
Abstract
Biomolecular chemical simulations have recently become a useful research method in the fields of organic chemistry and bioscience. In the last few years, we have been focusing on the biomolecular computational simulation on lipase enzyme and ligand complexes to predict the enantioselectivity and reactivity of lipases toward non-natural organic compounds. In this paper, we describe the molecular simulations including molecular dynamics (MD) and fragment molecular orbital (FMO) calculations for the complexes of Candida antarctica lipase type A (CALA) and trifluoromethylazulene alcohol derivatives. From the MD calculations, we found that the fast-reacting enantiomer of esters with high enantioselectivity stays in the vicinity of the active site of CALA, while the slow-reacting enantiomer leaves the active site of CALA. On the other hand, both (R)- and (S)-enantiomers of ester with low ensntioselectivity were found to keep near to near the active site of CALA. Further, for the esters that do not react with lipase enzyme, we found that both (R)- and (S)-enantiomers move away from the active site of lipase enzyme. From the FMO calculations, we found that each fast-reacting enantiomer of esters with high enantioselectivity strongly interacts with certain particular amino acid residues in CALA containing Asp95, while both (R)- and (S)-enantiomers of ester with low enantioselectivity interact with same amino acid residues in CALA including Asp95. These results suggest that it is possible to predict not only the enantioselectivity but also the reactivity of CALA and to identify the amino acid residues important to the enzymatic reaction. Therefore, we consider that our computational simulations would be a useful method for predicting and understanding the reactivity and the enantioselectivity of lipase-catalyzed biotransformations.
Keywords
Introduction
Today, the dramatic developments in computer technology and simulation software have made it possible to perform biomolecular chemical calculations not only for small organic compounds but also for biopolymers such as proteins. In Particular, simulation methods such as molecular dynamics (MD) calculation and fragment molecular orbital (FMO) calculation1–4 have been utilized as a research method in the fields of organic chemistry and bioscience. FMO calculation is a quantum chemical calculation method in which biopolymers such as proteins are divided into fragments (amino acid residues), and enable a detailed evaluation of the interaction energy between compounds and each amino acid residue in protein. From this fact, FMO calculation is expected to be an effective method for molecular design of drug discovery and functional new compounds and for modification of enzyme function. However, due to problems with the accuracy of simulations and the analytical environment, it seems that the manufacturing which connected computational simulations and experiments well is still insufficient. In other words, the methodology, such as the analytical validity and the creativity, to make good use of the computational simulations has not been established. Therefore, the computational simulation is not necessarily widespread enough in the industry for the actual manufacturing. As a first step toward solving these problems, we have aimed to reproduce and even to predict the experimental results by the computational simulations. In recent years, we are focusing on biomolecular chemical simulations on the complexes of lipases and organic compounds to predict the enantioselectivity and reactivity of lipase in organic synthesis, and have selected lipases such as Burkholderia cepacia lipase (BCL) and Candida antarctica lipase typeB (CALB) as target proteins.5,6
Lipase enzymes are widely used as environmentally friendly and efficient biocatalysts for chemoselective, regioselective, and enantioselective reactions under extremely mild conditions due to their ease of use. Lipases are very special enzymes having a peculiar mechanism of reaction.7–11 Lipases are capable of recognizing many different substrates, and can catalyze hydrolysis, acidolysis, esterification, transesterification, and amination reactions.8,12–22 Enzymes with excellent catalytic properties have been well studied as catalysts because of their unique physicochemical behavior and have been recently used as a potential biocatalyst15,23–25 in a large number of biotechnological sciences, such as dairy products, detergents, pharmaceuticals, chemicals, 26 agriculture products, and oil chemistry. 27 From these facts, the production and utilization of enzymes may enable enzymes to be a better alternative of chemical catalysts.8,9,26
Until a dozen years ago, our research group has investigated the enantioselectivity of such enzyme lipases as BCL, CALB, and Candida antarctica lipase typeA (CALA) through their biocatalytic synthetic reactions with lipases.28–31 The degree of enantioselectivity is given by an experimentally obtained E value that is an enantiomeric ratio for each lipase-catalyzed reaction. 32 As the E value increases, the enantioselectivity becomes higher. Although a large amount of experimental data is available for BCL, CALB, and CALA lipases, including our results, the mechanism responsible for their enantioselectivity is still not completely elucidated.
In the last few years, we have performed biomolecular chemical simulations, including MD calculation and FMO calculation, on the complexes of BCL and CALB with various primary and secondary alcohol esters.5,6 We have shown that our simulation is a useful tool for predicting the enantioselectivity of BCL and CALB for various alcohol esters.5,6
In this paper, we performed the biomolecular chemical simulations toward CALA complexes with the 7 different trifluoromethylazulene secondary alcohol esters (Figure 1). CALA is made from a single polypeptide chain bearing 431 amino acid residues. The active site amino acid residue of CALA is Ser184 and the catalytic triad comprises Ser184, Asp334, and His366. 33 CALA has 2 disulfide bonds, Cys101-Cys273 and Cys350-Cys394. The substrate esters, azulene and its derivatives, comprise “a characteristic fused 5-7 bicyclic aromatic ring system” and are typical “nonbenzenoid aromatic hydrocarbons.” 31 Azulenes are interesting substances due to their expected reaction specificity and physicochemical and pharmacological properties. 31 Especially, it is well known that the azulene derivative, sodium guaiazulene sulfonate, has been used as a blue dye in gargles. Over several decades, our research group Konan Chemical Industry Co., Ltd has been developing and producing the water-soluble azulene, sodium guaiazulene sulfonate. They have a lot of experimental data on the azulene derivatives and have reported a part of them in 2004. 31 In this work, we used their previous experimental data 31 for comparison with the present computational results. In Figure 1, we show the 7 different trifluoromethylazulene secondary alcohol esters and experimentally obtained E values of CALA toward each azulene alcohol ester. 31

Seven different trifluoromethylazulene secondary alcohol esters and experimentally obtained E values of CALA toward each azulene alcohol ester. 31 CALA shows high enantioselectivity toward esters 1-4 with a large E value, whereas for esters 5 with a very small E value, CALA gives low enantioselectivity. Esters 6 and 7 do not react with CALA.
As shown in Figures 1, CALA shows high enantioselectivity toward esters 1-4 with a large E value, whereas for esters 5 with a very small E value, CALA gives low enantioselectivity. It is observed that for the esters 1-4 with high enantioselectivity, (S)-enantiomer react faster than (R)-enantiomer. 31 Thus, for esters 1-4, (S)-enantiomer is “the fast-reacting enantiomer,” and (R)-enantiomer is “the slow-reacting enantiomer.” For ester 5, due to its low enantioselectivity, it is difficult to determine whether (R)- or (S)-enantiomer is “the fast-reacting enantiomer.” Here, the azulene alcohol esters 6 and 7 are not converted by CALA. In addition, azulene alcohol ester 1 has been found to react with CALA, but not with BCL and CALB.
Through a series of our work combining of experimental chemistry and computational simulations, we aim to obtain knowledge on predicting lipase enantioselectivity and the extent of the enantioselectivity, and on elucidating the mechanism for enantiomeric recognition and reactivity of enzyme lipases.
Materials and Methods
Construction of CALA Complexes with Azulene Derivatives and Structural Optimization
The structures of (R)- and (S)-enantiomers of 7 different azulene alcohol esters were modeled on a computer using GaussView. GaussView is a graphical user interface used with Gaussian, a quantum chemistry calculation program. Then, the structures of esters were optimized by 2 steps quantum chemical calculations at HF/6-31G* level and at B3LYP/6-31G* level using Gaussian03. 34 Next, we downloaded the x-ray crystallographic structure of CALA from Protein Data Bank 35 (PDB code: 3GUU). This pdb structure was obtained by x-ray structural analysis with resolution of 2.10 Å and does not include the structural information of hydrogen atoms. In this study, we assigned the protonation state of histidine as delta protonated state (HID). And all crystallization water was removed. We used AMBER11 software 36 both of construction of the CALA-ester complexes and the structural optimization of the complexes by the energy minimization. Here, N-and C-terminus of CALA were capped by NH3+ and COO−, respectively. 4 Each azulene alcohol ester is manually placed near the active site Ser184 of CALA (Figure 2(a)). We assigned the charge of esters by AM1-BCC of antechamber 37 in AMBER11. As a first step of our study, we focused on the enzymatic reactions in water. We solvated the structures of the CALA-ester complexes with TIP3P model water molecules within 8.0 Å of each complex. The number of TIP3P model water molecules placed around each complex is over 10,000. Counterions were placed to neutralize the systems. Subsequently, we carried out the energy minimizations for all atoms in the presence of TIP3P water molecules by a molecular mechanics (MM) calculation using AMBER11 with the ff99 and general AMBER force field for CALA and azulene alcohol esters, respectively.38,39 The obtained structure takes an atomic configuration that minimizes the energy of the system. Here, we took the disulfide bonds in CALA into account in our all calculations: MM, MD, and FMO calculations.
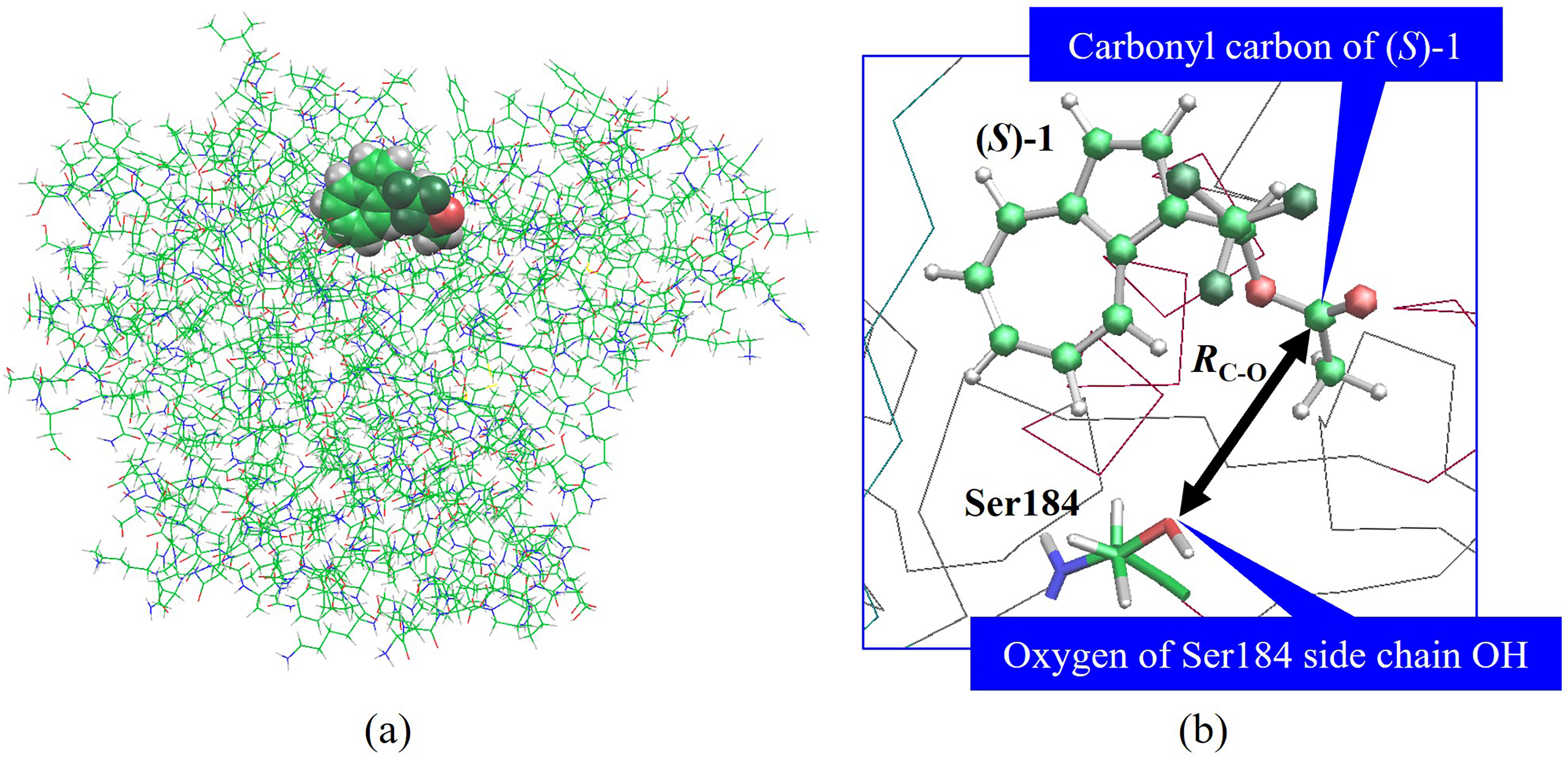
(a) Complex of CALA and (S)-enantiomer of ester 1 [(S)-1]. (b) Structure near the active site of CALA and (S)-1 complex. C-O interatomic distance, RC-O, between the carbonyl carbon of (S)-1 and the oxygen of the active site Ser184 side chain OH in CALA-ester complex is shown with a black double-headed arrow. Color code: green C, red O, blue N, white H.
Molecular Dynamics Calculations
After energy minimization, CALA-ester complexes with TIP3P model water molecules were subsequently subjected to a MD calculation over a period of 2000 ps at 300 K using AMBER11. We employed periodic boundary condition by controlling the pressure. The temperature was kept constant using the weak-coupling algorithm 40 with a coupling time of 1.0 ps. We constrained only bond lengths involving hydrogen atoms by the SHAKE method. 41 We calculated non-bonding interactions using the Particle Mesh Ewald method with 10.0 Å cutoff distance. The time step for the MD calculation was 1 fs. The whole system temperature was gradually increased by heating to 300 K for 50 ps and kept at 300 K for the next 2000 ps. Then, we estimated the C-O interatomic distance, RC-O, between the carbonyl carbon of azulene alcohol esters and the oxygen of the active site Ser184 side chain OH in each CALA-ester complex (Figure 2(b)) using VMD (Visual Molecular Dynamics) Version 1.8.6 42 and examined the time dependence of RC-O during the MD trajectory. Here, the C-O interatomic distance, so called RC-O, is also defined in Refs5,6. Moreover, to examine the reactivity of lipase enzymes in detail, we carried out the similar MD simulations on the complexes of BCL with azulene alcohol ester 1 and of CALB and azulene alcohol ester 1 which are prepared by above method. These complexes are unreactive ones.
Fragment Molecular Orbital Calculations
We performed the FMO calculations for the complexes in which the ligand remains in the active site. Since the depth of the active pocket of CALA, the distance from the entrance of the active pocket to Ser184 is about 9.0 Å, we concluded that the ligand is separated from the active site for complexes with RCO > 9.0 Å at 2000 ps of MD calculations. Therefore, we selected the complexes of CALA and (S)-enantiomer of esters 1-4 with high enantioselectivity, and the complexes of CALA and both (R)- and (S)-enantiomers of ester 5 with low enantioselectivity, for the FMO calculation. After MD calculations, for the structures at 2000 ps, we carried out the structural optimizations by MM calculations using AMBER11. Then, we removed the surrounding TIP3P model water molecules and counterions from the system. Subsequently, we performed an FMO calculation on the obtained complexes using ABINIT-MP/BioStation program43–45 at MP2/6-31G level. The Cholesky decomposition with adaptive metric method 46 was applied to the FMO calculations. In our FMO calculations, each amino acid residue except cysteines with disulfide bond of CALA and the enantiomer of azulene alcohol esters were treated as a single fragment, respectively. We treated 2 cysteines bonding with disulfide bond as one fragment. The computations were performed under vacuum conditions. We obtained the interaction energy (inter-fragment interaction energy: IFIE) between the azulene alcohol esters and the amino acid residues in CALA.
Results and Discussion
Molecular Dynamics Calculations
Figures 3 and 4 show the time dependence of RC-O in the complexes of CALA with the (R)- and (S)-enantiomer of azulene alcohol esters 1-4 and azulene alcohol ester 5, respectively. As shown in Figure 3, for the esters 1-4 with a large E value (high enantioselectivity), we found that RC-O of the fast-reacting (S)-enantiomer did not change almost around 5 Å, but RC-O of the slow-reacting (R)-enantiomer increased with time. This result indicates that the fast-reacting enantiomer of esters with high enantioselectivity stays in the vicinity of the active site of CALA, while the slow-reacting enantiomer leaves the active site of CALA. On the other hand, as shown in Figure 4, for ester 5 with a very small E value (low enantioselectivity), both (R)- and (S)-enantiomers were found to keep near to the active site of CALA. Similar MD calculations were performed for some initial structures, and we obtained the same results mentioned above.

Time dependence of the C–O interatomic distance, RC-O, obtained by MD calculations on the complexes of CALA with (R)- and (S)-enantiomers of esters 1-4 with a large E value (high enantioselectivity). For all complexes, RC-O of (R)-enantiomer is larger than that of (S)-enantiomer.

Time dependence of the C-O interatomic distance, RC-O, obtained by MD calculations on the complexes of CALA with (R)- and (S)-enantiomers of ester 5 with a very small E value (low enantioselectivity). RC-O of both (R)- and (S)-enantiomers are about 5 Å.
Then, we estimated the difference in RC-O between the (R)- and (S)-enantiomer complexes, ΔRC-O, at 2000 ps of the MD trajectory. Namely, ΔRC-O is defined by the following equation:


Relationship between the difference in the C-O interatomic distance of (R)- and (S)-enantiomer complexes, ΔRC-O, obtained by MD calculations and experimentally obtained E values for esters 1-5. For the esters 1-4 with a large E value, ΔRC-O is more than 7.0 Å, while that for the ester 5 with a very small E value, ΔRC-O is 0.2 Å.
In addition, we show the MD computations on the unreactive complexes such as CALA with ester 6, CALA with ester 7, BCL with ester 1, and CALB with ester 1 in Figure 6. For the esters that do not react with lipase enzyme, we found that both (R)- and (S)-enantiomers move away from the active site of lipase enzyme.

Time dependence of the C-O interatomic distance, RC-O, obtained by MD calculations on the unreactive complexes such as CALA with ester 6, CALA with ester 7, BCL with ester 1, and CALB with ester 1. For both (R)- and (S)-enantiomers, RC-O increases with time.
As a supplementary addition, we show the time dependence of root mean square deviation (RMSD) during the MD simulations on the complexes of lipases and trifluoromethylazulene alcohol esters in Supplemental Figures S1, S2, and S3.
Fragment Molecular Orbital Calculations
As shown in our previous works,5,6 the IFIE analysis is a useful method for gaining detailed information on protein-ligand binding. Figure 7 shows the IFIE values between the fast-reacting enantiomer of esters 1-4 with high enantioselectivity and the selected amino acid residues in CALA. We found that each fast-reacting enantiomer of esters with high enantioselectivity showed a large interaction energy with Asp95 in CALA. In addition to the interaction with Asp95, it was also found that each fast-reacting (S)-enantiomer interacted with certain particular amino acid residues in CALA such as Leu225, Phe230, Phe233, Glu336, and Ile336. These amino acid residues are located near the active site of CALA. In contrast, the interactions of each slow-reacting (R)-enantiomer with these amino acid residues were very weak. The reason for this result is considered to be that the slow-reacting enantiomer departs from the active site of CALA. In this paper, we omitted showing the IFIEs for the slow-reacting enantiomer. As an example, the structures of the fast-reacting enantiomers of esters 1, 2 and the interacting amino acid residues are illustrated in Figure 8. The IFIE between Asp95 and (S)-2 is larger than that between Asp95 and (S)-1. The distance between the oxygen atom in side chain of Asp95 and (S)-2 is about 2.4 Å, whereas that between the oxygen atom in side chain of Asp95 and (S)-1 is about 3.4 Å. The IFIE between Glu335 and (S)-1 is larger than that between Glu335 and (S)-2. The side chain of Glu335 orients toward (S)-1, but not toward (S)-2.
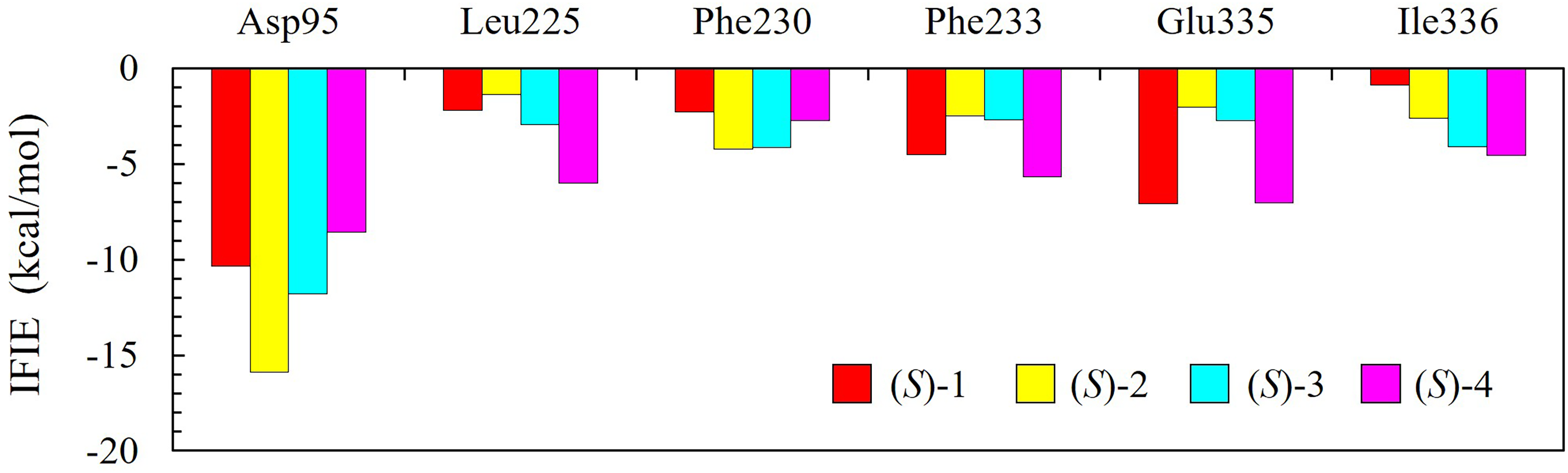
Results of FMO calculations: IFIE between the fast-reacting (S)-enantiomer of esters 1-4 with high enantioselectivity and the selected amino acid residues in CALA. (S)-enantiomer of esters show a large interaction energy with Asp95 in CALA. In addition to Asp95, each (S)-enantiomer interacts with certain particular amino acid residues in CALA such as Leu225, Phe230, Phe233, Glu335, and Ile336. These amino acid residues are located near the active site of CALA.

Structures of (S)-enantiomers of esters 1(a), 2(b), and the interacting amino acid residues. The distance between the oxygen atom in side chain of Asp95 and (S)-1 is about 3.4 Å (a), whereas that between the oxygen atom in side chain of Asp95 and (S)-2 is about 2.4 Å (b). The side chain of Glu335 orients toward (S)-1, but not toward (S)-2.
On the other hand, as shown in Figure 9, for ester 5 with low enantioselectivity, both (R)- and (S)-enantiomers were found to interact with the same amino acid residues containing Asp95. We also found that the interactions of both enantiomers with amino acid residues in CALA were similar to each other in their intensities and patterns. Figure 9 shows the IFIE values between (R)- and (S)-enantiomers of esters 5 and the selected amino acid residues in CALA.
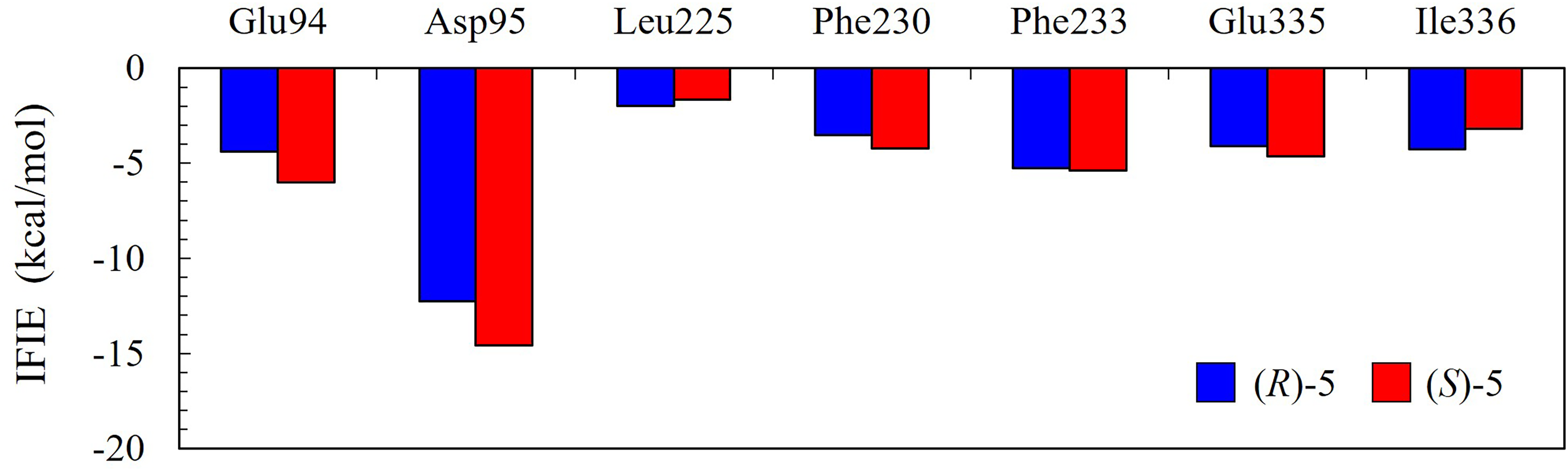
Results of FMO calculations: IFIE between the ester 5 with low enantioselectivity and the selected amino acid residues in CALA. Both (R)- and (S)-enantiomers interact with the same amino acid residues containing Asp95.
Interestingly, regardless of the extent of enantioselectivity, the interaction between each enantiomer and the active site amino acid residue Ser184 was very weak or hardly observed. These FMO computational results suggest that the IFIEs revealed in the present calculations can also be related to the enantioselectivity of CALA toward the azulene alcohol esters observed by synthetic investigations.
Conclusions
To predict the lipase enantioselectivity and the extent of enantioselectivity toward non-natural organic compounds, for the complexes of CALA and 7 different trifluoromethylazulene secondary alcohol esters, we performed the MD calculation over a period of 2000 ps and estimated the C-O interatomic distance, RC-O, between the carbonyl carbon of azulene alcohol esters and the oxygen of the active site Ser184 side chain OH for each CALA-ester complex. The FMO simulations on the CALA-ester complex structures obtained by MD calculations were subsequently carried out and the IFIE between esters and amino acid residues in CALA were calculated.
From the MD calculations, we found that the fast-reacting (S)-enantiomer of esters 1-4 with high enantioselectivity stays in the vicinity of the active site of CALA, while the slow-reacting (R)-enantiomer leaves the active site of CALA. On the other hand, both (R)- and (S)-enantiomers of ester 5 with low enantioselectivity were found to keep near to the active site of CALA. In addition, for the esters that do not react with lipase enzyme not only CALA but also BCL and CALB, both (R)- and (S)-enantiomers were found to leave the active site of lipase enzymes. The results strongly suggest that not only the enantioselectivity but also the reactivity of lipases such as CALA, BCL, and CALB can be predicted by the MD simulations.
Another aim of our study is to make clear the mechanism of enantiomeric recognition of enzyme lipases. For the esters showing high enantioselectivity, it is also found that each fast-reacting enantiomer shows strong interaction with the particular amino acid residues including Asp95 in CALA. On the contrary, each slow-reacting enantiomer hardly interacted with these amino acid residues. The reason for this result is considered to be that the slow-reacting enantiomer of esters with high enantioselectivity leaves the active site of CALA. In contrast, for the esters bearing low enantioselectivity, we observed that both (R)- and (S)-enantiomers interact with the same amino acid residues containing Asp95. The IFIE for the amino acid residue Asp95 is particularly noteworthy. It is predictable that Asp95 may play an important role in the chiral recognition of enantiomers by CALA. Alternatively, Asp95 might play the role of anchoring the esters to the active site of CALA during the reaction. This result is similar to that of His286 in BCL and Thr40 in CALB.5,6
The very weak interaction between esters and the active site amino acid residue Ser184 in CALA may be due to the fact that Ser184 is not yet activated. The additional more accurate FMO calculations that take into account the charge state of Ser184 might give more detailed information on the interactions between esters and amino acid residues in CALA. Otherwise, we may need to examine the structure changes of complexes and the time change of IFIE between esters and Ser184. Moreover, we made CALA-ester complexes manually using AMBER in the present work, but we are planning to carry out a docking simulation for constructing CALA-ester complexes. Moreover, in order to make our computational method more accurate and realistic, we have some issues to be solved in the future: (1) protonation states or charge states of the particular amino acid residues, such as histidine and the active site amino acids including Ser184, (2) identification of the amino acid residues involved in the molecular recognition mechanism or reaction mechanism/pathway of CALA and esters, (3) simulations under various situations, such as in organic solvents and exposure to the medium, (4) roles of surrounding water molecules or organic solvents (solvent effects) in CALA-ester binding, (5) comparison with new experiments with higher accuracy. Even though there are various issues at present, we believe that our computational approach including the MD calculation and FMO calculations may enable us to elucidate the mechanism responsible for enantiomeric recognition of enzyme lipases. Previously, we have performed similar computational simulations on the complexes of HIV-1 protease and its inhibitors, and found the following results: (1) a strong correlation between the experimentally obtained activity values and the calculated values such as binding energy, and (2) identification of the amino acid residues that strongly interact with the inhibitor. 47 Further, we are planning the computational simulations toward the complexes of a human protease and its inhibitor. We consider that our computational method is sufficiently applicable not only to the lipase-ester complexes in this work but also to other systems, such as the protease-inhibitor complexes and the enzyme-surrogate complexes. Through our continuous works, we have reached a conclusion that biomolecular chemical simulations are useful tools for predicting and understanding the reactivity and the enantioselectivity of lipase-catalyzed biotransformations.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221108572 - Supplemental material for Biomolecular Chemical Simulations on Enantioselectivity and Reactivity of Lipase Enzymes to Azulene Derivatives
Supplemental material, sj-docx-1-npx-10.1177_1934578X221108572 for Biomolecular Chemical Simulations on Enantioselectivity and Reactivity of Lipase Enzymes to Azulene Derivatives by Yoichiro Yagi, Takatomo Kimura and Makoto Kamezawa in Natural Product Communications
Footnotes
Acknowledgments
We would like to express our gratitude to Konan Chemical Industry Co., Ltd for their great cooperation from the early stages of this research. This research was performed as activities of the FMO drug design consortium (FMODD).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.