The isolation, structure elucidation, synthesis, biological properties, and biosynthesis of the homopurine alkaloids are reviewed, with an emphasis on the “victim-guardian” relationships between co-occurring alkaloids.
This review is focused on a small group of microbial metabolites in which the canonical 5:6 heterocyclic1 system of a purine (1) appears as an expanded 5:7 ring system, either an imidazo[4,5-d][1,3]diazepine (2) or an imidazo[4,5-e][1,4]diazepine (3) system.
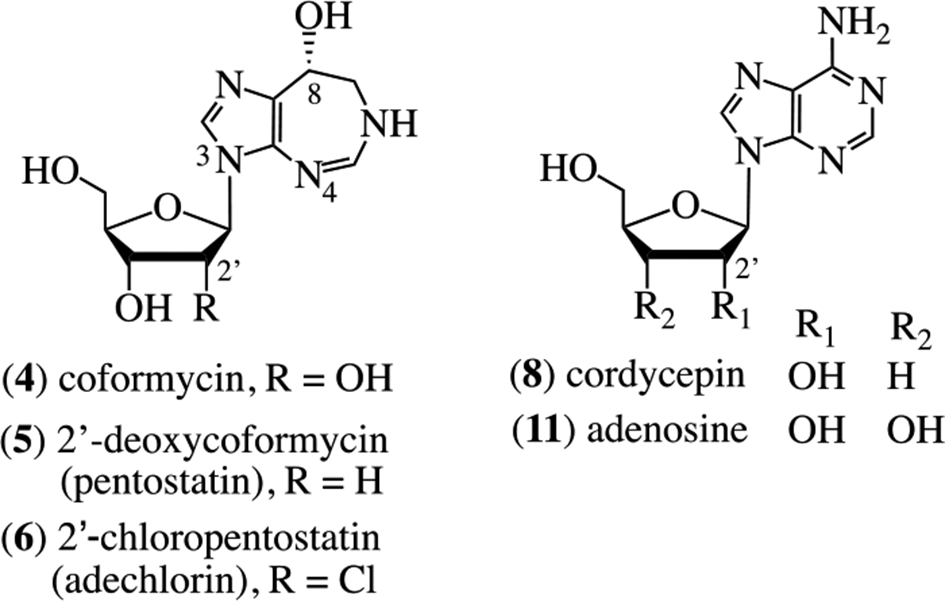
These alkaloids include metabolites in which the pyrimidine ring of adenine is expanded by a single carbon atom and becomes either C-8 or C-6 in the respective new ring system; collectively, these are known as the homopurine alkaloids. Here, the isolation, chemistry, biology, and biosynthesis of these compounds are discussed, mostly from a historical perspective. There are no detailed prior reviews of the homopurine alkaloids. A review from 2002 presented a discussion of the chemical modifications designed to explore their anticancer and antiviral activities.1 Four bacterial members of this series will be highlighted, namely, coformycin (4), pentostatin (also known as 2′-deoxy-coformycin) (5), adechlorin (2′-chloro-2′-deoxycoformycin, 2′-chloropentostatin) (6), and adecypenol (7). The intriguing biochemical relationships between structurally different, yet biosynthetically synchronous cometabolites, which result in protection from metabolism by adenosine deaminase (ADA) are discussed, as is a similar relationship discovered in the chemical ecology of the purine-related, fungal metabolite, cordycepin (8).
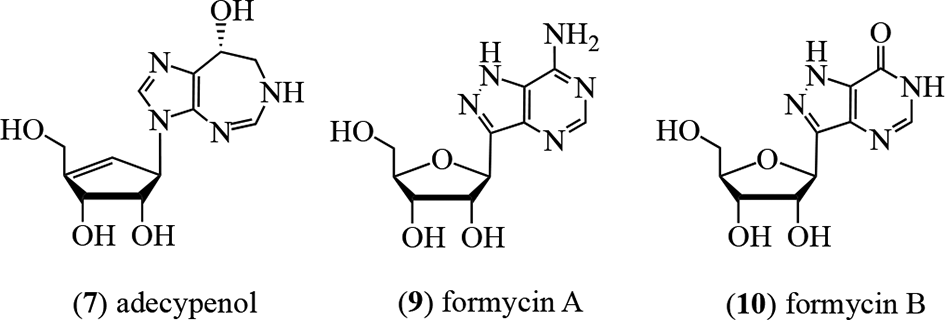
The history begins with the antibiotic formycin A (9),2 which was isolated from the bacterium Nocardia interforma and inhibits cancer cells, Mycobacterium sp. 607, Xanthomonas oryzae,3 and the influenza virus.4 Deamination of 9 in situ by the widely distributed enzyme ADA (adenosine aminohydrolase, EC 3.5.4.4) affords formycin B (10) which possesses significantly reduced activity. Niida et al isolated coformycin (4) from cultures of N. interforma and Streptomyces kaniharaensis SF-557 as a cometabolite of formycin A (9).5,6 They showed that 4 had a profound synergistic effect on the cytotoxicity of 9 on both bacterial and cancer cells.7 It was determined that the mechanism of the enhancement of activity for coformycin (4) was to act as a highly effective (89.1%) inhibitor of the deaminase, thereby extending the availability of 9 and retaining biological activity through maintaining intracellular concentrations of 9 rather than the less active 10. Coformycin (4) was assessed to be the strongest known inhibitor (98.1%) of the deamination of adenosine (11) by ADA.6 The structure of coformycin as 4 was proposed several years later following degradative studies which afforded d-ribose and by the way of an X-ray crystallographic analysis.8,9 Structure confirmation came through a concomitant synthesis (vide infra).10
Shortly thereafter, an important metabolite, the 2′-deoxy-derivative of 4, also known as pentostatin (5), was characterized chemically and spectroscopically from cultures of Streptomyces antibioticus by researchers at Parke, Davis and Co., Ann Arbor, MI, United States.11 This structure was also confirmed through synthesis.12,13 The natural 8R-isomer of pentostatin (5) is an exceptional inhibitor of calf intestine ADA with a dissociation constant of 2.5 × 10−12 M; the 8S-isomer is 1.3 × 107 fold less active.14 However, there is an inherent ambiguity in this assessment of bioactivity because epimerization at the C-8 locus may take place under neutral or mildly acidic aqueous conditions. Attempts to enhance the deaminase activity of 5 were explored through analog synthesis.15,16
Pentostatin (5) was also isolated from the fungus Aspergillus nidulans Y176-2, where it is a cometabolite of the nucleoside cordycepin (8).17,18 Cordycepin (8), which shows a range of biological properties (vide infra), was evaluated against refractory TdT-positive leukemia in combination with the ADA inhibitor pentostatin (5); the latter guarding against the facile deamination of the former.19 Interestingly, a significant biological and biosynthetic rationale exists for this “victim-guardian” co-existence, as discussed in more detail in the biosynthesis section.
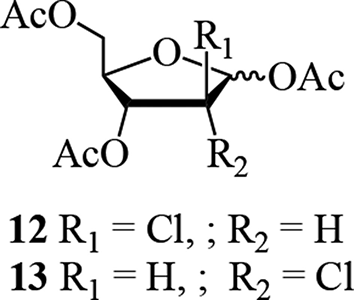
A flurry of isolations of homopurine alkaloids was reported in the mid-1980s, characteristically using ADA inhibition as a bioassay for directed fractionation. The homopurine alkaloid, 2′-chloropentostatin, also known as adechlorin (6), was initially obtained by the Parke-Davis group from Actinomadura sp. ATCC 39365.20,21 Detailed spectral comparison with 4 and 5 showed that C-2′ was shifted upfield 10 to 12 ppm by the chlorine substituent, and spectral interpretation of the tetraacetate supported this assignment for location of the chlorine atom. Characterization of the sugar moiety was accomplished through stereospecific synthesis of the 1,3,5-tri-O-acetyl-2-chloro-2-deoxy-α,β-d-arabinoses (12, 3:1 α/β anomeric mixture) and 1,3,5-tri-O-acetyl-2-chloro-2-deoxy-α,β-d-ribofuranoses (13); it was the latter which was identical to the corresponding derivative obtained from the natural product.20
Adechlorin (6) showed strong activity against Enterococcus faecalis PD 05045 and increased the sensitivity of this organism to the antiviral agent Ara-A (vidarabine, 14) probably due to its potent (1.1 × 10−10 M) ADA inhibition activity.21 The same alkaloid was reported by Ōmura et al from the strain Actinomadura sp. OMR-37 collected in Hodaka-cho, Nagano Prefecture, Japan,22 and demonstrated potent ADA inhibition activity. Based on the similarities of the proton and carbon-13 NMR spectra of adechlorin (6) with those of 4 and 5, the same aglycone moiety was established, thereby locating the chlorine atom in the sugar moiety. Comparison with these alkaloids revealed that C-2′ was the site of substitution, with the C-1 atom in an α (axial) configuration. No activity was observed against a variety of bacteria and fungi at 1.0 mg/mL. However, the Ki values for the inhibition of ADA were deduced to be 5.3 × 10−10, 2.1 × 10−10, and 7.6 × 10−11 M for adechlorin (6), coformycin (4), and pentostatin (5), respectively. As expected, each of these alkaloids enhanced the antiviral activity of Ara-A (14).22 Adechlorin (6) was the least toxic of these alkaloids in an acute mouse model by a factor of at least 4.
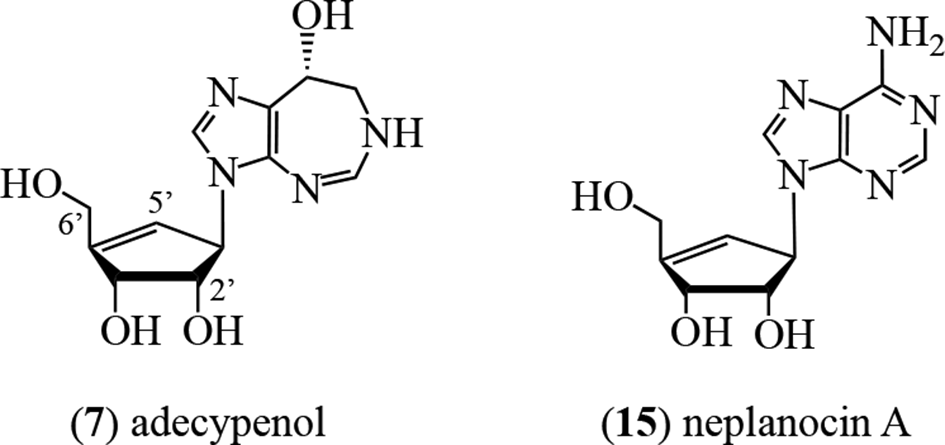
A further ADA inhibitor, adecypenol (7), was characterized by Ōmura’s group from cultures of the Streptomyces sp. strain OM-3223. The organism was derived from a soil sample collected from Inokashira Park in Musashino City, Tokyo, Japan, and also produced coformycin (4).23,24 The proton NMR data indicated that the aglycone moiety of 7 was identical to that in 4, 5, and 6, ie, it was the R-enantiomer of 3,6,7,8-tetrahydro-imidazo[4,5-d][1,3]diazepin-8-ol. Through the formation of a pentaacetate of the parent alkaloid, the sugar moiety was deduced to have 3 hydroxy groups. These were placed at the C-2′, C-3′, and C-6′ locations through proton NMR experiments. The sugar unit of 7 possessed a 4′,5′-double bond, with the olefinic proton appearing as a doublet of triplets at 5.83 ppm and C-4′ at 126.4 ppm.23 These spectral data correlated with those of the corresponding structural unit in neplanocin A (15).25
Adecypenol (7) and pentostatin (5) were also examined by Ōmura’s group for the ability to enhance the cytotoxic and in vivo antitumor activity of Ara-A (14).26 Fifty percent inhibition for 14 against HeLa cells occurred at 27 µg/mL, which was enhanced to 4.3 µg/mL by 0.28 µg/mL of 6 and to 3.9 µg/mL by 0.02 µg/mL of 5, reflecting the relative level of deaminase inhibition of the adjuvants. In a L1210 leukemia model in mice, neither 6 nor 14 were active separately; however, when administered together, a 94% increase in life span was observed at 2 mg/mL (no composition ratio was given).
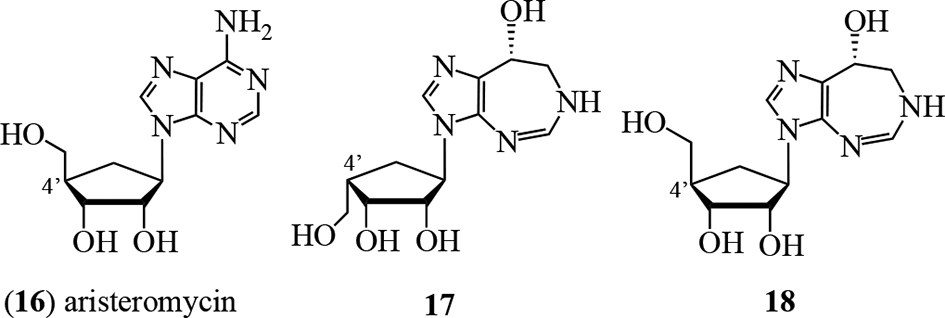
A culture of a Saccharothrix sp. NCIMB 40131, acquired from a soil sample collected in Évreux, France, showed herbicidal activity, and was examined by groups at Schering Agrochemicals and Glaxo Group Research in the United Kingdom.27 Bioactivity-directed fractionation for the growth inhibition of Polygonum lapathifolium L. (Polygonaceae) afforded 6, aristeromycin (16), and 3 new analogs of 6, namely, 17, 18, and 19. Proton and carbon-13 NMR spectral data established the aglycone moiety. Alkaloids 17 and 18 were shown to be isomers at the C-4′ position through nOe spectroscopy. Alkaloid 19 was considered to be a hexose (glucose?) derivative of 18, although the structure was not fully determined.27 In field tests, the most active against Hordeum vulgare, Polygonum lapathifolium, and Avena fatua growth was 18, while the C-4′ isomer 17 was almost inactive. The mechanism of action does not appear to have been determined.
Unlike other homopurine alkaloids, azepinomycin (20) isolated from cultures of Streptomyces sp. MF718-0328 lacks the N-ribofuranosyl moiety and has a different heterocyclic scaffold. The structure was unambiguously assigned through X-ray crystallographic analysis as 6-hydroxy-4,5,6,7-tetrahydro-3H-imidazo-[4,5-e][1,4]diazepin-8-one29 and was supported by several syntheses.29-33 It showed potent inhibitory activity against guanine deaminase, and cytotoxic activity against L5178Y cells was demonstrated.28
The synthetic approaches to the homopurine alkaloids were initiated with coformycin (4), using a route which began with 9-β-d-ribofuranosylpurine (nebularine) (21).10 Stereospecific, photochemical addition34 using a 10 W mercury lamp at 5°C for 3.5 hours on the 2′,3′,5′-tri-O-acetate derivative 22 of 21 in MeOH afforded 9-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-6-hydroxy-methyl-1,6-dihydro-purine (23) in 96% yield. Treatment with mesyl chloride, sodium hydride then potassium t-butoxide in DME followed by Dowex 1-X2 (OH−), provided, stereospecifically, coformycin (4) in 38% yield, identical with the natural product (Figure 1).10 The ring-expansion mechanism was considered to involve an aziridine intermediate 24. Cleavage through bond a affords 4 and indicates that the hydroxymethyl group in 23 also has the R-configuration.
The potent inhibition of ADA by pentostatin (5) garnered synthetic interest of the parent alkaloids and analogs in the late 1970s to 1990s. The first total synthesis of the natural product was reported in 1979 by Baker and Putt.12 The 7-step synthesis commenced with the benzylation of 5-nitro-4-styrylimidazole 25, followed by ozonolysis with an oxidative work-up which gave access to the acid 26. By employing the imidazoyl derivative of acid 26, elaboration of the -CH2NH2 was achieved in 75% yield to afford 27. With what was considered the most difficult transformation completed, reduction of the nitro group and subsequent debenzylation of 27 afforded the diamine hydrochloride 28. Cyclization of 28 to the penultimate compound, 1,3-diazepine 29, was accomplished using triethyl orthoformate. Glycosylation with pentofuranosyl chloride 30, followed by deacylation and reduction, afforded the natural product pentostatin (5) and its S isomer 31 in a 60:40 ratio (Figure 2).12
Baker-Putt synthesis of 8R- and 8S-pentostatins.12
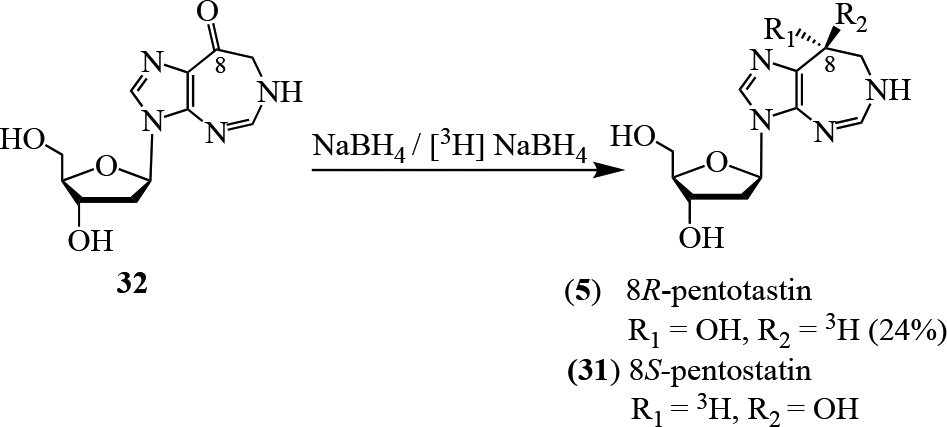
With the bioactivity of the natural product established, the synthesis of labeled pentostatin became of interest. Both 3H-35 and 14C-labeled36 pentostatin derivatives were prepared in order to explore the pharmacokinetic parameters of 5. Starting from diazepin-8-one 32, the synthesis of 8R-pentostatin (5) and its 8S-isomer 31 was achieved using tritium-labeled [3H]NaBH4. The isomers were separated using preparative reverse phase chromatography to yield the desired isomer, 8R-pentostatin (5) in 24% yield (Figure 3).35
In an effort to provide a cost efficient, C-14 labeled form of pentostatin for pharmacokinetic and metabolism studies, Woo and Lee36 constructed the [5-14C] diazepinone core in 60% yield, starting with triethyl ortho[14C]formate and 2‐amino‐1‐(5‐amino‐1H‐imidazol‐4‐yl)ethanone dihydrochloride (33) in the presence of 4A molecular sieves to afford 34. Persilylation followed by glycosylation gave the protected 35. Subsequent deprotective saponification and reduction afforded a R/S mixture, which was purified via preparative HPLC to give the desired C-5 labeled natural product 5 in 39% yield (Figure 4).36
Woo and Lee synthesis of 5-14C-labeled pentostatin (5).36
Azepinomycin (20) also acquired synthetic attention as a result of its similarities to pentostatin (5) and its biological activity. A straightforward synthesis was accomplished through N-alkylation of the amine 36. Umezawa et al found that the use of silver(I) oxide in the presence of DMF was necessary to increase the efficiency of the alkylation due to the poor reactivity of the amine. Reductive ring closure using DIBAL-H gave 37, and base hydrolysis, followed by deglycosidation, afforded a product with similar bioactivity to that reported for the isolated alkaloid (Figure 5).30
The synthesis of azepinomycin (20) was revisited in 1988 and 1994 by Fujii et al with the development of several approaches.31,33 One of the syntheses commenced with modification of the N(9)-benzyladenine (38) to the cleaved 2-amino-N(9)-benzyl-pyrazole 39. Alkylation of 39, followed by deformylation with 1 N NaOH, debenzylation through catalytic hydrogenation, and subsequent base hydrolysis, afforded amidine 40. A 2-step conversion of the amidine 40 to the natural product 20 was achieved in 45% overall yield (Figure 6).
A further synthesis of azepinomycin (20) was reported by Chakraborty et al in 2012,32 and was an outcome of investigations into the structure activity relationships of the synthetic natural product and some analogs. Following the protection of AICAR (41) as t-butyldimethylsilyl ethers, and N-acylation protection with acetic anhydride, the synthesis of 20 was accomplished through 42 (Figure 7) in a similar manner to that of Fujii’s synthesis.33
Chakraborty et al synthesis of azepinomycin (20).32
In a more efficient synthetic protocol, Coggins’ synthesis of 20 featured a 1 step, protecting group free synthesis in water.29 The pH-dependent coupling and Amadori rearrangement of carboxamide 43 and glycolaldehyde 44 resulted in the straightforward synthesis of azepinomycin (20) in 80% yield (Figure 8).
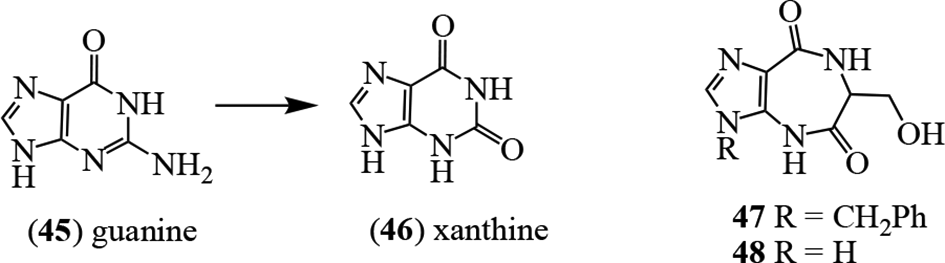
As an example of analog synthesis in the azepinomycin (20) series, a range of the so-called “fat” nucleoside derivatives was prepared in an effort to potentiate the cytotoxic and antiviral activities, and seek enhanced new guanase inhibitors.1,37,38 Guanine deaminase (guanase, EC 3.5.4.3) is of clinical relevance. It catalyzes the Zn2+-assisted deamination of guanine (45) to xanthine (46),39 and is found in human liver, brain, and kidney.40 High levels of guanase in serum are regarded as a biomarker for hepatitis and hepatoma41,42 and liver transplant rejection,43 and have also been observed in cancerous states related to the kidney44-46 and breast.45,47 Thus, inhibitors of guanase may be of value in understanding the biochemical relationships in these disease states and for developing potential treatments. Synthesis of 2 derivatives designed based on the Zn2+-mediated interaction led to 47 and 48, showing comparable activity to azepinomycin (20), although they were not tested simultaneously in the same assay.38
Pentostatin (5) binds tightly to ADA48 and was shown early in its history to enhance the antiviral activity of Ara-A (14).49,50 The mechanism of cytotoxic action was also studied in this time period.51,52 In 1991, pentostatin (5) was approved for the treatment of interferon-refractory hairy cell leukemia, and is currently marketed under the trade name Nipent by Pfizer (https://www.pfizer.com/products/product-detail/nipent_trade). The toxicity of 5 relates to immunosuppression, as well as impacts on liver, renal, and pulmonary functions, and the central nervous system53; the toxicity can be mollified by dosage reduction. Early clinical data suggested effectiveness against lymphocytic leukemia54,55 and hairy cell leukemia (HCL).56-58 A summary of the clinical effects of 5 reported response rates of 62% for Sézary syndrome and 45% for T-prolymphocytic leukemia, together with a 60% survival rate for 165 patients with HCL,59,60 which followed earlier successful clinical results.61,62 Further clinical data on 241 patients with HCL (154 having no prior interferon treatment) revealed mean 5- and 10-year survival rates of 90% and 81%, respectively, for patients with a complete response.63 More recent clinical data have been reviewed, and the current practices and the development of future therapeutic combinations discussed.64,65 Pentostatin (5) has also been used in the treatment of acute graft-versus-host disease,66,67 and chronic lymphocytic leukemia (CLL).68,69 Comparison for CLL with fludarabine indicated that 5 provides better clinical outcomes.69
The adenosine deaminase of Plasmodium falciparum (PfADA) parasites catalyzes the deamination of both adenosine (11) and 5′-methylthioadenosine (49).70 As a follow-up, the 5′-methylthiocoformycins, 50 and 51, were synthesized and showed selective, low nanomolar (0.43 and 0.79 nM, respectively), inhibition of PfADA, with no effect on human ADA.71 Since ADA is essential for purine salvage in P. falciparum,72,73 and inhibition of purine salvage kills the parasite,74 these compounds are of interest as antimalarial agents which will not inhibit human ADA, thereby reducing potential toxicity. They are also of interest in the wider sense, since, as mentioned earlier, the activity of pentostatin (5) on human ADA is a dose-limiting factor in therapy.53 The isolation, chemistry, and biology of these nucleoside antibiotics have been reviewed.75-77
As the clinical demand for pentostatin (5) increased, the need to enhance production levels became evident. The original isolation protocol had yielded only 8 g of 5 from 9500 L of culture beer.78 Researchers at Parke-Davis/Warner-Lambert in Ann Arbor, MI, United States made major modifications to the procedure. Water-insoluble Ara-A (14) is the dominant metabolite of S. antibioticus NRRL 3238,79 and was removed (by 80%) initially, and the filtrate processed through a series of resins. These steps allowed the separation of contaminating small quantities of 4, 2′-deoxyguanosine, and the 8S-diastereoisomer of 5. The isolation of 5 was finalized with charcoal processing and recrystallization; no yield enhancement was cited, however.78
In consideration of the biosynthetic pathways for the homopurine alkaloids, 2 scaffolds are present: the ring-expanded, adenine-like moiety and the pentofuranosyl unit. In adecypenol (7), the sugar unit is replaced by a polyhydroxy-cyclopentene moiety at the N-3 locus. Several crucial biosynthetic questions need to be addressed with respect to the homopurine alkaloids. First, are the 2 core units established separately and then united? Second, if not, then when and how do the respective ring expansion and, where relevant (ie, 5), the loss of the 2′-OH group occur? Furthermore, how and when is the chlorine atom in adechlorin (6) introduced? Details of the biosynthesis of cyclopentene moiety in adecypenol (7) are discussed elsewhere in relation to the biosynthesis of neplanocin A (15).80-82 In the instance of azepinomycin (20), how is the additional carbon atom at C-6 introduced?
The homologs of adenosine (11) in the homopurine alkaloids have incorporated an additional carbon atom in the pyrimidine ring of the nucleus. It was considered that a clue to the biosynthetic pathway may come from the cometabolites. For example, in cultures of S. antibioticus, the co-, and dominating, metabolite with pentostatin (5) is 9-β-d-arabinofuranosyladenine, familiarly known as Ara-A (14), a clinical antiviral (herpes zoster, herpes simplex, etc.) and anticancer (chronic myelogenous leukemia) agent.83 Unusually, Ara-A (14) was established as a synthetic compound prior to its isolation from natural sources.83 As expected, it is metabolized by ADA to afford arabinofuranosyl hypoxanthine (Ara-I) (52).78
Studies on the demise of the 2′-hydroxy group of adenosine (11) began with the demonstration that 11 was a direct precursor of Ara-A (14) in cultures of S. antibioticus.84 To assess the potential for intact nucleoside involvement in the pathway, d-[1-14C]ribofuranose (53), [U-14C]adenine (54), and [U-14C]adenosine (11) were each incorporated into 14; other sugar units were not effectively taken up into the product. Importantly, the N-ribonucleoside bond of 11 was not broken in the transformation process, suggesting an intact incorporation of the complete skeleton of 11 into 14. As a result, it was proposed that an epimerase was operational on adenosine (11) for the inversion of the C-2′ stereocenter to afford Ara-A (14).84 This notion was counter to earlier data85,86 and references therein which had indicated that exogenous nucleosides would be cleaved to the purine base prior to nucleotide formation.
Baker and coworkers speculated that a common precursor, based on an intact adenosine (11) moiety, could serve in the formation of both Ara-A (14) and pentostatin (5).87 To evaluate this idea, both [U-14C]adenosine (11) and [U-14C]adenine (54) were incorporated into 14 and 5 in cultures of S. antibioticus.87 Through degradation studies, it was shown that the ratio of labels from 11 and 54 between the aglycon and sugar units remained the same subsequent to incorporation. Thus, like the biosynthesis of Ara-A (14), intact 11 served as a precursor of 5, and there was no scission of the N-glycosidic bond. The direct precursor involvement of 11 into 5 was demonstrated with all 5 of the carbons of the aglycon of 11 being incorporated into the imidazole-1,3-diazepine ring system.87 [U-14C]Glycine, as a potential source of a C1 fragment, was not incorporated into either 5 or 14, and thus is not a precursor of the C-7 methylene carbon in 5. The conclusion was that 2 pathways could be operational for the further intact metabolism of adenosine (11), one route leading to 14 and the other to 5.87
So what was the source of carbon-7 in pentostatin (5) and how is it inserted into the aminopyrimidine ring? Several proposals were made,87 however, it was not until 1987 that carbon-13 labeling studies revealed the origin of the methylene carbon (C-7) in 5.88 When [1-13C]serine and [1-13C]ribofuranose (53) were examined as potential precursors, serine labeled C-5 and C-5′ (and to a lesser extent C-2). Consequently, the tetrahydrofolate C1-pool was not the source of C-7. In parallel with the early stages in histidine formation,89 [1-14C]ribofuranose [53] showed circa 50:50 split in labeling between the aglycon and sugar units in 5. The regiospecificity of incorporation was assured using [1-13C]ribofuranose (53) which indicated almost equal enrichment (2.82:2.71-fold) at C-1′, and, more importantly, exclusively at C-7 in the diazepine ring. In this way, the intact incorporation of a purine scaffold into 5 and identification of the source of the methylene carbon were revealed in a single experiment.89 It is worth mentioning that this study was a very early use of 2D-NMR for the exploration of biosynthetic processes, following the unambiguous assignment of the proton and carbon resonances.
Baker’s group also examined the stereochemical development of a hydroxy group at C-8 in 5.90 Extracts of S. antibioticus provided an enzyme capable of affecting the stereospecific reduction of the C-8 carbonyl function of 32 to the corresponding 8R-hydroxy group of 5, as the final pathway step. In addition, it was demonstrated that the 4-pro-S hydrogen of NADPH was delivered stereospecifically at C-8 in the 8-oxo derivatives 55 and 56 as the pyrophosphates. As a result, a mechanism was proposed for the formation of pentostatin (5)90 in which the first step is condensation of α-5-phosphoribosyl-l-pyrophosphate (PRPP) (57) at N-1 of ATP (58). Ring cleavage between N-1 and C-6 is then followed by an Amadori-type rearrangement at C-1′ and C-2′ in the ribose unit. An aldol-type reaction and subsequent elimination of the tetronic acid fragment leads to 55 (or 56) as the phosphates. The monophosphates of coformycin (4) and pentostatin (5) are then produced through cleavage (Figure 9).90
Biosynthesis of coformycin (4), pentostatin (5), and adechlorin (7).90-92
The 2′-hydroxy group of 4 is replaced stereoselectively by a chlorine atom in adechlorin (6), the homopurine alkaloid from Actinomadura sp.22 As anticipated, based on the previous studies, [U-14C]adenosine (11) was incorporated intact into 6, and the addition of Na36Cl to the cultures generated labeled 6.93 Mechanistic alternatives were offered for this transformation, however, the precise pathway remains to be deduced. In a parallel series of experiments, the transformation and intact incorporation of adenosine (11) into the cometabolite 2′-amino-2′-deoxyadenosine (59)22 was examined. After feeding a mixture of [2′-18O]- and [U-14C]adenosine (11), the 18O:14C ratio did not change in 14, but with [3′-18O]- and [U-14C]adenosine (11) all of the original 18O was lost.94
Using an enzyme preparation from S. antibioticus, it was also shown that the label from [2′-3 H]adenosine (11) was lost as 3H2O, while added 3H2O efficiently incorporated 3H at C-2′ in 14. A mechanistic explanation of these observations was presented (Figure 10).94 Adenosine (11) was therefore established as the precursor of Ara-A (14), coformycin (4), and pentostatin (5).
Twenty-eight years later, some paradoxical aspects of homopurine alkaloid biosynthesis were described by Chen’s group.91,92 During their investigations, an exceptional biosynthetic relationship was revealed, whereby, within a single biosynthetic gene cluster, the formation of 1 metabolite “protected” the development of a cometabolite; described by the authors as a “protector-protégé” strategy,91 most notably for pentostatin (5) biosynthesis.
The most significant observation was the guardian or “protector” effect of 5. It was shown earlier that Ara-A (14) is susceptible to deamination by ADAs, and that conversion of Ara-A (14) to Ara-I (52) could be accomplished with SanADA3 from S. antibioticus.91 On the other hand, inhibition of 52 formation through deamination was complete in the presence of 0.1 M 5. Kinetic studies revealed both the high specificity and the potency for protection from deamination of the dominant metabolite in the culture medium, Ara-A (14) by the cometabolite pentostatin (5).91 Quite remarkably, Nature has made available an internal guardian which allows the formation of 14 in the presence of a deactivating deamination process.
Following the sequencing of the genomic DNA of S. antibioticus, and using HisG from l-histidine biosynthesis90 as a probe, a unified, 10-gene biosynthetic cluster, designated as pen, was identified and confirmed as adequate for the formation of both 14 and pentostatin (5) through the expression of penA-J in Streptomyces aureochromogenes CXR14.
Deactivation of penA, penB, or penC completely mitigated the biosynthesis of 5, while the formation of 14 was substantially reduced.91 In retrospect, this was to be expected, since the ability of 5 to provide protection for 14 from an available deaminase was no longer present. Functional attributions were made for the encoded enzymes of the cluster based on in silico analysis. PenA was identified as an ATP phosphoribosyltransferase corresponding to the first step in l-histidine biosynthesis, and PenB was assigned as a short-chain dehydrogenase. PenC indicated identity at the 80% level to a phosphoribosyl-aminoimidazole-succinocarboxamide (SAICAR) synthetase, although its definitive role in pentostatin (5) biosynthesis is yet to be ascribed. The 6 genes, penDFGHIJ, were assigned to be involved in encoding enzymes for the biosynthesis of 14. S-Adenosyl-l-homocysteine (SAH) hydrolase functionality was ascribed to PenD and PenG; these enzymes reversibly convert SAH to adenosine (11).95 PenF was assigned as a metal-dependent phosphohydrolase, and the enzymes PenH, PenI, and PenJ were attributed to act collaboratively to function as a dehydrogenase. PenE was identified as a transporter enzyme. The biochemical function of PenB, after expression in Escherichia coli BL21, was to establish a reversible reduction/oxidation reaction involving the 8-oxo-derivative 32 and the final product 5, as demonstrated by following the fate of either substrate after addition to the culture medium. This process effectively limits the availability of pentostatin (5).
Genetic studies on homopurine biosynthesis were then expanded. From Actinomadura sp. ATCC 39365, a single, 14.4 kb, 13 gene cluster, adaA-M, was identified as responsible for the biosynthesis of both adechlorin (6) and 2′-amino-2′-deoxyadenosine (59).92 The cluster was identified through a bioinformatics search for homologs of the 3 enzymes PenA, PenB, and PenC. The encoded enzymatic products AdaA, AdaB, and AdaC displayed high identity (53%-70%) with the enzymes PenC, PenB, and PenA, respectively. Further downstream was a series of genes which encoded for an aminotransferase (AdaF) and a dehydrogenase (AdaG), as required for 59 formation, and genes producing a phosphoribosyl isomerase (AdaK), an ATP phosphoribosyltransferase (AdaL), and a hydrolase (AdaM) were identified. When the adaJKLM operon was inactivated, the formation of 5, 6, and 59 was each abrogated, indicating that the single gene cluster is responsible for the biosynthesis of all 3 metabolites. When the ada gene cluster was cloned and expressed in S. aureochromogenes CXR14, the formation of all 3 metabolites was restored.92
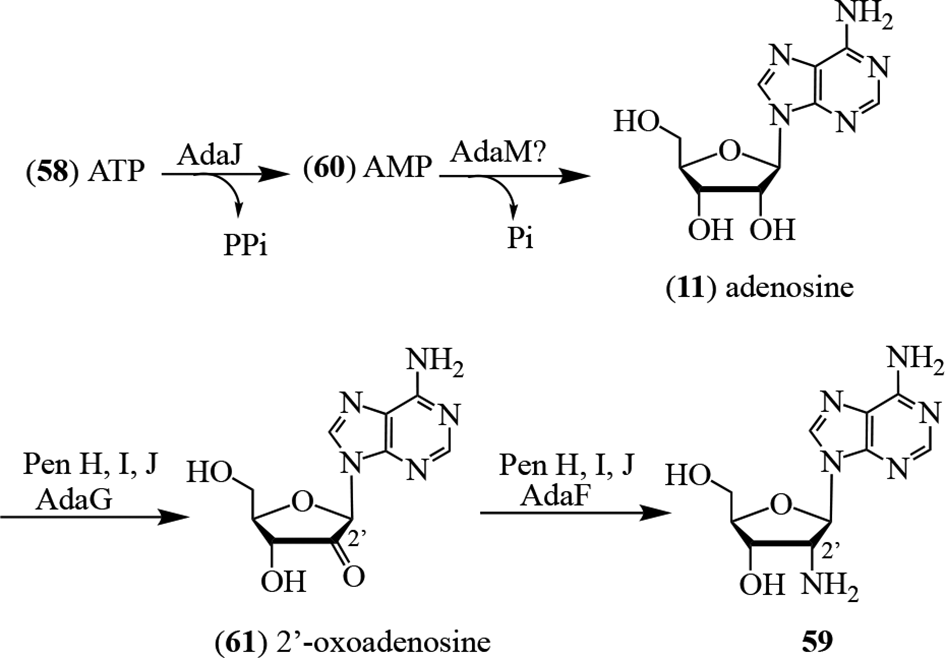
Through selective mutation studies, the 5 enzymes AdaA, AdaB, AdaC, AdaK, and AdaL were disclosed as crucial for the pathway to 5.92 No pentostatin (5) was produced when either AdaC or AdaL was inactivated. Interestingly, in silico analysis did not identify a specific halogenase-encoding gene in the cluster which could carry out the conversion of pentostatin (5) to coformycin (6). It was proposed that AdaE, a cation/H+-antiporter, was involved in this biosynthetic step, together with a free-standing halogenase. This remains to be clarified and the mechanism of halogenation firmly established. For the biosynthesis of 59, it was proposed that AdaJ, which affects the release of pyrophosphate in the conversion of ATP (57) to AMP (60), was the initiating enzyme. Dephosphorylation of 60 to 11 then occurs. The pathway concludes through the oxidation of C-2′ by AdaG to produce the 2′-oxo-derivative 61, which undergoes transamination through the action of AdaF to afford 59 (Figure 11).92
Biosynthesis of 2′-amino-2′-deoxyadenosine (59).92
Cooperative “victim-guardian” antibiotic pairs continue to be noted as additional microbial genomes are examined. The pentostatin (5)/Ara-A (14), coformycin (4)/formycin (9), 2′-chloropentostatin (6)/2′-amino-2′-deoxyadenosine (59), and the aristeromycin (16)/coformycin (4) systems are well established. Additional occurrences of these relationships were found when Micromonospora haikouensis DSM 45626 and Streptomyces citricolor NBRC 13005 were studied at the genome level.96 The 25.3 kb mac cluster from M. haikouensis DSM 45626 comprised 24 open reading frames, and after introduction into S. aureochromogenes CXR14 produced neplanocin A (15) and aristeromycin (16), in addition to coformycin (4). Streptomyces citricolor is also a producer of both 16 and 4, and was identified as such by using MacM (SAICAR synthetase) and MacN (short-chain dehydrogenase) as genomic DNA probes. When the resulting gene cluster, com, was expressed in S. aureochromogenes CXR14, it also produced 4.96
Although the data supported the intermediacy of neplanocin A (15) in the biosynthesis of aristeromycin (16), important aspects of the pathway remain unresolved, including (i) the structure(s) of any intermediate metabolites between d-glucose (62) and the cyclopentenone 63, (ii) the timing and the mechanism for the inversion of the C-4-hydroxy group of d-glucose (62); (iii) whether there are intermediates between 63 and 15; and (iv) mechanistic aspects of the reduction of neplanocin A (15) to aristeromycin (16) (Figure 12).
Biogenesis of neplanocin A (15) and aristeromycin (16).
An additional “victim-guardian” alkaloid pair, mentioned earlier, is the cordycepin (8) and pentostatin (5) relationship which occurs in the fungus Cordyceps militaris (L.) Link. Cordycepin (8) was initially reported in 1950 and is one of the oldest purine nucleosides. The discovery was derived from a remarkable ecological observation that the host tissue of the fungus was resistant to decay.97 The original source of cordycepin A was a sporophore of C. militaris obtained from Tollymore Forest Park, Newcastle, County Down, Ireland, and the isolation was based on the inhibition of Bacillus subtilis NCTC 6752. Fascinatingly, the molecular weight “247 ± 10” was established through X-ray analysis by the third woman to win the Nobel Prize in Chemistry, Dorothy Hodgkin. The UV spectrum indicated the presence of an adenosine derivative, which was confirmed through degradative studies.98 Unlike adenosine (11), cordycepin (8) was not cleaved by periodate, and thus a 3′-deoxypentose attached at N-9 of adenine was assigned, and the sugar named as cordycepose.
Many years later, Folker’s group at Merck, Sharpe, & Dohme in Rahway, NJ, United States characterized 3′-deoxyadenosine (8) from cultures of the fungus Aspergillus nidulans (Eidam) Wint. as a cytotoxic metabolite.99 When this alkaloid was compared with synthetic material100 and with natural cordycepin, the samples were found to be identical101 thereby determining the structure of cordycepin as 8. These structural identities were subsequently confirmed through mass spectrometric analysis102 and eventually by X-ray crystallography.103 In another part of the world, particularly China, cordycepin (8) is highly valued.
Ophiocordyceps species, particularly the entomopathogenic fungus Ophiocordyceps sinensis (Berk.) Sacc. acquired in the Himalayas, are known as Himalayan Viagra and yartsa gunbu. They are a prized and very expensive commodity in traditional Chinese medicine104,105 and are recognized for the diversity of their bioactive metabolites.106 Cordycepin (8), as a major metabolite, has demonstrated antibiotic, anti-inflammatory, and anticancer activities,107,108 and affects intracellular signal transduction.109 Consequently, significant efforts have been made to enhance production levels. Both ammonium ion110 and ferrous ion,111 when added to the culture medium, improved the culture yields. Other species are also likely producers of 8. Transcriptomic analysis of the fruiting body of O. sinensis112 and of the endoparasitic fungus of O. sinensis, Paecilomyces hepiali Q.T.Chen & R.T.Dai (Aspergillaceae),113 indicated that they too carried genes which could encode for the biosynthesis of 8, but this has not been biochemically demonstrated.
The initial biosynthetic studies on cordycepin (8)114 established a pathway from [8-14C]adenine (52) and [6-14C]glucose (62), and revealed an apparent loss of C-1 from [1-14C]glucose (62). The potential precursors acetate and isovalerate were not incorporated. Subsequently, using [U-14C]adenosine (11) and unlabeled ribofuranose (53), it was affirmed that intact incorporation of the adenosine (11) framework into 8 occurred without cleavage of the N-ribose bond.115 Further support for this came several years later, when it was demonstrated that activity from [3-3H]ribofuranose (53) was retained at C-3′ in 8. However, the stereospecific location of the 3H was not identified.116 The mechanistic pathway and the cofactors involved in the selective removal of the 3′-hydroxy group, apparently without proton loss, also remain unknown.
The 4-gene cluster, cns1 to cns4, responsible for the biosynthesis of cordycepin (8) in C. militaris, provided evidence for the intimate relationship of 2 entwined biosynthetic systems, for 8, and for pentostatin (5).117 Cns4 was a selective transporter enzyme for 8 and not for 5. While Cns1/Cns2 were tightly bound to produce 8, they could not be overexpressed in E. coli. Cns3 was a HisG domain-containing enzyme for the formation of 5. Thus, although in bacteria 3 genes are necessary for the formation of 5, only 1 is apparently required in fungi. Notably, there was little sequence similarity between the enzymes PenA, PenB, and PenC with Cns3, implying that although the structural outcome was the same, the pathways appear to have evolved over time in a divergent manner. Whether they are different mechanistically remains to be resolved. The deaminase-protecting effect (“victim-guardian” synergy) of coproducing 5 was observed when the Δcns3 mutant was cultured. The dominant product was not cordycepin (8), rather it was the deaminated analog 3′-deoxyinosine (64), since the ADA protector, 5, was no longer available.117 The remarkable 8/5 4 gene system (ckk1-ckk4) was also found in Cordyceps kyushuensis A. Karam.,118 a fungus which grows on the larvae of the edible insect Clanis bilineata Walker (Lepidoptera).119
Conclusion
The history of the isolation, structure elucidation, synthesis, biological properties, clinical relevance, and biosynthesis of the homopurine class of alkaloids has been reviewed. The group reflects clinically significant biological relevance, fascinating pathways for synthesis and biosynthesis, and the observation of single gene clusters which produce metabolites with different, albeit related, skeletons functioning in a “victim-guardian” relationship. These studies have mirrored the development of natural products research in the past 70 years. Revealing the significant and fascinating ecological relationships which have been disclosed through the assessment of how natural, clinically important metabolites have evolved gives voice to the stunning potential of natural products in human health utilizing the contemporary biotechnological tools that are available. Areas for further development of the homopurine alkaloids include enhanced synthetic procedures, mechanistic explorations of the biosynthetic pathways in bacteria and fungi at the enzyme level, and potentiation of the biological significance of deaminase inhibitors with reduced side effects in the clinical setting.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD
Geoffrey A. Cordell
References
1.
HosmaneRS. Ring-expanded ("fat") nucleosides as broad-spectrum anticancer and antiviral agents. Curr Top Med Chem. 2002;2(10):1093-1109.doi:10.2174/1568026023393147http://www.ncbi.nlm.nih.gov/pubmed/12173969
2.
KoyamaG.MaedaK.UmezawaH.IitakaY. The structural studies of formycin and formycin B. Tetrahedron Lett. 1966;7(6):597-602.doi:10.1016/S0040-4039(01)99671-6http://www.ncbi.nlm.nih.gov/pubmed/5905317
3.
HoriM.TakitaT.KoyamaG.TakeuchiT.UmezawaH. A new antibiotic, formycin. J Antibiot. 1964;17(5):96-99.http://www.ncbi.nlm.nih.gov/pubmed/14171222
4.
TakeuchiT.IwanagaJ.AoyagiT.UmezawaH. Antiviral effect of formycin and formycin B. J Antibiot. 1966;19(6):286-287.http://www.ncbi.nlm.nih.gov/pubmed/6013241
5.
NiidaT.NiwaT.TsuruokaT.EzakiN.ShomuraT.UmezawaH. Isolation and characteristics of coformycin. The 153rd Scientific Meeting of Japan Antibiotics Research Association, Tokyo, Japan, Jan 27. 1967.
6.
SawaT.FukagawaY.HommaI.TakeuchiT.UmezawaH. Mode of inhibition of coformycin on adenosine deaminase. J Antibiot. 1967;20(4):227-231.http://www.ncbi.nlm.nih.gov/pubmed/6072897
7.
UmezawaH.SawaT.FukagawaY. Studies on formycin. The 153rd Scientific Meeting of Japan Antibiotics Research Association, Tokyo, Japan, Jan. 27. 1967.
8.
NakamuraH.KoyamaG.IitakaY.OnoM.YagiawaN. Structure of coformycin, an unusual nucleoside of microbial origin. J Am Chem Soc. 1974;96(13):4327-4328.doi:10.1021/ja00820a049http://www.ncbi.nlm.nih.gov/pubmed/4854435
9.
NakamuraH.KoyamaG.UmezawaH.IitakaY. The crystal and molecular structure of coformycin. Acta Crystallogr B. 1976;32(4):1206-1212.doi:10.1107/S0567740876004949
10.
OhnoM.YagisawaN.ShibaharaS.KondoS.MaedaK.UmezawaH. Synthesis of coformycin. J Am Chem Soc. 1974;96(13):4326-4327.doi:10.1021/ja00820a048http://www.ncbi.nlm.nih.gov/pubmed/4854576
11.
WooPWK.DionHW.LangeSM.DahlLF.DurhamLJ. A novel adenosine and Ara-A deaminase inhibitor, (R)-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d]diazepin-8-ol. J Het Chem. 1974;11(4):641-643.
12.
BakerDC.PuttSR. A total synthesis of pentostatin, the potent inhibitor of adenosine deaminase. J Am Chem Soc. 1979;101(20):6127-6128.doi:10.1021/ja00514a048
13.
ChanE.PuttSR.ShowalterHDH.BakerDC. Total synthesis of (8R)-3-(2-deoxy-β-D-erythropentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][l,3]diazepin-8-ol, the potent inhibitor of adenosine deaminase. J Org Chem. 1982;47(18):3457-3464.doi:10.1021/jo00139a015
14.
SchrammVL.BakerDC. Spontaneous epimerization of (S)-deoxycoformycin and interaction of (R)-deoxycoformycin, (S)-deoxycoformycin, and 8-ketodeoxycoformycin with adenosine deaminase. Biochemistry. 1985;24(3):641-646.doi:10.1021/bi00324a016
15.
BakerDC.HawkinsLD. Synthesis of inhibitors of adenosine deaminase. A total synthesis of erythro-3-(adenin-9-yl)-2-nonanol and its isomers from chiral precursors. J Org Chem. 1982;47(11):2179-2184.doi:10.1021/jo00132a039
16.
ShowalterHD.PuttSR.BorondyPE.ShillisJL. Adenosine deaminase inhibitors. Synthesis and biological evaluation of (+/-)-3,6,7,8-tetrahydro-3-[(2-hydroxyethoxy)methyl]imidazo[4,5-d] [1,3]diazepin-8-ol and some selected C-5 homologues of pentostatin. J Med Chem. 1983;26(10):1478-1482.doi:10.1021/jm00364a022http://www.ncbi.nlm.nih.gov/pubmed/6604819
17.
KodamaK.KusakabeH.MachidaHet al. Isolation of 2’-deoxycoformycin and cordycepin from wheat bran culture of Aspergillus nidulans Y176-2. Agr Biol Chem. 1979;43(11):2375-2377.
18.
KanbeT.EndoA.HashizumeT. Production of 2’-deoxycoformycin by the fungus Emericella nidulans and its inhibitory effect on adenosine deaminase. Nucl Acids Symp Ser. 1983;12:115-118.
19.
TuliHS.SharmaAK.SandhuSS.KashyapD. Cordycepin: a bioactive metabolite with therapeutic potential. Life Sci. 2013;93(23):863-869.doi:10.1016/j.lfs.2013.09.030
20.
SchaumbergJP.HokansonGC.FrenchJC.SmalE.BakerDC. 2'-Chloropentostatin, a new inhibitor of adenosine deaminase. J Org Chem. 1985;50(10):1651-1656.doi:10.1021/jo00210a018
21.
TunacJB.UnderhillM. 2'-Chloropentostatin: discovery, fermentation and biological activity. J Antibiot. 1985;38(10):1344-1349.doi:10.7164/antibiotics.38.1344
22.
ŌmuraS.ImamuraN.KugaHet al. Adechlorin, a new adenosine deaminase inhibitor containing chlorine. Production, isolation and properties. J Antibiot. 1985;38(8):1008-1015.doi:10.7164/antibiotics.38.1008
23.
ŌmuraS.TanakaH.KugaH.ImamuraD. Adecypenol, a unique adenosine deaminase inhibitor containing homopurine and cyclopentene rings. J Antibiot. 1986;39(2):309-310.doi:10.7164/antibiotics.39.309http://www.ncbi.nlm.nih.gov/pubmed/3957791
24.
ŌmuraS.IshikawaH.KugaHet al. Adecypenol, a unique adenosine deaminase inhibitor containing homopurine and cyclopentene rings. taxonomy, production and enzyme inhibition. J Antibiot. 1986;39(9):1219-1224.doi:10.7164/antibiotics.39.1219
25.
HayashiM.YaginumaS.YoshiokaH.NakatsuK. Studies on neplanocin A, new antitumor antibiotic. II. J Antibiot. 1981;34(6):675-680.
26.
TanakaH.KawakamiT.YangZB.KomiyamaK.ŌmuraS. Potentiation of cytotoxicity and antitumor activity of adenosine analogs by the adenosine deaminase inhibitor adecypenol. J Antibiot. 1989;42(11):1722-1724.doi:10.7164/antibiotics.42.1722
27.
BushBD.FitchettGV.GatesDA.LangleyD. Carbocyclic nucleosides from a species of Saccharothrix. Phytochemistry. 1993;32(3):737-739.doi:10.1016/S0031-9422(00)95163-X
28.
UmezawaH.TakeuchiT.IinumaH.HamadaM.NishimuraS. Physiologically active agent azepinomycin. Japan Kokai Tokkyo Koho. 1983.
29.
CogginsAJ.TocherDA.PownerMW. One-step protecting-group-free synthesis of azepinomycin in water. Org Biomol Chem. 2015;13(11):3378-3381.doi:10.1039/C5OB00210A
30.
IsshikiK.TakahashiY.IinumaHet al. Synthesis of azepinomycin and β-D-ribofuranoside. J Antibiot. 1987;40(10):1461-1463.doi:10.7164/antibiotics.40.1461
31.
FujiiT.SaitoT.FujisawaT. Alternative syntheses of azepinomycin. Heterocycles. 1988;27(5):1163-1166.doi:10.3987/COM-88-4519
32.
FujiiT.SaitoT.FujisawaT. Purines. LXIII. Syntheses of azepinomycin, an antitumor antibiotic from Streptomyces species, and its 3-β-D-ribofuranoside and their 8-imino analogs. Chem Pharm Bull. 1994;42(6):1231-1237.doi:10.1248/cpb.42.1231http://www.ncbi.nlm.nih.gov/pubmed/8069974
33.
ChakrabortyS.ShahNH.FishbeinJC.HosmaneRS. Investigations into specificity of azepinomycin for inhibition of guanase: discrimination between the natural heterocyclic inhibitor and its synthetic nucleoside analogues. Bioorg Med Chem Lett. 2012;22(23):7214-7218.doi:10.1016/j.bmcl.2012.09.053
34.
LinschitzH.ConnollyJS. The photochemical addition of alcohols to purine. J Am Chem Soc. 1968;90(11):2979-2980.
35.
PuttSR.HartmanJD.ShowalterHDH.KeplerJA.TaylorG. Synthesis of [8-3H]Pentostatin. J Label Compd Radiopharm. 1981;18(7):925-931.doi:10.1002/jlcr.2580180702
36.
PWKW.LeeHT. Synthesis of [5-14C]pentostatin. J Labelled Compd Rad. 1990;28(4):445-454.
37.
RajappanVP.HosmaneRS. Analogs of azepinomycin as inhibitors of guanase. Nucleos Nucleot. 1998;17(7):1141-1151.doi:10.1080/07328319808004227
38.
UjjinamatadaRK.BhanA.HosmaneRS. Design of inhibitors against guanase: synthesis and biochemical evaluation of analogues of azepinomycin. Bioorg Med Chem Lett. 2006;16(21):5551-5554.doi:10.1016/j.bmcl.2006.08.033
39.
LiawS-H.ChangY-J.LaiC-T.ChangH-C.ChangG-G. Crystal structure of Bacillus subtilis guanine deaminase the first domain-swapped structure in the cytidine deaminase superfamily. J Biol Chem. 2004;279(34):35479-35485.
ShiotaG.FukadaJ.ItoT.TsuizawaM.YamadaM.SatoM. Clinical significance of serum guanase activity in various liver diseases. Jpn J Med. 1989;28(1):22-24.doi:10.2169/internalmedicine1962.28.22
42.
ItoS.TsujiY.KitagawaNet al. Clinical value of the guanase screening test in donor blood for prevention of posttransfusional non-A, non-B hepatitis. Hepatology. 1988;8(2):383-384.doi:10.1002/hep.1840080233
43.
CraryGS.YasminehWG.SnoverDC.VineW. Serum guanase: a biochemical indicator of rejection in liver transplant recipients. Transplant Proc. 1989;21(1 Pt 2):2315-2316.http://www.ncbi.nlm.nih.gov/pubmed/2652749
44.
DurakI.BedükY.KavutcuMet al. Activity of the enzymes participating in purine metabolism of cancerous and noncancerous human kidney tissues. Cancer Invest. 1997;15(3):212-216.doi:10.3109/07357909709039717http://www.ncbi.nlm.nih.gov/pubmed/9171854
45.
YuanG.BinJC.McKayDJ.SnyderFF. Cloning and characterization of human guanine deaminase purification and partial amino acid sequence of the mouse protein. J Biol Chem. 1999;274(12):8175-8180.doi:10.1074/jbc.274.12.8175http://www.ncbi.nlm.nih.gov/pubmed/10075721
46.
PaletzkiRF. Cloning and characterization of guanine deaminase from mouse and rat brain. Neuroscience. 2002;109(1):15-26.doi:10.1016/S0306-4522(01)00352-9
47.
CanbolatO.DurakI.ÇetinR.KavutcuM.DemirciS.ÖztürkS. Activities of adenosine deaminase, 5′-nucleotidase, guanase, and cytidine deaminase enzymes in cancerous and non-cancerous human breast tissues. Breast Cancer Res Treat. 1996;37(2):189-193.doi:10.1007/BF01806500
48.
AgarwalRP.SpectorT.ParksRE. Tight-binding inhibitors—IV. Inhibition of adenosine deaminases by various inhibitors. Biochem Pharmacol. 1977;26(5):359-367.doi:10.1016/0006-2952(77)90192-7
49.
BorondyPE.ChangT.MaschewskeE.GlazkoAJ. Inhibition of adenosine deaminase by co-vidarabine and its effect on the metabolic disposition of adenine arabinoside (vidarabine). Ann N Y Acad Sci. 1977;284(1 Third Confere):9-20.doi:10.1111/j.1749-6632.1977.tb21932.xhttp://www.ncbi.nlm.nih.gov/pubmed/280160
50.
SloanBJ.KieltyJK.MillerFA. Effect of a novel adenosine deaminase inhibitor (co-vidarabine, co-V) upon the antiviral activity in vitro and in vivo of vidarabine (Vira-Atm) for DNA virus replication. Ann N Y Acad Sci. 1977;284(1 Third Confere):60-80.doi:10.1111/j.1749-6632.1977.tb21937.xhttp://www.ncbi.nlm.nih.gov/pubmed/212990
51.
VennerPM.GlazerRI. The metabolism of 2'-deoxycoformycin by L1210 cells in vitro. Biochem Pharmacol. 1979;28(21):3239-3242.doi:10.1016/0006-2952(79)90070-4http://www.ncbi.nlm.nih.gov/pubmed/526329
52.
SiawMF.ColemanMS. In vitro metabolism of deoxycoformycin in human T lymphoblastoid cells. Phosphorylation of deoxycoformycin and incorporation into cellular DNA. J Biol Chem. 1984;259(15):9426-9433.http://www.ncbi.nlm.nih.gov/pubmed/6204981
53.
MargolisJ.GreverMR. Pentostatin (Nipent): a review of potential toxicity and its management. Semin Oncol. 2000;27(2 Suppl 5):9-14.10877045
54.
KeffordRF.FoxRM. Deoxycoformycin-induced response in chronic lymphocytic leukaemia: deoxyadenosine toxicity in non-replicating lymphocytes. Br J Haematol. 1982;50(4):627-636.doi:10.1111/j.1365-2141.1982.tb01963.xhttp://www.ncbi.nlm.nih.gov/pubmed/6978147
55.
HoAD.ThalerJ.StryckmansPet al. Pentostatin in refractory chronic lymphocytic leukemia: a phase II trial of the European organization for research and treatment of cancer. J Natl Cancer Inst. 1990;82(17):1416-1420.doi:10.1093/jnci/82.17.1416http://www.ncbi.nlm.nih.gov/pubmed/2388293
56.
O'DwyerPJ.MarsoniS.AlonsoMT.WittesRE. 2'-Deoxycoformycin: summary and future directions. Cancer Treat Symp. 1984;2(9):1-5.
57.
SpiersASD.RuckdeschelC.HortonJ. 2'-deoxycoformycin) in refractory lymphoid neoplasms. Scand J Haematol. 1984;32(2):130-134.
DeardenCE.MatutesE.CatovskyD. Clinical overview of pentostatin (Nipent) use in lymphoid malignancies. Semin Oncol. 2000;27(2 Suppl 5):22-26.http://www.ncbi.nlm.nih.gov/pubmed/10877047
60.
CannonT.MobarekD.WeggeJ.TabbaraIA. Hairy cell leukemia: current concepts. Cancer Invest. 2008;26(8):860-865.doi:10.1080/07357900801965034http://www.ncbi.nlm.nih.gov/pubmed/18798068
ChesonBD.VenaDA.FossFM.SorensenJM. Neurotoxicity of purine analogs: a review. J Clin Oncol. 1994;12(10):2216-2228.doi:10.1200/JCO.1994.12.10.2216
63.
FlinnIW.KopeckyKJ.FoucarMKet al. Long-term follow-up of remission duration, mortality, and second malignancies in hairy cell leukemia patients treated with pentostatin. Blood. 2000;96(9):2981-2986.
64.
RobakT. Hairy-cell leukemia variant: recent view on diagnosis, biology and treatment. Cancer Treat Rev. 2011;37(1):3-10.doi:10.1016/j.ctrv.2010.05.003
65.
SarvariaA.ToppZ.SavenA. Current therapy and new directions in the treatment of hairy cell leukemia. J Am Med Assoc Oncol. 2016;2(1):123-129.doi:10.1001/jamaoncol.2015.4134
ParmarS.AnderssonBS.CourielDet al. Prophylaxis of graft-versus-host disease in unrelated donor transplantation with pentostatin, tacrolimus, and mini-methotrexate: a phase I/II controlled, adaptively randomized study. J Clin Oncol. 2011;29(3):294-302.doi:10.1200/JCO.2010.30.6357
68.
KayNE.GeyerSM.CallTGet al. Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood. 2007;109(2):405-411.doi:10.1182/blood-2006-07-033274
69.
SauterC.LamannaN.WeissMA. Pentostatin in chronic lymphocytic leukemia. Expert Opin Drug Met. 2008;4(9):1217-1222.doi:10.1517/17425255.4.9.1217
70.
TingL-M.ShiW.LewandowiczAet al. Targeting a novel Plasmodium falciparum purine recycling pathway with specific immucillins. J Biol Chem. 2005;280(10):9547-9554.doi:10.1074/jbc.M412693200
71.
TylerPC.TaylorEA.FröhlichRFG.SchrammVL. Synthesis of 5’-methylthiocoformycins: specific inhibitors for malarial adenosine deaminase. J Am Chem Soc. 2007;129(21):6872-6879.doi:10.1021/ja0708363
72.
WebsterHK.WiesmannWP.PaviaCS. Adenosine deaminase in malaria infection: effect of 2'-deoxycoformycin in vivo. Adv Exp Med Biol. 1984;165 Pt A:225-229.doi:10.1007/978-1-4684-4553-4_44http://www.ncbi.nlm.nih.gov/pubmed/6609525
73.
WiesmannWP.WebsterHK.LambrosC.KelleyWN.DaddonaPE. Adenosine deaminase in malaria infected erythrocytes: unique parasite enzyme presents a new therapeutic target. Prog Clin Biol Res. 1984;165:325-342.http://www.ncbi.nlm.nih.gov/pubmed/6334315
74.
KicskaGA.TylerPC.EvansGB.FurneauxRH.SchrammVL.KimK. Purine-less death in Plasmodium falciparum induced by immucilin-H a transition state analogue of purine nucleoside phosphorylase. J Biol Chem. 2002;277(5):3226-3231.doi:10.1074/jbc.M105906200
75.
NiuG.TanH. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol. 2015;23(2):110-119.doi:10.1016/j.tim.2014.10.007
76.
SheltonJ.LuX.HollenbaughJA.ChoJH.AmblardF.SchinaziRF. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem Rev. 2016;116(23):14379-14455.doi:10.1021/acs.chemrev.6b00209http://www.ncbi.nlm.nih.gov/pubmed/27960273
77.
SerpiM.FerrariV.PertusatiF. Nucleoside derived antibiotics to fight microbial drug resistance: new utilities for an established class of drugs?J Med Chem. 2016;59(23):10343-10382.doi:10.1021/acs.jmedchem.6b00325http://www.ncbi.nlm.nih.gov/pubmed/27607900
78.
ShowalterHD.BungeRH.FrenchJCet al. Improved production of pentostatin and identification of fermentation cometabolites. J Antibiot. 1992;45(12):1914-1918.doi:10.7164/antibiotics.45.1914http://www.ncbi.nlm.nih.gov/pubmed/1490883
79.
HowellsJD.RyderA. Fermentation process for 9-(beta-D-arabinofuranosyl) adenine. U.S. Patent 3,616,208, issued October 26. 1971.
80.
HillJM.JenkinsGN.RushCPet al. Revised pathway for the biosynthesis of aristeromycin and neplanocin A from D-glucose in Streptomyces citricolor. J Am Chem Soc. 1995;117(19):5391-5392.doi:10.1021/ja00124a035
81.
HanrahanJ.NavratilovaH.ShoberuKAet al. Biosynthesis of aristeromycin: the role of (1R,2R,3S,4S)-1-hydroxymethylcyclopentane-2,3,4-triol. J Chem Soc Perkin 1. 1994(24):3533-3536.doi:10.1039/p19940003533
82.
ParryRJ.JiangY. The biosynthesis of aristeromycin. conversion of neplanocin A to aristeromycin by a novel enzymatic reduction. Tetrahedron Lett. 1994;35(52):9665-9668.doi:10.1016/0040-4039(94)88354-8
83.
LeeWW.BenitezA.GoodmanL.BakerBR. Potential anticancer agents. XL. Synthesis of the β-anomer of 9-(D-arabinofuranosyl)adenine. J Am Chem Soc. 1960;82(10):2648-2649.doi:10.1021/ja01495a070
84.
FarmerPB.SuhadolnikRJ. Nucleoside antibiotics. Biosynthesis of arabinofuranosyladenine by Streptomyces antibioticus. Biochemistry. 1972;11(5):911-916.doi:10.1021/bi00755a034
85.
HoffmeyerJ.NeuhardJ. Metabolism of exogenous purine bases and nucleosides by Salmonella typhimurium. J Bacteriol. 1971;106(1):14-24.doi:10.1128/JB.106.1.14-24.1971http://www.ncbi.nlm.nih.gov/pubmed/4928005
86.
Hochstadt-OzerJ.StadtmanER. The regulation of purine utilization in bacteria III. The involvement of purine phosphoribosyltransferases in the uptake of adenine and other nucleic acid precursors by intact resting cells. J Biol Chem. 1971;246(17):5312-5320.
87.
HanveyJC.HardmanJK.SuhadolnikRJ.BakerDC. Evidence for the conversion of adenosine to 2'-deoxycoformycin by Streptomyces antibioticus. Biochemistry. 1984;23(5):904-907.doi:10.1021/bi00300a017
88.
HanveyJC.HawkinsES.TunacJB.DechterJJ.BakerDC.SuhadolnikRJ. Biosynthesis of 2'-deoxycoformycin: evidence for ring expansion of the adenine moiety of adenosine to a tetrahydroimidazo[4,5-d][1,3]diazepine system. Biochemistry. 1987;26(18):5636-5641.doi:10.1021/bi00392a008
89.
AmesBN.MartinRG.GarryBJ. The first step of histidine biosynthesis. J Biol Chem. 1961;236(7):2019-2026.
90.
HanveyJC.HawkinsES.BakerDC.SuhadolnikRJ. 8-Ketodeoxycoformycin and 8-ketocoformycin as intermediates in the biosynthesis of 2'-deoxycoformycin and coformycin. Biochemistry. 1988;27(15):5790-5795.doi:10.1021/bi00415a059
91.
WuP.WanD.XuGet al. An unusual protector-protege strategy for the biosynthesis of purine nucleoside antibiotics. Cell Chem Biol. 2017;24(2):171-181.doi:10.1016/j.chembiol.2016.12.012
92.
GaoY.XuG.WuPet al. Biosynthesis of 2′-chloropentostatin and 2′-amino-2′-deoxyadenosine highlights a single gene cluster responsible for two independent pathways in Actinomadura sp. strain ATCC 39365. Appl Environ Microbiol. 2017;83(10):e-00078-00017.doi:10.1128/AEM.00078-17
93.
SuhadolnikRJ.PornbanlualapS.BakerDC.TiwariKN.HebblerAK. Stereospecific 2′-amination and 2′-chlorination of adenosine by Actinomadura in the biosynthesis of 2′-amino-2'-deoxyadenosine and 2′-chloro-2′-deoxycoformycin. Arch Biochem Biophys. 1989;270(1):374-382.doi:10.1016/0003-9861(89)90040-4
94.
SuhadolnikRJ.PornbanlualapS.WuJM.BakerDC.HebblerAK. Biosynthesis of 9-β-d-arabinofuranosyladenine: hydrogen exchange at C-2′ and oxygen exchange at C-3′ of adenosine. Arch Biochem Biophys. 1989;270(1):363-373.doi:10.1016/0003-9861(89)90039-8
95.
ReddyMC.KuppanG.ShettyND.OwenJL.IoergerTR.SacchettiniJC. Crystal structures of Mycobacterium tuberculosis S‐adenosyl‐L‐homocysteine hydrolase in ternary complex with substrate and inhibitors. Protein Sci. 2008;17(12):2134-2144.doi:10.1110/ps.038125.108
96.
XuG.KongL.GongRet al. Coordinated biosynthesis of the purine nucleoside antibiotics aristeromycin and coformycin in actinomycetes. Appl Environ Microbiol. 2018;84(22):e01860-18.doi:10.1128/AEM.01860-18
97.
CunninghamKG.MansonW.SpringFS.HUTCHINSONSA. Cordycepin, a metabolic product isolated from cultures of Cordyceps militaris (Linn.) link. Nature. 1950;166(4231):949.doi:10.1038/166949a0http://www.ncbi.nlm.nih.gov/pubmed/14796634
98.
BentleyHR.CunninghamKG.SpringFS. 509. Cordycepin, a metabolic product from cultures of Cordyceps militaris(Linn.) link. Part II. The structure of cordycepin. J Chem Soc. 1951:2301-2305.doi:10.1039/jr9510002301
99.
KaczkaEA.DulaneyEL.GittermanCO.WoodruffHB.FolkersK. Isolation and inhibitory effects of KB cell cultures of 3′-deoxyandenosine from Aspergillus nidulans (Eidam) Wint. Biochem Biophys Res Commun. 1964;14(5):452-455.doi:10.1016/0006-291X(64)90085-3http://www.ncbi.nlm.nih.gov/pubmed/5836540
100.
LeeWW.BenitezA.AndersonCD.GoodmanL.BakerBR. Potential anticancer agents. LV. Synthesis of 3'-amino-2',3'-dideoxyadenosine and related analogs. J Am Chem Soc. 1961;83(8):1906-1911.doi:10.1021/ja01469a030
101.
KaczkaEA.TrennerNR.ArisonB.WalkerRW.FolkersK. Identification of cordycepin, a metabolite of Cordycepsmilitaris, as 3′-deoxyadenosine. Biochem Biophys Res Commun. 1964;14(5):456-457.doi:10.1016/0006-291X(64)90086-5
102.
HanessianS.DeJonghDc.McCloskeyJA. Further evidence on the structure of cordycepin. Biochim Biophys Acta. 1966;117(2):480-482.doi:10.1016/0304-4165(66)90101-2
103.
RadwanMM.WilsonHR. The structure of cordycepin. Acta Crystallogr B. 1980;36(9):2185-2187.doi:10.1107/S056774088000831X
PatersonRR. Cordyceps: a traditional Chinese medicine and another fungal therapeutic biofactory?Phytochemistry. 2008;69(7):1469-1495.doi:10.1016/j.phytochem.2008.01.027http://www.ncbi.nlm.nih.gov/pubmed/18343466
106.
YueK.YeM.ZhouZ.SunW.LinX. The genus Cordyceps: a chemical and pharmacological review. J Pharm Pharmacol. 2013;65(4):474-493.doi:10.1111/j.2042-7158.2012.01601.xhttp://www.ncbi.nlm.nih.gov/pubmed/23488776
107.
RoseKM.BellLE.JacobST. Specific inhibition of chromatin-associated poly(A) synthesis in vitro by cordycepin 5’-triphosphate. Nature. 1977;267(5607):178-180.doi:10.1038/267178a0http://www.ncbi.nlm.nih.gov/pubmed/16073440
108.
LiaoY.LingJ.ZhangGet al. Cordycepin induces cell cycle arrest and apoptosis by inducing DNA damage and up-regulation of P53 in leukemia cells. Cell Cycle. 2015;14(6):761-771.doi:10.1080/15384101.2014.1000097http://www.ncbi.nlm.nih.gov/pubmed/25590866
109.
WongYY.MoonA.DuffinRet al. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J Biol Chem. 2010;285(4):2610-2621.doi:10.1074/jbc.M109.071159http://www.ncbi.nlm.nih.gov/pubmed/19940154
110.
MaoX-B.ZhongJ-J. Significant effect of NH4+ on cordycepin production by submerged cultivation of medicinal mushroom Cordyceps militaris. Enzyme Microb Technol. 2006;38(3-4):343-350.doi:10.1016/j.enzmictec.2004.10.010
111.
D-dF.WangW.ZhongJ-J. Enhancement of cordycepin production in submerged cultures of Cordyceps militaris by addition of ferrous sulfate. Biochem Eng J. 2012;60(1):30-35.
112.
XiangL.LiY.ZhuYet al. Transcriptome analysis of the Ophiocordyceps sinensis fruiting body reveals putative genes involved in fruiting body development and cordycepin biosynthesis. Genomics. 2014;103(1):154-159.doi:10.1016/j.ygeno.2014.01.002http://www.ncbi.nlm.nih.gov/pubmed/24440419
113.
PangF.WangL.JinYet al. Transcriptome analysis of Paecilomyces hepiali at different growth stages and culture additives to reveal putative genes in cordycepin biosynthesis. Genomics. 2018;110(3):162-170.doi:10.1016/j.ygeno.2017.09.008http://www.ncbi.nlm.nih.gov/pubmed/28935392
114.
KredichNM.GuarinoAJ. Studies on the biosynthesis of cordycepin. Biochim Biophys Acta. 1961;47(3):529-534.doi:10.1016/0006-3002(61)90546-7http://www.ncbi.nlm.nih.gov/pubmed/13754189
115.
SuhadolnikRJ.WeinbaumG.MelocheHP. The biosynthesis of cordycepin. J Am Chem Soc. 1964;86(5):948-949.doi:10.1021/ja01059a057
116.
LennonMB.SuhadolnikRJ. Biosynthesis of 3'-deoxyadenosine by Cordyceps militaris: mechanism of reduction. Biochim Biophys Acta. 1976;425(4):532-536.doi:10.1016/0005-2787(76)90017-4http://www.ncbi.nlm.nih.gov/pubmed/1083247
117.
XiaYL.LuoFF.ShangYF.ChenPL.LuY.WangCS. Fungal cordycepin biosynthesis is coupled with the production of the safeguard molecule pentostatin. Cell Chem Biol. 2017;24(12):1479-1489.doi:10.1016/j.chembiol.2017.09.001http://www.ncbi.nlm.nih.gov/pubmed/29056419
118.
ZhaoX.ZhangG.LiC.LingJ. Cordycepin and pentostatin biosynthesis gene identified through transcriptome and proteomics analysis of Cordyceps kyushuensis Kob. Microbiol Res. 2019;218(1):12-21.doi:10.1016/j.micres.2018.09.005http://www.ncbi.nlm.nih.gov/pubmed/30454654
119.
LingJ-Y.SunY-J.ZhangH.LvP.ZhangC-K.J-yL.Y-jS.C-kZ. Measurement of cordycepin and adenosine in stroma of Cordyceps sp. by capillary zone electrophoresis (CZE). J Biosci Bioeng. 2002;94(4):371-374.doi:10.1263/jbb.94.371http://www.ncbi.nlm.nih.gov/pubmed/16233320