Recent advances in the total syntheses of cyclic natural products and related compounds from 2005 to 2021, which employ domino Michael reactions as key steps, have been reviewed, focusing mainly on the domino Michael reactions catalyzed by organocatalysts.
A domino reaction is a chain and cascade reaction, in which new active species generated after the first reaction reacts in the same manner as the first reaction without participating in another reaction and without adding extra substrates or reagents, thereby making multiple bonds successively in a one-pot operation,1,2 just like successive toppling down of a row of self-standing domino pieces. Such aspects of the domino reaction satisfy step-, time- and pot-economy,3,4 which are useful for a target-oriented multistep synthesis such as a total synthesis of natural products. Reduction of the amount of reagents, wastes and experimental operations in a domino reaction make organic syntheses more sustainable and attractive.
Among a variety of basic reactions as a candidate for a domino reaction as a key step for natural product syntheses, a Michael reaction has been utilized often,5–7 because the species generated on the Michael acceptor has an ability to achieve a next Michael reaction furthermore. Since both a Michael donor and an acceptor are installed in a product without loss of atoms, a Michael reaction is superior in atom-economy,8 which provides an additional merit for a domino process. A Michael reaction is a thermodynamically controlled reaction. In order to shift the equilibrium into thermodynamically stable product and to push the domino Michael reaction forward, several reaction systems were designed, among which kinetically controlled carbanion species were utilized to initiate the domino Michael reactions,5–7 whereas, the use of a strong base such as a stoichiometric amount of lithium diisopropylamide (LDA) to generate a kinetically controlled carbanion for the initiation of the first Michael reaction is not suitable for the reactions of a labile Michael acceptor such as an αβ-unsaturated aldehyde. However, recent developments in the chemistry of organocatalysts9,10 changed the game, in which an unmodified aldehyde as well as an unmodified αβ-unsaturated aldehyde can be applicable as a Michael donor and a Michael acceptor, respectively, to extend substrate scope. Moreover, high potential of an organoatalyst for enantioselective reaction is suitable for asymmetric synthesis.
As the result, a variety of domino Michael reactions catalyzed by organocatalysts have been reported to construct a variety of frameworks.11–14 Some of them are suitable as the key steps for construction of cyclic natural products by inserting an intramolecular Michael addition at a proper stage of the domino process.15,16 Formation of the cyclic compound might be helpful for termination of the domino process to shift the equilibrium of the Michael reaction toward the product. Moreover, mild reaction condition of organocatalyst is favorable for total syntheses of natural products having sensitive functional groups.
In this review, recent representative advances employing domino Michael reactions as key steps for natural product syntheses were picked up, mainly focusing on the reactions triggered by organomolecular catalysts intermolecularly.
Domino Michael Reactions Catalyzed by Organocatalysts
The diphenylprolinol silyl ether 1 and 2 (Figure 1) developed by Hayashi and Jørgensen17–19 opened a door as a very versatile organocatalyst especially to the enantioselective nucleophilic reactions of unmodified aldehydes, so far not easy to access. Catalytic versatility of the diphenylprolinol silyl ethers 1 and 2 was expanded to a domino Michael reaction.
Diphenylprolinol silyl ethers 1 and 2.
Hong et al. described a total synthesis of (+)-galbulin (15),20 a natural lignan isolated from Himantandra baccata and belgraveana via [4 + 2] organocatalytic domino Michael reaction, which was initiated by the attack of the enamine 6 to the Re-face of the s-trans vinylogous iminium cation 7 diastereoselectively, which was followed by an intramolecular Michael reaction leading to the six-membered keto-aldehyde 9. The reaction was non-equilibrated by the final intramolecular aldol condensation to afford the bicyclic enone 10 in 82% yield and 99% enantioselectivity. The enantiomer of the enamine 6 might suffer steric repulsion between the R-methyl group and the proton indicated in the transition state recovering the enantiomer. Reduction of the enone 10 with sodium borohydride and subsequent selective oxidation with manganese dioxide led to the hydroxy-enone 11 in 80% yield. Treatment of the enone 11 gave the unstable epoxide 12, which without purification was transformed with potassium hydroxide in methanol to the methoxy-enone 13. Aromatization and subsequent methylation of the phenol led to the bis-methylether 14. Finally, (+)-galbulin (15) was furnished by mesylation of the primary alcohol followed by super hydride reduction of the derived mesylate in 80% overall yield. Present total synthesis was carried out in 11% overall yields in 12 steps with 7 isolations and purifications (Figure 2).
Total synthesis of (+)-galbulin (15) by Hong et al.20
Hayashi and Umekubo established an efficient total synthesis of Corey lactone (24) via [3 + 2] domino Michael reaction, a key intermediate for the synthesis of prostaglandins (Figure 3).21,22 The initial Michael reaction began by an attack of the methyl ketone moiety of the keto-ester 16 to the Re-face of the s-trans vinylogous iminium cation 18, which was followed by an intramolecular second Michael reaction of the intermediary enamine 19 to the αβ-unsaturated ester group in 90% yield over 99% enantioselectivity. An intramolecular Michael reaction in the enamine 19 leading to a six-membered ring by an attack to the αβ-unsaturated ketone moiety was not observed, probably because the 5-exo mode attack was favorable than 6-endo cyclization due to the open chain conformation of the enamine 19. An intermolecular aldol condensation between the methylketone 16 and the aldehyde 17 was suppressed by employing the catalyst 2a. Addition of p-nitrophenol was essential for better yield and selectivity.23 Presence of a small amount of water was crucial, whereas excess amount of water decreased the enantioselectivity. Diastereoselectivity in the reduction of the ketone 20 was improved by lithium tri-t-butoxyaluminum hydride. Transformation of Si–Ph bond into Si–F bond took 4 h at elevated temperature in the presence of fluoroboric acid (HBF4). Oxidative rearrangement of the C–Si bond to the C–OH bond proceeded smoothly, which resulted in five pot total synthesis of Corey lactone (24) in 58% overall yield.
Total synthesis of (–)-Corey lactone (24) by Umekubo and Hayashi.21,22
Furthermore, Hayashi et al. established a remarkably efficient pot- and time-economical synthesis of Corey lactone (24), in which all reactions toward Corey lactone (24) starting from the domino Michael reaction were accomplished in one pot in 152 min in 50% overall yield and over 99% enantioselectivity (Figure 4).21,22 The whole procedure was carried out in a gram scale. In the previous synthesis in Figure 3, long reaction times are required for the conversion of the Si–Ph into the Si–F bond. They solved the issue by switching the solvent from 1,2-dichloroethane to THF and evaporating at 80 °C in the presence of HBF4, which played a quadruple role, namely protonation of lithium alkoxide, decomposition of excess reducing agent, lactonization and transformation of the Si–Ph bond into the Si–F bond.
One-pot total synthesis of (–)-Corey lactone (24) by Hayashi et al.21,22
Hayashi and Umekubo accomplished the total synthesis of (+)-latanoprost (38) also, an analogue of the prostaglandin PGF2, which is used to treat glaucoma (Figure 5).21,22 After the [3 + 2] domino Michael reaction, a Horner-Wadsworth-Emmons (HWE) reaction was carried out in situ to give the enone 29 in 72% yield and over 99% enantioselectivity, which was reduced diastereoselectively with (–)-B-chlorodiisopinocamphenylborane [(–)-DIP-Cl] (30) followed by lactonization to afford the lactone 32 after acidic work-up. Transformation of dimethylphenylsilyl group to a hydroxyl group led to the alcohol 34, in which the lactonic moiety was reduced to give the acetal 35. A Wittig condensation and subsequent esterification furnished (+)-latanoprost (38) in 25% overall yield in seven pots.
Total synthesis of (+)-latanoprost (38) by Umekubo and Hayashi.21,22
Clinprost (58) in Figure 6 is a stable analogue of prostacyclin (PGI2) and exhibits a potent neuroprotective activity in animal studies. According to the same [3 + 2] domino Michael strategy as in the Figure 5, Umekubo and Hayashi demonstrated a total synthesis of clinprost (58) (Figure 7).24 After the domino Michael reaction with 3-triphenylsilylpropenal (39), protection of the ketone and aldehyde moieties of the domino product 40 as the bis-acetal 41 followed by selective deprotection of the ketone acetal with HCl in situ afforded the acetal 42 in a one-pot operation. The ketone moiety of 42 was temporarily protected as the enolate 43 toward a reaction with the anion of dimethyl methylphosphonate. Selective protonation of the ketone enolate 45 was successful by the addition of acetic acid, which culminated in an intramolecular HWE reaction to provide the enone 47 in 86% yield from the ketone 42. The conjugate reduction of 47 was accompanied by the trap of the derived enolate with Tf2NPh leading to the enol triflate 49.
Total synthesis of clinprost (58) by Umekubo and Hayashi.24
Synthesis of the bicyclo[3.3.0]framework 49 of clinprost (58) by Umekubo and Hayashi.24
The side chain was combined by a Suzuki-Miyaura coupling with the borane 50 to afford the ester 51, which was subjected to a Tamao-Fleming reaction25,26 and subsequent esterification to provide the hydroxy ester 54 in 81% overall yield in a one-pot operation. Deprotection of the acetal of the ester 54 followed by a HWE reaction furnished the unsaturated ketone 57 in 81% overall yield. Reduction of the enone 57 with (–)-DIP-Cl 30 completed the total synthesis of clinprost (58) in a seven-pot sequence in 17% overall yield.
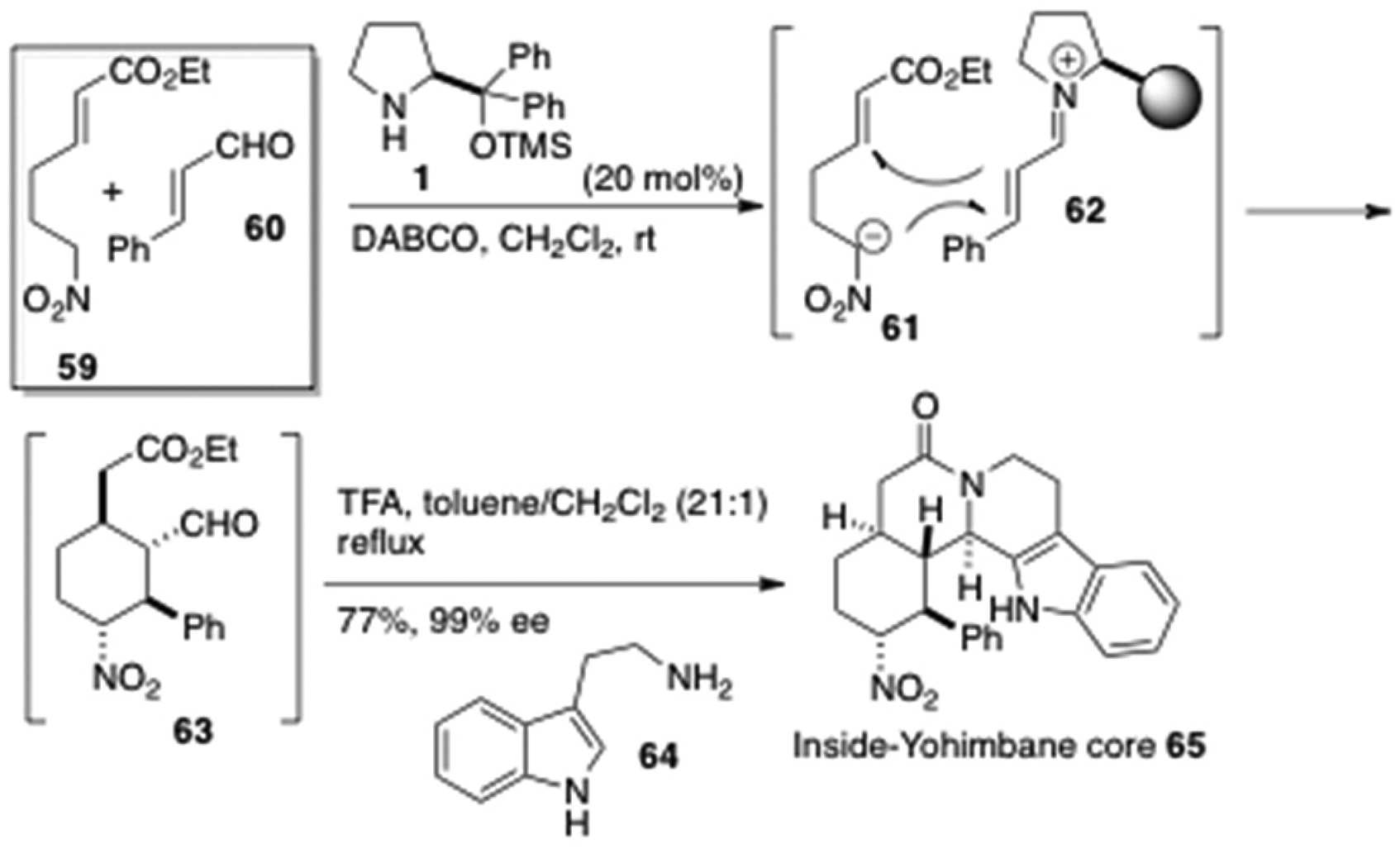
Alkaloids having a dodecahydrobenz[a]indolo[3,2-h]-quinolizine framework are called inside-yohimbane, which are known to show a wide spectrum of biological activities and have been attracted intense pharmacological synthetic attentions. Hong et al. synthesized the pentacyclic core 65 with five contiguous stereogenic centers in a one-pot operation by an organocatalytic enantioselective [4 + 2] domino Michael reaction followed by Pictet-Spengler lactamization27 reaction (Figure 8).28 The Michael reaction was initiated by an attack of the nitronium anion 61 to the Si-face of the vinylogous s-trans iminium cation 62, which was followed by a second Michael addition of an intermediary enamine to the Si-face of the αβ-unsaturated moiety leading to the cyclohexane derivative 63 in a highly enantioselective manner. Without isolation of 63, addition of 2-(1H-indol-3-yl)-ethanamine (64) resulted in the Pictet-Spengler lactamization to afford the inside-yohimbane 65 having five contiguous stereogenic centers in 77% yield and 99% enantioselectivity in a one-pot operation.
One-pot synthesis of the inside-yohimbane core 65 by Hong et al.28
The same domino Michael product 63 in the Figure 8 was employed for syntheses of the pro-azayohimbane 67 and the inside-azayohimbane 68 systems (Figure 9).29
One-pot synthesis of the inside-azayohimbane cores 67 and 68 by Yang et al.29
Furthermore, the domino Michael product 63 in Figures 8 and 9 was subjected to a Mannich reaction and reduction, which was followed by a Bischler-Napieralski reaction30 and reduction to afford the yohimbane analogue 70 in 54% yield and 99% enantioselectivity in a one-pot operation (Figure 10).31
One-pot synthesis of the yohimbane core 70 by Kao et al.31
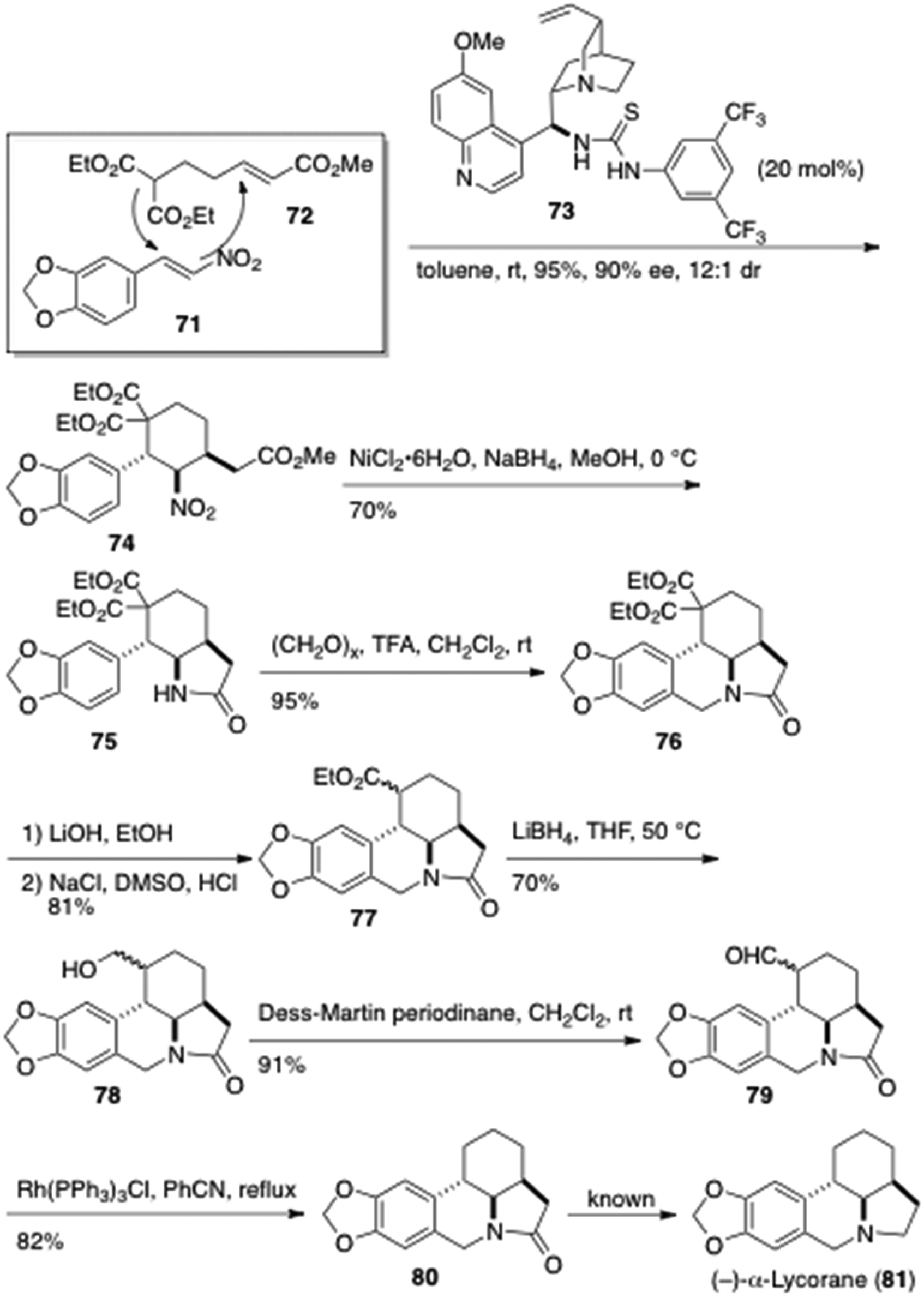
Xu et al. reported a formal total synthesis of (–)-α-lycorane (81) via a bifunctional thiourea-catalyzed [4 + 2] domino Michael reaction (Figure 11).32 Activation of the nitro group of the unsaturated nitroolefin 71 by the coordination of the quinine-thiourea (QNT) 73 promoted the domino Michael reaction with the unsaturated ester 72. The reaction proceeded with high diastereoselectivity and enantioselectivity to afford the cyclohexane derivative 74 with three contiguous stereogenic centers. Reduction of the nitro group of 74 with nickel boride led to the lactam 75, which was treated with paraformaldehyde to provide the tetracyclic structure 76. After hydrolysis and decarboxylation of the bis-ester 76, the derived monoester was removed by the transformation into the aldehyde 79 followed by a rhodium-catalyzed decarbonylation to afford the lycorane type compound 80, which has been reduced to (–)-α-lycorane (81).
Formal total synthesis of (–)-α-lycorane (81) by Wang et al.32
Subsequently, Zhao et al. carried out the domino Michael reaction of the nitroolefin 71 and the aldehyde 82 also by a self-assembled organocatalyst, the quinidine-thiourea (QDT) 83 and L-proline (84) system, in which a 1,2-syn isomer was first obtained and subsequent epimerization of the formyl group at 40 °C provided the cyclohexane derivative 85 in a highly diastereoselectively and enantioselectively manner (Figure 12).33 The first intermolecular Michael reaction involves L-proline (84) catalysis via an enamine intermediate and the second intramolecular Michael reaction would be catalyzed by the thiourea moiety of QDT 83 through a noncovalent catalysis. The cyclohexane 85 was transformed into (–)-β-lycorane (90) according to the similar procedure in the Xu synthesis in the Figure 11.
Formal total synthesis of (–)-β-lycorane (90) by Rana et al.33
The domino Michael reaction catalyzed by a combination of the quinine-thiourea (QNT) 91 and L-proline (84) afforded the diastereomeric cyclohexane derivative 92, which was converted to the precursor 93 of (–)-α-lycorane (81) (Figure 13).33
Formal total synthesis of (–)-α-lycorane (81) by Rana et al.33
Hajos-Parrish-type ketones have been very versatile starting materials for the total syntheses of natural products such as sesquiterpenoids or steroids. By employing two Michael acceptors 94 and 95 having different modes of reactivity, Hong et al. established a [1 + 2 + 2] inter/intra-domino Michael reaction, which was catalyzed by the prolinol 1 and terminated in Henry reaction to shift the equilibrium to the indanone derivative 100 with five contiguous stereogenic centers in 75% yield and 91% enantioselectivity (Figure 14).34 Authors proposed a reaction pathway, which goes through the dihydro-oxazine N-oxide intermediate 98 followed by the nitro-enamine derivative 100. The equilibrium in the Figure 14 might enable the reaction of acrolein (94) and the nitroolefin 95 in the order. The water/acetonitrile co-solvent system was chosen due to the low solubility of 2-methylcyclopentan-1,3-dione (96) in the bulk of organic solvent.
Synthesis of the functionalized Hajos-Parrish-type ketone 101 by Raja et al.34
Hong et al. prepared the steroidal framework 106 in a one-pot operation in 47% yield with 99% enantioselectivity via diastereomeric mixture of the cyclohexane derivative 104 by the [4 + 2] domino Michael reaction, which was catalyzed by the prolinol 1 in the similar manner as in Figures 8–10 (Figure 15).35 Subsequent addition of excess p-toluenesulfonic acid in situ resulted in an intramolecular aldol condensation to give the enone 105, which after evaporation of the solvent underwent the Henry reaction with diazabicycloundecene (DBU) and then tetrabutylammonium fluoride (TBAF) to afford the steroidal derivative 106 having the six contiguous stereogenic centers. TBAF mediated isomerization of the nitro group into β-position prior to the Henry reaction, though the reaction only with TBAF was sluggish.
One-pot synthesis of the steroid system 106 by Jhuo et al.35
Hexahydrophenanthrene cores constitute key frameworks of polycyclic natural products such as steroids or pharmaceutical compounds. An unusual [4 + 2 + 2] threefold domino Michael reaction, which terminated at a subsequent aldol condensation, was reported by Hong et al. to afford the highly functionalized hexahydrophenanthrene 112 with five contiguous stereogenic centers enantioselectively in a one-pot operation (Figure 16).36 The first intermolecular Michael reaction to the iminium cation 62 was initiated by the attack of the benzylic anion 108 generated in situ from the dinitrobenzene 107 with diisopropylethylamine, which proceeded on the Si-face of the iminium cation 62 under the control of the prolinol catalyst 1. The second Michael reaction of the resulting enamine 109 occurred intramolecularly from the Re-face of the nitroolefin moiety, which was followed by the third intermolecular Michael reaction with the iminium cation 62 on its Si-face. Final aldol condensation provided the hexahydrophenanthrene 112 in 54% yield and 99% enantioselectivity in one pot.
One-pot synthesis of the functionalized hexahydrophenanthrene 112 by Raja et al.36
A Domino Michael Reaction Promoted by a Stoichiometric Amount of Base
Homodimericin A (122), isolated from a metabolite of the fungus Trichoderma harzianum WC13, is a racemic hexacyclic polyketide with a carbon backbone containing eight contiguous stereogenic carbon atoms (including three contiguous quaternary centers) in a highly sterically demanding cage like core. Yang et al. designed a biogenetic type synthesis of 122 through [3 + 2] domino Michael reaction (Figure 17).37 The quinone 116 dimerized in the presence of five equivalent of sodium hydride via domino Michael pathway leading to the quinol 117 in 47% overall yield from the trimethoxybenzene 115, which was prepared by a palladium-catalyzed Kuwajima-Urabe arylation38 of the bromide 113 with the vinyl ether 114. Oxidation of the hydroquinone moiety of 117 with silver oxide was carried out in the presence of anhydrous sodium sulfate due to the sensitivity of the product quinone 118, which was subjected to intramolecular Diels-Alder reaction to afford the penta- and the hexacyclic cores 120 and 121. Double-bond isomerization of the intermediate 119 led to the pentacyclic compound 120, whereas the hexacyclic compound 121 was derived by the 5-exo-trig-ene cyclization of 119. Deprotection of the methyl ether in 120 led to racemic homodimericin A (122).
Synthesis of racemic homodimericin A (122) by Huang et al.37
Domino Michael Reactions by Mukaiyama-Type Group Transfer Reactions
A Mukaiyama-type Michael reaction39 is a very powerful tool for construction of C–C bonds with wide applicability and had been used also for the domino Michael reaction in natural product syntheses,6,40 in which a trimethylsilyl group is transferred from the starting substrate to the intermediate and to the product.
Scabronine G (141) is a cyathane diterpene isolated from the fruit body of mushroom Sarcodon scabrosus, which is known to enhance the secretion of neurotrophic factors from 1321N1 human astrocytoma cells (Figure 18).41 Iwabuchi et al. accomplished the total synthesis of scabronine G (141) via an intramolecular Mukaiyama-type group transfer [4 + 2] domino Michael reaction as a key step. The reaction of the enone 125 was promoted by trimethylsilyl iodide to provide the tricyclic compound 126 in 81% yield and 86% diastereoselectivity after hydrolysis of the derived silylenol ether, while the reaction of 125 with LDA resulted in low yield. Protection of the carbonyl group as an acetal followed by reduction of the ester group led to the alcohol 127, which was subjected to 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO)-catalyzed oxidation and subsequent deprotection of the TBS-ether to provide the lactol 128via epimerization of the formyl group with TBAF. The keto-alcohol 129 was obtained by reduction, selective protection of the resulting primary alcohol and 2-azaadamantane N-oxyl (AZADO)-catalyzed oxidation. Regioselective enol triflate formation followed by a Kumada coupling42,43 and deprotection of the silyl-ether led to the compound 130, which was oxidized with 2-iodoxybnzoic acid (IBX) followed by a Nozaki-Hiyama coupling44 with the allyliodide 131 in the presence of chromous chloride to afford a 1:1 epimeric mixture of the coupling products 132. The secondary alcohol was removed by a Barton-McCombie procedure. After deprotection of the ketal and hydroxylation of the derived ketone with Davis reagent 134, the derived acyloin was cleaved to give the aldehyde 135. Intramolecular Prins cyclization was effected with dimethylaluminum chloride to afford the tricyclic alcohols 136 as a separable 1:1 E/Z mixture. The protecting group of the primary alcohol of the aldehyde 135 was necessary for the Prins reaction. A Dess-Martin oxidation of the epimeric mixture of the alcohols 136 followed by treatment with phenylselelenyl chloride and hydrogen peroxide oxidation led to the enones 138 and 139. After isomerization of the enone 138 to thermodynamically more stable enone 139, protection of the formyl group, hydrolysis of the ester and deprotection furnished (–)-scabronine G (141).
Total synthesis of (–)-scabronine G (141) by Kanoh et al.41
(+)-Chatancin (154) and its congeners are cembrane diterpenoids, which were isolated from Sarcophyton elegans as well as Sinularia pavida and revealed a potent platelet-activating factor (PAF) antagonist. Their total syntheses by Ding et al. started from a [4 + 2] domino Michael reaction between the enone 142 and Rawal diene 143 to proceed at 70 °C in good diastereoselectivity (>20:1) without promoter, which was followed by the addition of hydrogen fluoride for the β-elimination to afford the requisite cis-bicyclic diketone 145 (Figure 19).45 Treatment of the diketone 145 with dimethyl carbonate in the presence of potassium t-butoxide proceeded presumably via transesterification with benzoate moiety followed by intramolecular Dieckmann condensation to result in the formation of the lactone 146. In spite of the enol form of the lactone 146, the conjugate addition of the Grignard reagent 147 was successful and subsequent intramolecular aldol condensation provided the tetracyclic enone 149 as a common synthetic intermediate for chatancin family. Reduction of 149 with sodium cyanoborohydride proceeded regio- and diastereoselectively, and then removal of the carbonyl group via Wolff-Kishner reaction led to the hydroxy-lactone 150. Hydration of the olefin was carried out by Shenvi's Mn(dpm)3-catalyzed protocol46 to give the bis-hydroxy lactone 151 in a 6.5:1 diastereoselectivity. β-Hydroxy-αβ-unsaturated moiety was introduced by mesylation of 151 and methanolysis of the derived epoxide 152 in situ afforded the ester 153. Dess-Martin oxidation resulted in spontaneous hemi-acetalization to accomplish a total synthesis of (+)-chatancin (154). Starting from the diketone 149, syntheses of (–)-3-oxochatancin (157) and (+)-sarcohytin (158) via racemization of the isopropyl group and translactonization were also achieved.
Total syntheses of (+)-chatancin (154) (–)-3-oxochatancin (157) and (+)-sarcohytin (158) by He et al.45
Hetero-Domino Michael Reaction
So far, examples of domino Michael reactions initiated by the anions of hetero-atoms are not enough due to their relative stability to carbanions in the thermodynamically controlled Michael reactions. However, development of organocatalysts has enabled a variety of hetero-domino Michael reactions for the construction of a heterocyclic compounds14 and inspired total syntheses of natural products also.
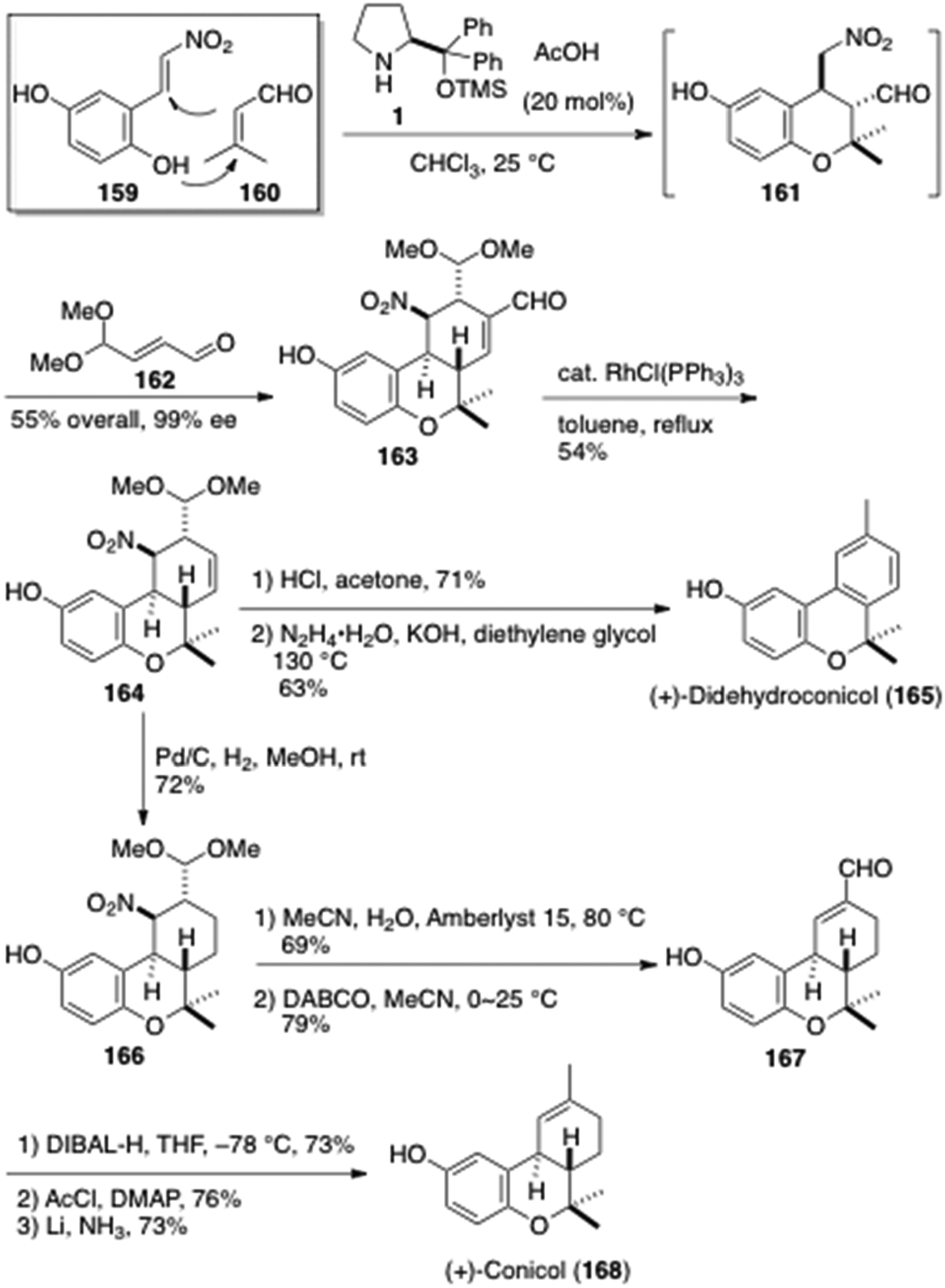
(+)-Conicol (168) is a meroterpenoid isolated from a marine invertebrate, the ascidian Aplidium conicum. The core hexahydro-6H-benzo[c]chromene architecture 161 was constructed by a [4 + 2] oxa-domino Michael reaction between the nitroolefin 159 and the unsaturated aldehydes 160 catalyzed by the prolinol 1 by Hong et al. (Figure 20).47 After the oxa-domino Michael reaction, the unsaturated aldehyde 162 was added furthermore in situ to result in stereoselective Michael reaction followed by a aldol condensation to provide the tricyclic compound 163, in which highly enantioselective three component [4 + 2 + 2]-oxo-domino Michael-Michael-aldol reaction was realized in 55% yield and 99% enantioselectivity in a one-pot operation. Decarbonylation of the aldehyde 163 with Wilkinson's catalyst gave the olefin 164. Hydrolysis of 164 accompanied β-elimination of the nitro group and aromatization to provide aromatic aldehyde, which was transformed to (+)-didehydroconicol (165) after Wolff-Kishner reaction. Hydrogenation of the unsaturated acetal 164 to the acetal 166 followed by hydrolysis and subsequent β-elimination led to the αβ-unsaturated aldehyde 167. Transformation of the formyl group of 167 to a methyl group required three steps, diisobutylaluminum hydride reduction, acetylation and metal reduction in liquid ammonia to accomplish the total synthesis of (+)-conicol (168). Wolff-Kishner reaction of the unsaturated aldehyde 167 was not fruitful. The absolute configuration of (+)-conicol (168) was thereby elucidated through this work, which was ambiguous so far.
Total syntheses of (+)-didehydroconicol (165) and (+)-conicol (168) by Hong et al.47
Racemic incarviditone (173) along with rengyolone (171) and incarvilleatone (177) was isolated from Incarvillea delevayi and Incarvillea younghusbandii, respectively, in which racemic rengyolone (171) was postulated to be a biogenetic precursor of incarvilleatone (177). Brown et al.48 and at the same time Zhao et al.49 reported syntheses of these compounds (Figure 21). Rengyolone (171) was prepared in a large quantity through monoprotection of the primary alcohol 169, (diacetoxyiodo)benzene (PIDA) oxidation to the quinol 170 and subsequent intramolecular oxa-Michael reaction. Alternative one-pot synthesis of rengyolone (171) employing Oxone® oxidation resulted in low yield. Lawrence et al. treated rengyolone (171) with potassium carbonate to furnish racemic incarviditone (173) as a result of a homochiral [3 + 2] oxa-domino Michael reaction, and racemic incarvilleatone (177) as a result of heterochiral [3 + 2] oxa-domino Michael reaction and subsequent aldol reaction, whereas Tang et al. used in the reaction sodium hydride in dichloromethane resulting better yield in 40% for 173 and 38% for 176. The originally proposed structure of incarviditone (173) was revised through this synthesis.
Total syntheses of racemic incarviditone (173) and incarvilleatone (175) by Brown et al.48 and Zhao et al.49
(+)-Angiopterlactone B (an enantiomer of 183) is a structurally complex bis-lactone metabolite isolated from the rhizome of Angiopteris caudatiformis. A concise total synthesis of (–)-angiopterlactone B (183) was promoted based on a proposed biogenesis of angiopterlactones by Thomson et al. (Figure 22).50 The known (S)-alcohol 17951 prepared by Noyori's transfer hydrogenation was transformed to the δ-lactone 180 as a mixture of diastereomers by Achmatowicz rearrangement52 using N-bromosuccinimide. Dynamic kinetic isomerization of the δ-lactone 180 mediated by Brønsted acid and iridium catalyst53 gave the isomeric δ-lactone 181 as a single diastereomer on a multigram scale. Treatment of the δ-lactone 181 with 20 mol% of potassium carbonate at 70 °C promoted ring contraction to the γ-lactone 182, which was reacted with the remaining δ-lactone 181 in a stereoselective [3 + 2] oxa-domino Michael manner to complete a scalable protecting free total synthesis (–)-angiopterlactone B (183) in four steps. Selective formation of the sterically congested cis-syn-cage structure is worthy of note. Since the sign of optical rotation of synthetic angiopterlactone B (183) was opposite to that of natural product, the originally proposed absolute stereostructure of (+)-angiopterlactone B was revised to be the antipode of structure 183 in the Figure 22. An intermediary single Michael product, which corresponds to angiopterlactone A, was not obtained.
Total synthesis of (–)-angiopterlactone B (183) by Thomson et al.50
Bhattacharya et al. described a parallel study on the total synthesis of (–)-angiopterlactone B (183) according to the same biomimetic strategy as Lawrence et al. in the Figure 22, by employing the [3 + 2] oxa-domino Michael reaction between the γ-lactone 182 and the δ-lactone 181, which were synthesized separately from 3,4-di-O-acetyl-L-rhamnal (180) (Figure 23).54 Epoxidation of the rhamnal 184 with Lewis acid was followed by rearrangement and β-elimination to yield the δ-lactone 185. Treatment with barium hydroxide resulted in hydrolysis and subsequent translactonization to afford the γ-lactone 182, which was protected as the silyl-ether 186, whereas treatment of the rhamnal 184 with borontrifluoride etherate effected elimination of acetic acid and subsequent acetalization to deliver ethyl-ether, which was hydrolyzed to give the alcohol 187. Mitsunobu inversion of the secondary alcohol of 187 and subsequent hydrolysis afforded the alcohol 189. After protection of the secondary alcohol, Jones oxidation and deprotection led to the δ-lactone 181. The designed [3 + 2] oxa-domino Michael reaction between the γ-lactone 186 and the δ-lactone 181 was attempted with sodium hydride to provide angiopterlactone B (183) in 32% yield recovering the γ-lactone 186, which suggested the sensitive nature of the δ-lactone 181 to bases. Then, the treatment of the δ-lactone 181 with TBAF induced partial isomerization to the more stable γ-lactone 182, which reacted in situ with the remaining δ-lactone 181 to furnish angiopterlactone B (183) in 62% yield.
Total synthesis of angiopterlactone B (183) by Kotammagari et al.54
Takasu et al. described a formal synthesis of racemic deplancheine (197), an indolo[2,3-a]quinolizidine alkaloid isolated from the New Caledonian plant Alstonia deplanchei (Figure 24).55 Treatment of the acrylamide 190 with tert-butyldimethylsilyl triflate and hexamethyldisilazane yielded the piperidinone 191 in 90% yield without a single Michael product as a result of [4 + 2] aza-domino Michael dimerization. After protection of the piperidinone 191 as a tri-Boc derivative, transesterification led to the ester 192. Bischler-Napieralski reaction of 192 with phosphorous oxychloride in acetonitrile followed by sodium borohydride reduction afforded a diastereomeric mixture of the tetracyclic compounds 193 and 194 in 55% and 26% yield, respectively. The ester 193 was transformed to the Weinreb amide 195, which was subjected to Grignard addition to furnish the known ketone 196, a precursor of deplancheine (187) synthesis.
Formal total synthesis of racemic deplancheine (187) by Takasu et al.55
In this review, the recent progress from 2005 to 2021 in the domino Michael reaction is summarized focusing on its application to syntheses of cyclic natural products as key steps. In the initial studies on domino Michael reactions, kinetic enolates generated by strong bases such as LDA were mainly used to initiate the domino reactions.6,56,57 In these reactions, the ketone enolate underwent Michael reactions intermolecularly or intramolecularly, and the new enolate generated on the Michael acceptor underwent subsequent Michael reactions intramolecularly to give thermodynamically stable cyclic compounds. In the reactions using strong bases, aldehydes and αβ-unsaturated aldehydes could not be used as Michael donors or Michael acceptors due to the highly electrophilic nature of the formyl groups. Another problem is the use of a stoichiometric amount of strong base in spite of the plausible reusability of the base in a Michael reaction. Example of the domino Michael reaction using a catalytic amount of base is rare.58 Recent developments in the area of organocatalysts has changed the game in the domino Michael reaction. The use of organocatalysts has widely expanded the degree of freedom in the design of reaction and the substrate scope and has greatly advanced the domino Michael reaction, in which a variety of frameworks have been constructed11–16,59 under mild reaction conditions employing a variety of organocatalysts. Among organocatalysts, the Hayashi-Jørgensen catalysts 1 and 2 are preferable to a variety of substrates due to easy availability of both enantiomers by easy transformations compared to other organocatalysts such as bifunctional thiourea- and squaramide-derived catalysts or quinine derivatives. Further development of the Hayashi-Jørgensen catalysts 1 and 2 such as reduction of amount of catalyst, immobilization on solid surface to give robustness and recyclability, are desired. In spite of abundant protocols of organocatalytic domino Michael reactions, application of the reaction to natural product syntheses as a key step, which finally led to final total syntheses, is limited as shown in this review. The potential of the domino Michael reaction is still high owing to a variety of chemical economies cited above, which leads to sustainability in synthetic organic chemistry. Development of more multifold domino Michael reaction in natural product synthesis is expected. We are looking forward to see further development and emergence of novel trends in this area.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
ORCID iD
Hisahiro Hagiwara
Trial Registration
Not applicable, because this article does not contain any clinical trials.
Supplemental Material
Supplemental material for this article is available online.
References
1.
TietzeLF. Domino reactions in organic synthesis. Chem Rev. 1996;96(1):115–136. doi:10.1021/cr950027e
2.
TietzeLFBrascheGGerickeKM. Domino reactions in organic synthesis. Wiley-VCH; 2006.
3.
HayashiY. Time economy in total synthesis. J Org Chem. 2021;86(1):1–23. doi:10.1021/acs.joc.0c01581
4.
HayashiY. Time and pot economy in total synthesis. Acc Chem Res. 2021;54(6):1385–1398. doi:10.1021/acs.accounts.0c00803
5.
HagiwaraHUdaHKodamaT. Synthetic study on several eremophilane sesquiterpenes using a common intermediate. J Chem Soc, Perkin Trans. 1980;1980(0):963–977. doi:10.1039/P19800000963
6.
IharaM. Syntheses of biologically active natural products and leading compounds for new pharmaceuticals employing effective construction of a polycyclic skeleton. Chem Pharm Bull. 2006;54(6):765–774. doi:10.1248/cpb.54.765
7.
GaugeleDMaierME. Approach to the core structure of the polycyclic alkaloid palhinine A. Synlett. 2013;24(8):955–958. doi:10.1055/s-0032-1316899
8.
TrostB. M. Atom economy-A challenge for organic synthesis: homogeneous catalysis leads the way. Angew Chem Int Ed. 1995;34(3):259–281. doi:10.1002/anie.199502591
9.
ListB (ed). Asymmetric organocatalysis 1: Lewis base and acid catalysts. Thieme; 2012.
10.
MaruokaB (ed). Asymmetric organocatalysis 2: Brønsted base and acid catalysts, and additional topics. Thieme; 2012.
GrondalCJeantyMEndersD. Organocatalytic cascade reactions as a new tool in total synthesis. Nat Chem. 2010;2(3):167–178. doi:10.1038/nchem.539
16.
HongB-CRajaAShethV-M. Asymmetric synthesis of natural products and medicinal drugs through one-pot-reaction strategies. Synthesis (Mass). 2015;47(21):3257–3285. doi:10.1055/s-0035-1560344
17.
MarigoMWabnitzTCFielenbachDJørgensenKA. Enantioselective organocatalyzed α-sulfenylation of aldehydes. Angew. Chem., Int Ed Engl. 2005;44(5):794–797. doi:10.1002/anie.200462101
18.
HayashiYGotohHHayashiTShojiM. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew Chem Int Ed. 2005;44(5):4212–4215. doi:10.1002/anie.200500599
19.
FranzenJMarigoMFielenbachDWabnitzTCKjarsgaardAJørgensenKA. A general organocatalyst for direct α-functionalization of aldehydes: stereoselective C−C, C−N, C−F, C−Br, and C−S bond-forming reactions. Scope and mechanistic insights. J Am Chem Soc. 2005;127(5):18296–18304. doi:10.1021/ja056120u
20.
HongB-CHsuaC-SLeeG-H. Enantioselective total synthesis of (+)-galbulin via organocatalytic domino Michael-Michael-aldol condensation. Chem Commun. 2012;48(18):2385–2387. doi: 10.1039/c2cc16682h
21.
UmekuboNHayashiY. Asymmetric synthesis of Corey lactone and latanoprost. Eur J Org Chem. 2020;2020(39):6221–6227. doi:10.1002/ejoc.202001063
22.
UmekuboNSugaYHayashiY. Pot and time economies in the total synthesis of Corey lactone. Chem Sci. 2020;11(5):1205–1209. doi:10.1039/c9sc05824a
23.
HayashiYUmekuboN. Direct asymmetric Michael reaction of α,β-unsaturated aldehydes and ketones catalyzed by two secondary amine catalysts. Angew Chem Int Ed. 2018;57(7):1958–1962. doi:10.1002/anie.201710085.
24.
UmekuboNHayashiY. Pot-economical total synthesis of clinprost. Org Lett. 2020;22(23):9365–9370. doi: 10.1021/acs.orglett.0c03616
25.
TamaoKIshidaNTanakaTKumadaM. Hydrogen peroxide oxidation of the silicon-carbon bond in organoalkoxysilanes. Organometallics. 1983;2(11):1694–1696. doi:10.1021/om50005a041
26.
FlemingIHenningRPlautH. The phenyldimethylsilyl group as a masked form of the hydroxy group. J Chem Soc, Chem Commun. 1984;1984(1):29–31. doi:10.1039/C39840000028
27.
LarghiELKaufmanTS. Synthesis of oxacycles employing the oxa-Pictet-Spengler reaction: recent developments and new prospects. Eur J Org Chem. 2011;2011(27):5195–5231. doi:10.1002/ejoc.201100271
28.
HongB-CLiaoW-KDangeNSLiaoJ-H. One-pot organocatalytic enantioselective domino double-Michael reaction and Pictet-Spengler lactamization reaction. A facile entry to the “inside yohimbane” system with five contiguous stereogenic centers. Org Lett. 2013;15(3):468–471. doi:10.1021/ol3032329
29.
YangV-WHongB-CKaoH-K,et al.One-pot dichotomous construction of inside-azayohimban and pro-azayohimban systems via an enantioselective organocatalytic cascade; their use as a model to probe the (aza-)indole local solvent environment. Org Lett. 2015;17(23):5816–5819. doi: 10.1021/acs.orglett.5b02949
30.
HeraviMMNazariN. Bischler-Napieralski reaction in total synthesis of isoquinoline based natural products. An old reaction, a new application. Curr Org Chem. 2015;19(24):2358–2408. doi:10.2174/138527281966150730214506
31.
KaoH-KLinX-JHongB-CYangV-WLeeG-H. Enantioselective synthesis of yohimbine analogues by an organocatalytic and pot-economic strategy. J Org Chem. 2019;84(18):12138–12147. doi:10.1021/acs.joc.9b01193
32.
WangYLuoY-CZhangH-BXuP-F. Concise construction of the tetracyclic core of lycorine-type alkaloids and the formal synthesis of α-lycorane based on asymmetric bifunctional thiourea-catalyzed cascade reaction. Org Biomol Chem. 2012;10(41):8211–8215. doi:10.1039/c2ob26422f
33.
RanaNKHuangHZhaoJC-G. Highly diastereodivergent synthesis of tetrasubstituted cyclohexanes catalyzed by modularly designed organocatalysts. Angew Chem, Int Ed Engl. 2014;53(29):7619–7623. doi:10.1002/anie.201404072
34.
RajaAHongB-CLiaoJ-HLeeG-H. Organocatalytic enantioselective Michael-Michael-Henry reaction cascade. An entry to highly functionalized Hajos-Parrish-type ketones with five to six contiguous stereogenic centers and two quaternary carbons. Org Lett. 2016;18(8):1760–1763. doi:10.1021/acs.orglett.6b00459
35.
JhuoD-HHongB-CChangC-WLeeG-H. One-pot organocatalytic enantioselective Michael-Michael-aldol-Henry reaction cascade. A facile entry to the steroid system with six contiguous stereogenic centers. Org Lett. 2014;16(10):2724–2727. doi:10.1021/ol501011t
36.
RajaAHongB-CLeeG-H. Organocatalytic enantioselective Michael-Michael-Michael-aldol condensation reactions: control of five stereocenters in a quadruple-cascade asymmetric synthesis of highly functionalized hexahydrophenanthrenes. Org Lett. 2014;16(21):5756–5759. doi:10.1021/ol502821e
37.
HuangJGuYGuoK,et al.Bioinspired total synthesis of homodimericin A. Angew Chem Int Ed. 2017;56(27):7890–7894. doi:10.1002/anie.201702768
38.
KuwajimaIUrabeH. Regioselective arylation of silyl enol ethers of methyl ketones with aryl bromides. J Am Chem Soc. 1982;104(24):6831–6833. doi: 10.1021/ja00388a083
39.
NarasakaKSoaiKAikawaYMukaiyamaT. The Michael reaction of silyl enol ethers with αβ-unsaturated ketones and acetals in the presence of titanium tetraalkoxide and titanium tetrachloride. Bull Chem Soc Jpn. 1976;49(3):779–783. doi: 10.1246/bcsj.49.779
40.
HagiwaraHOkanoAUdaH. One-step synthesis of tricyclo[5.3.1.03,8]undecan-4,11-diones by three consecutive Michael reaction. A formal synthesis of (±)-seychellene. J Chem Soc, Chem Commun. 1985;1985(15):1047. doi:10.1039/C39850001047
41.
KanohNSakanishiKIimoriENishimuraKIwabuchiY. Asymmetric total synthesis of (–)-scabronine G via intramolecular double Michael reaction and Prins cyclization. Org Lett. 2011;13(11):2864–2867. doi:10.1021/ol200873y
42.
TamaoKSumitaniKKumadaM. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J Am Chem Soc. 1972;94(12):4374–4376. doi:10.1021/ja00767a075
43.
EnquistJAStoltzBM. The total synthesis of (–)-cyanthiwigin F by means of double catalytic enantioselective alkylation. Nature2008;453(7199):1228–1231. doi:10.1038/nature07046
44.
OkudeYHiranoSHiyamaTNozakiH. Grignard-type carbonyl addition of allyl halides by means of chromous salt. Chemospecific synthesis of homoallyl alcohols. J Am Chem Soc. 1977;99(9):3179–3181. doi:10.1021/ja00451a061
45.
HeCXuanJRaoP,et al.Total syntheses of (+)-sarcophytin, (+)-chatancin, (–)-3-oxochatancin, and (–)-pavidolide B: a divergent approach. Angew Chem, Int Ed Engl. 2019;58(15):5100–5104. doi:10.1002/anie.201900782
46.
CrossleySWMObradorsCMartinezRMShenviRA. Mn–, Fe–, and Co-catalyzed radical hydroformylations of olefins. Chem Rev. 2016;116(15);8912–9000. doi: 10.1021/acs.chemrev.6b00334
47.
HongB-CKotamePTsaiC-WLiaoJ-H. Enantioselective total synthesis of (+)-conicol via cascade three-component organocatalysis. Org Lett. 2010;12(4):776–779. doi:10.1021/ol902840x
48.
BrownPDWillisACSherburnMSLawrenceAL. Total synthesis of incarviditone and incarvilleatone. Org Lett. 2012;14(17):4537–4539. doi:10.1021/ol302042u
49.
ZhaoKChengG-JYangH,et al.Total synthesis of incarvilleatone and incarviditone: insight into their biosynthetic pathways and structure determination. Org Lett. 2012;14(18):4878–4881. doi:10.1021/ol302205w
50.
ThomsonMINicholGSLawrenceAL. Total synthesis of (−)-angiopterlactone B. Org Lett. 2017;19(9):2199–2201. doi:10.1021/acs.orglett.7b00929
51.
PatersonIHaslettGW. Synthesis of the C1C11 western fragment of madeirolide A. Org Lett. 2013;15(6):1338–1341. doi:10.1021/ol400280b
52.
CroattMPCarreiraEM. Proving the role of the mycosamine C2’-OH on the activity of amphotericin B. Org Lett. 2011;13(6):1390–1393. doi: 10.1021/ol2000765
53.
WangH-YYangKBennettSRGuoS-rTangW. Iridium-catalyzed dynamic kinetic isomerization: expedient synthesis of carbohydrates from Achmatowicz rearrangement products. Angew Chem, Int Ed Engl. 2015;54(30):8756–7859. doi:10.1002/anie.201503151
54.
KotammagariTKGonnadeRGBhattacharyaAK. Biomimetic total synthesis of angiopterlactone B and other potential natural products. Org Lett. 2017;19(13):3564–3567. doi:10.1021/acs.orglett.7b01525
55.
TakasuKNishidaNTomimuraAIharaM. Convenient synthesis of substituted piperidinones from αβ-unsaturated amides: formal synthesis of deplancheine, tacamonine, and paroxetine. J Org Chem. 2005;70(10):3957–3962. doi:10.1021/jo050261x
56.
LeeRA. Reactions of α‘-dienolates with Michael acceptors. Synthesis of bicyclo[2.2.2]octan-2-ones. Tetrahedron Lett. 1973;14(35):3333–3336. doi:10.1016/S0040-4039(01)86908-2
57.
HagiwaraHNakayamaKUdaH. Michael Reactions of α-phenylthio and α-phenylsulfinyl crotonic esters with cyclic enones. Bull Chem Soc Jpn. 1975;48(12):3769–3770. doi:10.1246/bcsj.48.3769
58.
HagiwaraHEndouSFukushimaMHoshiTSuzukiT. The autocatalytic domino Michael reaction leading to bicyclo[2.2.2]octane-2,5-dione derivatives. Org Lett. 2004;6(7):1115–1118. doi: 10.1021/ol049948e
59.
VeticaFde FigueiredoRMOrsiniMTofaniDGasperiT. Recent advances in organocatalytic cascade reactions toward the formation of quaternary stereocenters. Synthesis (Mass). 2015;47(15):2139–2184. doi:10.1055/s-0034-1378742