups were removed simultaneously to give alcohol 168. Although the substitution of the hydroxyl group to a bromine atom was a formidable task, the radical protocol employed in their previous synthesis of aplydactone (144) was fruitful to deliver 10-bromo-β-chamigrene (126) in higher diastereoselectivity than in Figure 15 as a result of an equatorial attack of a bromine atom to secondary radical. Regioselective dihydroxylation of the bromide 126 led to 10-bromo-3,4-dihydroxy-β-chamigrene (127) and subsequent IBX oxidation followed by dehydration afforded dactylone (128). Irradiation to dactylone (128) furnished aplydactone (144), which is in good agreement with the result of Burns et al in Figure 16. From 10-bromo-β-chamigrene (126), epi-aplydactone (131), and subsequently 8-epi-isoaplydactone (171) were also furnished according to a similar procedure. Authors suggested that 8-epi-isoaplydactone (171) may be found in nature since 10-epi-aplydactone (131) exists in the sea hare.
Syntheses of (±)-10-bromo-β-chamigrene (126), (±)-dactylone (128), aplydactone (144), (±)-10-epi-dactylone (131), and (±)-8-epi-isoaplydactone (171) by Meier et al.43
The intriguing complex structure and biological activities of aplydactone (144) have been stimulating the interests of synthetic organic chemists, and Zhang et al challenged to a concise protecting group-free synthesis of aplydactone (144) and epi-aplydactone (153) in 11 steps without passing dyctylone (128) synthesis (Figure 18).45 The Barbier-type addition of prenylbromide 173 to aldehyde 172 led to cyclopentenone 174, whose [2 + 2]-photocycloaddition afforded tricyclic ketone 175. The ladderane scaffold was installed by the photochemical Wolff ring contraction of diazoketone 176, which was prepared via formylation of the ketone 175 followed by a diazo transfer reaction. The ring contraction proceeded smoothly and ester 177 was obtained after methylation from the less hindered α-face. Owing to the difficulty to substitute the hydroxyl group into a bromine atom at C-8, Meier and Trauner employed the radical reaction in Figure 15. In turn, Zhang et al introduced the bromine atom at the very end of total synthesis by hydrogenation of vinylbromide 182. To this end, oxidation of the hydroxyl group of 177 followed by transformation to tosylhydrazone and subsequent bromination provided vinylbromide 179, which was saponified and transformed to diazoketone 180 by Shioiri Arndt-Eistert reaction.46 Treatment of the diazoketone 180 with a catalytic amount of dirhodium complex resulted in intramolecular remote C–H insertion to give cyclopentane 181 and desired cyclohexane 182 carbocycles in a 1.5:1 ratio. The use of highly electrophilic Rh2(tfa)4 in refluxing hexane was suitable to increase the formation of six-membered C–H insertion product 182. After several attempts by conventional hydrogenation, aplydactone (144) along with 8-epi-aplydactone (153) were successfully furnished by Shenvi's hydrogen atom transfer (HAT) protocol in a 1.5:1 ratio.36
Syntheses of (±)-aplydactone (144) and (±)-epi-aplydactone (153) by Zhang et al.45
Recently, List et al developed highly efficient organomolecular catalyst 183 for asymmetric intermolecular spirocyclizing Diels-Alder reactions of α-methylene-cycloalkanones with dienes for the first time and applied it to concise enantioselective syntheses of several spirocyclic sesquiterpenoids such as α-chamigrene (185), β-chamigrene (186), laurencenone C (188), omphalic acid (121), and colletoic acid (14) (Figure 19).47 Among a variety of Brönsted acidic catalysts having binaphthol scaffold investigated, the strongly acidic and confined imidodiphosphorimidate catalyst 183 was the best, showing reasonable yield, good enantioselectivity, regioselectivity, and wide substrates applicability.48 The Diels-Alder reaction of α-methylenecyclohexanone 111 with isoprene 112 proceeded at sterically demanding neopentyl position highly enantio- and regioselectively in good yield to afford spirocyclic ketone 113, which was a common key intermediate for compact syntheses of sesquiterpenoids having spiro[5.5]undecane scaffolds. The catalyst 183 was recovered in 95% yield without losing its catalytic activity. (+)-α-Chamigrene (185) was obtained by transformation of the spirocyclic ketone 113 to enoltriflate 184 followed by an iron cross-coupling reaction with methylmagnesium iodide. Wittig methylenation of the spirocyclic ketone 113 led to (+)-β-chamigrene (186). Synthesis of (+)-laurencenone C (188), an enantiomer of the natural product, was accomplished by Saegusa reaction of the spirocyclic ketone 113, through methylation and subsequent transposition of resulting tertiary alcohol. Synthesis of omphalic acid (121) from the spirocyclic ketone113 was already reported in Figure 12. A kinetic resolution was observed in the intermolecular spirocyclizing Diels-Alder reaction of racemic exo-methylenecyclopentanone 189 with isoprene 112, in which spirocyclic product 191, a precursor of (+)-colletoic acid (14) synthesis,49 was obtained in 82% ee leaving the (R)-exo-methylenecyclopentanone 190. The imidodiphosphorimidate catalyst 183 is remarkably efficient for the Diels-Alder reaction to construct spirocarbocyclic scaffold, which might be a powerful tool not only for enantioselective syntheses of dactylone (128) and aplydactone (144) in Figures 13, 16 and 17 directly but also for a variety spirocyclic compounds.
Syntheses of (+)-α-chamigrene (185), (+)-β-chamigrene (186), (+)-laurencenone C (188), omphalic acid (121), and (+)-colletoic acid (14) by List et al.47
Conclusion
The significance of the synthetic study of sesquiterpenoids is still unchanged even now when total syntheses of natural products are blooming. As summarized in this review, much effort has been devoted and realized the total syntheses of sesquiterpenoids involving spiro-carbocyclic scaffold, in which diverse creativity was presented not only in the construction of spirocyclic frameworks but also asymmetric inductions as well as functional group transformations. The quaternary stereogenic spiro-carboncenter is sterically demanding in addition to the congestion by an adjacent substituent. Installation of the spiro-carboncenter was elaborated based on the individual concept and strategy of authors.
The first total synthesis of sesquiterpenoid involving a spirocyclic scaffold may be the synthesis of cedrene by Corey et al,50 in which intramolecular alkylation at the p-position of phenol with pendant alkyl chain at the p-position was employed. Then, several procedures were exploited such as the aldol reaction of two carbonyl units on quaternary carbon, Claisen rearrangement of enol ether with an olefinic side chain or intermolecular Diels-Alder reaction, and so on.8 RCM reaction of two olefinic side chains on quaternary carbon in the carbocyclic compound is now an established protocol,8 while the stereoselective introduction of a quaternary carbon center becomes a key.
In addition to these protocols, novel protocols appeared since then. The intramolecular Heck-type cyclization was effective to make congested quaternary carbon center diastereoselectively constructing spirocyclic scaffold directly. The sequential 1,3-acyloxy shift/cyclization/semipinacol rearrangement of acetylenic acetate derived from Castro-Stephens coupling was interesting and unique, though issues on stereoselectivity remained. Ring-opening of cyclopropane ring of cubebane framework derived from intramolecular keto-carbenoid addition was successfully applied to the construction of carbocycles. Photochemical transformation such as rearrangement of a cross-conjugated dienone, 1,3-shift of an acyl group, and [2 + 2]-cycloaddition played a key role to embed the spirocyclic scaffold. RCM and Diels-Alder reactions are now common protocols but still occupy an important position. Especially, the success of organocatalytic enantioselective Diels-Alder reaction is quite noteworthy in view of the atom- and step-economy with wide applicability. For the transformation of functional groups, the introduction of a labile C–Br bond by radical reaction or HAT reduction and the introduction of amino acid side chain by Ugi reaction attracted attention.
The discovery of new structures and novel biological activities in sesquiterpenoid offers new challenging synthetic targets not only to inspire total syntheses but also to develop synthetic methodologies. It is hopeful that the present mini-review gives some inspiration on the total synthesis of natural products involving spirocyclic carbon frameworks.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
Statement of Human and Animal Rights
Not applicable.
Statement of Informed Consent
Not applicable.
ORCID iD
Hisahiro Hagiwara
Abbreviations
References
1.
HeathcockCLGrahamSLPirrungMCPlavacFWhiteCT. The total synthesis of sesquiterpenes, 1970‐1979. In: ApSimonJ, ed. In the total synthesis of natural products. Vol 5. John Wiley & Sons; 1983:264‐313.
2.
WelschMESnyderSAStockwellBR. Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol. 2010;14(3):347‐361. doi:10.1016/j.cbpa.2010.02.018
3.
ZhengYTiceCMSinghSB. The use of spirocyclic scaffolds in drug discovery. Bioorg Med Chem Lett. 2014;24(16):3673‐3682. doi:org/10.1016/j.bmcl.2014.06.081
4.
RiosR. Enantioselective methodologies for the synthesis of spiro compounds. Chem Soc Rev. 2012;41(3):1060‐1074. doi:org/10.1039/C1CS15156H
5.
D’yakonovVATrapeznikovaOAde MeijereADzhemilevUM. Metal complex catalysis in the synthesis of spirocarbocycles. Chem Rev. 2014;114(11):5775‐5814. doi:org/10.1021/cr400291c
XuPWYuJSChenCCaoZ-YZhouFZhouJ. Catalytic enantioselective construction of spiro quaternary carbon stereocenters. ACS Catal. 2019;9(3):1820‐1882. doi:org/10.1021/acscatal.8b03694
8.
SmithLKBaxendaleIR. Total syntheses of natural products containing spirocarbocycles. Org Biomol Chem. 2015;13(39):9907‐9933. doi:10.1039/c5ob01524c
9.
AoyagiAIto-KobayashiMOnoY, et al.Colletoic acid, a novel 11(-hydroxysteroid dehydrogenase type 1 inhibitor from colletotrichum gloeosporioides SANK 21404. J Antibiot. 2008;61(3):136‐141. doi:10.1038/ja.2008.122
10.
LingTGautamLNGriffithE,et al.Synthesis and evaluation of colletoic acid core derivatives. Eur J Med Chem. 2016;110(3):126‐132. doi:org/10.1016/j.ejmech.2016.01.027
11.
LingTGriffithEMitachiKRivasF. Scalable and divergent total synthesis (+)-colletoic acid, a selective 11(-hydroxysteroid dehydrogenase type 1 inhibitor. Org Lett. 2013;15(22):5790‐5793. doi:org/10.1021/ol402842u
12.
GorobetsEWongNEPatonRSDerksenDJ. Divergent photocyclization/1,4-sigmatropic rearrangements for the synthesis of sesquiterpenoid derivatives. Org Lett. 2017;19(4):484‐487. doi:10.1021/acs.orglett.6b03635
13.
RojasGWagenerKB. Avoiding olefin isomerization during decyanation of alkylcyano α,ω-dienes: a deuterium labeling and structural study of mechanism. J Org Chem. 2008;73(13):4962‐4970. doi:org/10.1021/jo800640j
14.
OzakiTKobayashiY. Concise synthesis of (–)-axenol by using stereocontrolled allylic substitution. Synlett. 2015;26(8):1085‐1088. doi:10.1055/s-0034-1380273
15.
BlanchetteMAChoyWDavisJT,et al.Horner-wadsworth-emmons reaction: use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett. 1984;25(21):2183‐2186. doi:org/10.1016/S0040-4039(01)80205-7
16.
KanekoYKiyotsukaYAcharyaHPKobayashiY. Construction of a quaternary carbon at the carbonylcarbon of the cyclohexane ring. Chem Commun. 2010;46(30):5482‐5484. doi:org/10.1039/C0CC00653J
17.
AtheSGhoshSMehtaG. A unified, RCM anchored approach to spiro[4.5]decane-based sesquiterpenoids: collective synthesis of (±)-( & (-vetispirenes,(±)-(-vetivone, (±)-agarospirol and (±)-hinesol. Tetrahedron Lett. 2019;60(24):1570‐1573. doi:org/10.1016/j.tetlet.2019.05.011
18.
NakazakiAEraTKobayashiS. Total synthesis of (+/-)-gleenol and (+/-)-axenol via a functionalized spiro[4.5]decane. Chem Pharm Bull. 2007;55(11):1606‐1609. doi:10.1248/cpb.55.1606
19.
UyMMOhtaSYanaiMOhtaEHirataTIkegamiS. Exiguamide, a new spirocyclic sesquiterpene from the marine sponge Geodia exigua that inhibits cell fate specification during sea urchin embryogenesis. Bioorg Med Chem Lett. 2002;12(21):3037‐3039. doi:org/10.1016/S0960-894X(02)00662-5
20.
KonishiSMitaniYMoriNTakikawaHWatanabeH. First enantioselective synthesis of exiguamide, a nitrogen-containing spirocyclic sesquiterpene isolated from the marine sponge Geodia exigua. Tetrahedron. 2019;75(5):652‐657. doi:org/10.1016/j.tet.2018.12.046
21.
UyMMOhtaSYanaiMOhtaEHirataTIkegamiS. New spirocyclic sesquiterpenes from the marine sponge Geodia exigua. Tetrahedron. 2003;59(6):731‐736. doi:org/10.1016/S0040-4020(02)01619-8
22.
HosokawaSNakanishiKUdagawaY,et al.Total synthesis of Exigurin: Ugi reaction in a hypothetical biosynthesis of natural products. Org Biomol Chem. 2020;18(4):687‐693. doi:org/10.1039/C9OB02249J
23.
UgiIDemharterAHörlWSchmidT. Ugi Reactions with trifunctional α-amino acids, aldehydes, isocyanides and alcohols. Tetrahedron. 1996;52(35):11657‐11664. doi:org/10.1016/0040-4020(96)00647-3
24.
BagchiAOshimaYHikinoH. Spirojatamol, a new skeletal sesquiterpenoid of roots. Tetrahedron. 1990;46(5):1523‐1530. doi:org/10.1016/S0040-4020(01)81961-X
25.
PathiranaCFenicalWCorcoranEClardyJ. Erythrodiene: a new spirobicyclic sesquiterpene of a rare skeletal class from the Caribbean gorgonian coral Erythropodium caribaeorum. Tetrahedron Lett. 1993;34(21):3371‐3372. doi:org/10.1016/S0040-4039(00)79158-1
26.
ZhangYZhengT-LChengF, et al.Facile access to diverse all-carbon quaternary center containing spirobicycles by exploring a tandem Castro–Stephens coupling/acyloxy shift/cyclization/semipinacol rearrangement sequence. Chem Sci. 2020;11(15):3878‐3884. doi:10.1039/D0SC00102C
27.
ChengFDuanD-SZhangY,et al.Total synthesis of (±)-erythrodiene and (±)-spirojatamol. Tetrahedron Lett. 2021;85:153291‐153293. doi:org/10.1016/j.tetlet.2021.153291
28.
YanT-HTsaiC-CChienC-TChoC-CHuangP-C. Dichloromethane activation. Direct methylenation of ketones and aldehydes with CH2Cl2 promoted by Mg/TiCl4/THF. Org Lett. 2004;6(26):4961‐4963. doi:org/10.1021/ol0478887
29.
LiWHeJFengT,et al.Antroalbocin A, an antibacterial sesquiterpenoid from higher fungus Antrodiella albocinnamomea. Org Lett. 2018;20(24):8019‐8021. doi:org/10.1021/acs.orglett.8b03595
30.
KawamotoYOzoneDKobayashiTItoH. Total synthesis of (±)-chondrosterin I using a desymmetric aldol reaction. Org Biomol Chem. 2018;16(44):8477‐8480. doi:10.1039/C8OB02557F
31.
SiekmeyerBLübkenDBajerkeKBernhardtBSchreinerPRKalesseM. Total synthesis of (‐)-antroalbocin A enabled by a strain release-controlled photochemical 1,3-acyl shift. Org Lett. 2022;24(31):5812‐5816. doi:org/10.1021/acs.orglett.2c02347
32.
ZhouPZhangLLuoSChengJ-P. Asymmetric synthesis of Wieland-Miescher and Hajos-Parrish ketones catalyzed by an amino-acid-derived chiral primary amine. J Org Chem. 2012;77(5):2526‐2530. doi:org/10.1021/jo202433v
33.
XuCZhangLZhouPLuoSChengJ-P. A practical protocol for asymmetric synthesis of Wieland-Miescher and Hajos-Parrish ketones catalyzed by a simple chiral primary amine. Synthesis (Mass).2013;45(14):1939‐1945. doi:10.1055/s-0033-1338891
34.
ToriMNakashimaKAsakawaY. Sesquiterpenes and a phenolic compound from the liverwort Omphalanthus filiformis. Phytochem. 1995;38(3):651‐653. doi:org/10.1016/0031-9422(94)00682-J
35.
ChenRQiuDLeiX,et al.Total synthesis and assignment of the absolute configuration of (+)-omphalic acid. Org Lett. 2021;23(17):6972‐6976. doi:org/10.1021/acs.orglett.1c02599
36.
ObradorsCMartinezRMShenviRA. Ph(i-PrO)SiH2: an exceptional reductant for metal-catalyzed hydrogen atom transfers. J Am Chem Soc. 2016;138(14):4962‐4971. doi:org/10.1021/jacs.6b02032
37.
FedorovSNRadchenkoOSShubinaLK,et al.Aplydactone, a new sesquiterpenoid with an unprecedented carbon Skeleton from the Sea Hare Aplysia dactylomela, and its Cargill-like rearrangement. J Am Chem Soc. 2001;123(3):504‐505. doi:org/10.1021/ja003254t
38.
ShenMKretschmerMBrillZGSnyderSA. Strategies for the total synthesis of diverse bromo-chamigrenes. Org Lett. 2016;18(19):5018‐5021. doi:10.1021/acs.orglett.6b02478
39.
MeierRTraunerD. A synthesis of (±)-aplydactone. Angew Chem Int Ed Engl. 2016;55(37):11251‐11255. doi:10.1002/anie.201604102
40.
BurckleAJVasilevVHBurnsNZ. A unified approach for the enantioselective synthesis of the brominated chamigrene sesquiterpenes. Angew Chem Int Ed Engl. 2016;55(38):11476‐11479. doi:10.1002/anie.201605722
41.
HuDXSeidlFJBucherCBBurnsNZ. Catalytic chemo-, regio-, and enantioselective bromochlorination of allylic alcohols. J Am Chem Soc. 2015;137(11):3795‐3798. doi:org/10.1021/jacs.5b01384
42.
DenmarkSEBurkMTHooverAJ. On the absolute configurational stability of bromonium and chloronium ions. J Am Chem Soc. 2010;132(4):1232‐1233. doi:org/10.1021/ja909965
43.
MatsuuraBSKöllePTraunerDde Vivie-RiedleRMeierR. Unravelling photochemical relationships among natural products from aplysia dactylomela. ACS Cent Sci. 2017;3(1):39‐46. doi:10.1021/acscentsci.6b00293
44.
MatsubaraSSugiharaMUtimotoK. Methylenation of carbonyl compounds by means of Nysted reagent. Synlett. 1998;1998(3):313‐315. doi:10.1055/s-1998-36073
45.
LiuCChenRShenYLiangZHuaYZhangY. Total synthesis of aplydactone by a conformationally controlled C–H functionalization. Angew Chem Int Ed Engl. 2017;56(28):8187‐8190. doi:10.1002/anie.201703803
46.
AoyamaTShioiriT. New methods and reagents in organic synthesis. 17. Trimethylsilydiazomethane (TMSCHN2) as a stable and safe substitute for hazardous diazomethane. Its application to the Arndt-Eistert synthesis. Chem Pharm Bull. 1981;29(11):3249‐3255. doi.org/10.1248/cpb.29.3249
47.
GhoshSErchingerJEMajiRListB. Catalytic asymmetric spirocyclizing Diels-Alder reactions of enones: stereoselective total and formal syntheses of α-chamigrene, β-chamigrene, laurencenone C, colletoic acid, and omphalic acid. J Am Chem Soc. 2022;144(15):6703‐6708. doi:.org/10.1021/jacs.2c01971
48.
GhoshSDasSDeCK,et al.Strong and confined acids control five stereogenic centers in catalytic asymmetric Diels‐Alder reactions of cyclohexadienones with cyclopentadiene. Angew Chem Int Ed Engl. 2020;59(30):12347‐12351. doi:org/10.1002/anie.202000307
49.
SawadaTNakadaM. Enantioselective total synthesis of (+)-colletoic acid via catalytic asymmetric intramolecular cyclopropanation of an α-diazo-β-keto diphenylphosphine oxide. Org Lett. 2013;15(5):1004‐1007. doi:org/10.1021/ol303459x
50.
CoreyEJGirotraNNMathewCT. Total synthesis of dl-cedrene and dl-cedrol. J Am Chem Soc. 1969;91(6):1557‐1559. doi:org/10.1021/ja01034a062