Five new ent-atisene diterpene glycosides (1-5) have been isolated from Stevia rebaudiana Bertoni. This adds to a list of only 2 new ent-atisene diterpene glycosides isolated previously from S. rebaudiana. Diterpene glycosides isolated from S. rebaudiana, such as the rebaudiosides, are typically associated with the ent-kaurene core and glycosylation at the C-13 and C-19 positions. Extensive application of nuclear magnetic resonance spectroscopy techniques (1H, 13C, 1D-TOCSY, COSY, HSQC-DEPT, HMBC, and ROESY) as well as high-resolution mass spectrometry demonstrated that the ent-atisenes 1 to 5 had glycosylation patterns identical to rebaudiosides J, N, O, K, and T, respectively. We have named these compounds stevatisene J, N, O, K, and T, respectively.
Stevia rebaudiana Bertoni (Asteraceae), a perennial herb indigenous to Brazil and Paraguay, is an important source of low-calorie highly potent sweeteners known as steviol glycosides.1 These diterpenes are characterized by an ent-kaurene aglycone and a wide variety of glycosylation patterns at the C-13 and C-19 positions.2 To date, there are dozens of published naturally occurring steviol glycosides, and efforts to isolate new steviol analogs continue.3 Indeed, the availability of large quantities of commercially available S. rebaudiana leaf extracts has facilitated efforts to uncover new minor diterpene glycoside components.4 Here, we report on the isolation and structure elucidation of 5 new ent-atisene diterpene glycosides.
Five new compounds (1-5, Figures 1 and 2) were isolated from a commercial supply of S. rebaudiana leaf extract. 1H and 13C NMR data suggested that these compounds were all diterpene glycosides containing an ent-atisene core glycosylated at the C-13 and C-19 positions. The presence of this class of chemistry in S. rebaudiana was recently reported.5
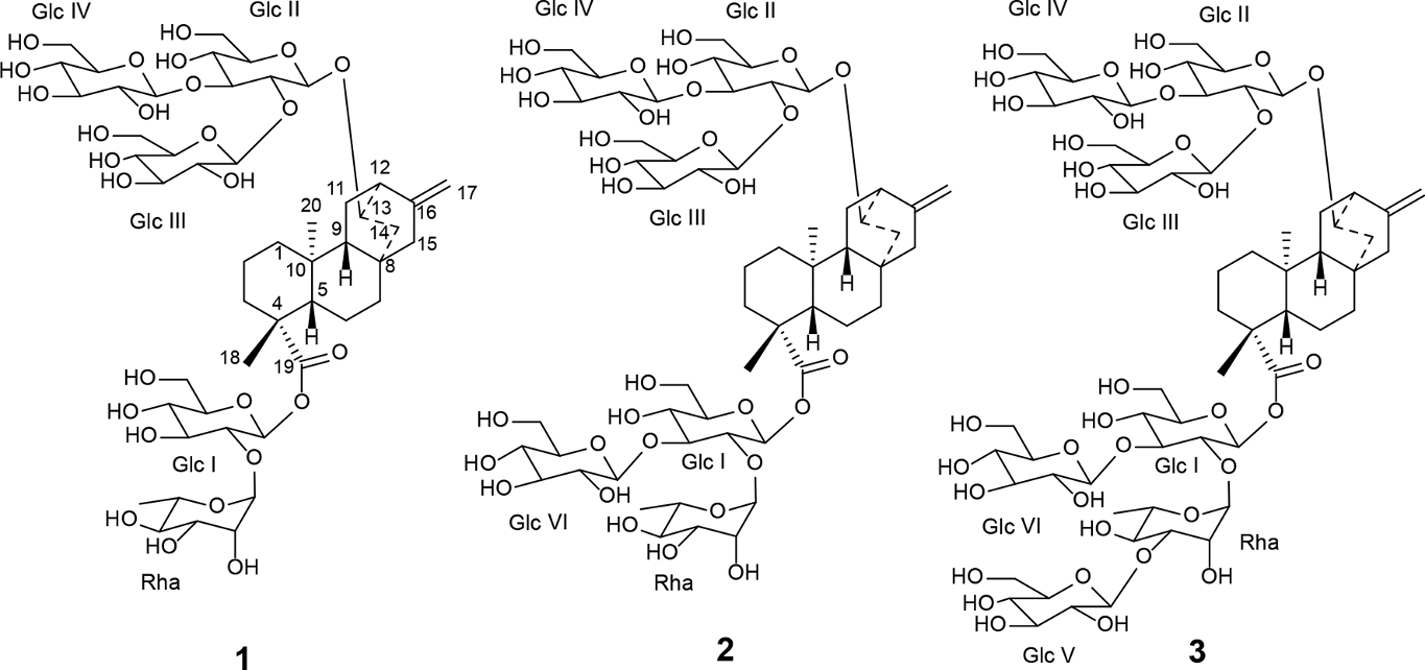
Structures of new ent-atisene glycosides 1 to 3.
Structures of new ent-atisene glycosides 4 and 5 with key HMBC correlations shown for the sugar connectivity.
Extensive analysis of 1D and 2D NMR data, including that based on 1H, 13C, 1D-TOCSY, 1H-1H COSY, 1H-13C HSQC-DEPT, 1H-13C HMBC, and 1H-1H ROESY experiments, confirmed that 1 to 5 contained the ent-atisene core with a varying number of sugar units attached to that core (see Supporting Information for representative 1D and 2D NMR spectra). The complete 1H and 13C NMR assignments of compounds 1 to 5 are provided in Tables 1 and 2, respectively. The following discussion describes key steps in establishing the ent-atisene core together with the glycosylation substitution patterns for each of these new compounds.
1H NMR Assignments for Compounds 1-5 (In CD3OD; Compounds 1-4 500 MHz and Compound 5 600 MHz; Compounds 1-3 and 5 at 300 K and Compound 4 at 286 K).
Several methine and methylene resonances of the sugar units are either partially or completely overlapped in the region ~3.11-3.96 ppm. These protons were assigned based on 1D-TOCSY and 2D NMR data.
Resonance obscured by H2O, assignment based on HSQC-DEPT data, multiplicity could not be determined.
Two protons.
Resonance obscured by H2O, assignment based on HSQC-DEPT data and coupling constant determined based on 1D-TOCSY data.
Broad doublet (J = ~1.7 Hz) based on 1D-TOCSY data.
Partially overlapped with H-18 (1.23 ppm), thus multiplicity could not be determined.
13C NMR Assignments for Compounds 1-5 (In CD3OD; Compounds 1-3 125 MHz and Compounds 4 and 5 150 MHz; Compounds 1-3 and 5 at 300 K and Compound 4 at 286 K).
Resonance obscured by CD3OD, assignment based on HSQC-DEPT data.
Chemical shifts could not be unequivocally assigned due to very close chemical shifts or overlapping resonances.
Two carbon resonances observed at 71.6 ppm (71.55 and 71.60 ppm).
Three carbon resonances in the range of 62.5-62.6 ppm (62.51, 62.55, and 62.57 ppm), hence chemical shifts could not be unequivocally assigned.
The structure of compound 1 was elucidated in the same manner described previously in our discovery of other new Stevia glycosides.4 While most of the observed resonances in the 1H and 13C NMR spectra for the diterpene core of compound 1 were similar to that of ent-kaurene Stevia glycosides,1-4 significant deviations were noted. Furthermore, HSQC-DEPT data for 1 revealed only 8 methylene units instead of the 9 commonly observed in the central diterpene core of related Stevia glycosides. Also, in addition to 2 methine units at the C-5 and C-9 positions, 2 additional methine units at δH 2.48/δC 42.3 and δH 4.04/δC 79.0 were observed, indicating structural differences in the central diterpene core. In the COSY spectrum, correlations were observed from H-9 (δH 1.09) to H-11 (δH 1.43 and 1.60) and from H-11 to a methine proton at δH 2.48, confirming that a methine proton is present at C-12 instead of the more commonly present methylene protons. The proton at δH 2.48 (H-12) showed COSY correlations to another methine proton at δH 4.04 which was assigned to H-13. The carbon chemical shift of C-13 was then assigned as δC 79.0 based on HSQC-DEPT data indicating a hydroxyl or substituted hydroxyl unit at C-13. In the COSY spectrum, correlations from H-13 (δH 4.04) to δH 1.15 and 2.43 were then assigned to H-14. In the HMBC spectrum, correlation observed from H-13 to an anomeric carbon at δC 101.7 and a reciprocal correlation from the anomeric proton at δH 4.53 to δC 79.0 (C-13) confirmed the attachment of sugar unit at C-13 position. Analysis of available NMR data thus confirmed that C-13 was attached to C-12, C-14, a sugar unit, and a methine proton, but not to C-16 commonly found in other Stevia glycosides. Furthermore, analysis of NMR data confirmed that C-16 is attached to C-12 (Figure 3).
Key HMBC and COSY correlations used to assign the aglycone region of 1.
These data thus confirmed that compound 1 is an ent-atisene diterpenoid glycoside (Figure 3). In the ROESY spectrum, NOE correlations were observed between H-20 and one of the H-14 protons (δH 2.43) indicating that H-14 and H-20 are on the same face of the ring. This assignment was further supported by NOE correlations between H-20 and H-13. Similarly, NOE correlations were observed between H-18 and H-5/H-9 but no NOE correlations were observed with H-13, H-14, and H-20 indicating that H-5, H-9, and H-18 were on the opposite face of the ring compared to H-13, H-14, and H-20 (Figure 3).
Analysis of the 1H-13C HSQC-DEPT data for 1 confirmed the presence of 5 anomeric protons at δH 5.57 (δC 94.2), 5.28 (δC 101.8), 4.79 (δC 103.5), 4.63 (δC 104.5), and 4.53 (δC 101.7). NMR data indicated that 4 sugar units were glucose and 1 unit was rhamnose.
The anomeric proton at δH 5.57 showed an HMBC correlation to C-19 (δC 177.2) and was assigned to Glc I. The Glc I proton spin system was established through COSY and 1D-TOCSY data and 13C NMR shift assignments were assigned based on HSQC-DEPT data. The relatively high frequency chemical shift of Glc I C-2 (δC 78.1 or 78.2 or 78.3) implied substitution at that position. An HMBC correlation from the anomeric Rha proton observed at δH 5.28 to Glc I C-2 (δC 78.1 or 78.2 or 78.3) and a reciprocal correlation from Glc I H-2 (δH 3.60) to the anomeric carbon of Rha (δC 101.8) confirmed placement of the rhamnose unit at this position. The COSY and 1D-TOCSY data allowed the assignments of Rha H-1 (δH 5.28), H-2 (δH 3.91), H-3 (δH 3.62), H-4 (δH 3.37), H-5 (δH 3.75), and H-6 (δH 1.24). The HSQC-DEPT data then allowed the assignments of the carbons as Rha C-1 (δC 101.8), C-2 (δC 72.0 or 72.1 or 72.2), C-3 (δC 72.0 or 72.1 or 72.2), C-4 (δC 73.8), C-5 (δC 70.3), and C-6 (δC 18.3). These assignments were further confirmed by HMBC correlations of H-1/C-2 and/or C-3 and C-5; H-2/C-3; H-3/C-4; H-4/C-6; and H-6/C-4, C-5. The characteristic H-6 methyl resonance at δH 1.24 (d, J = 6.2 Hz) in 1H NMR spectrum together with other NMR data discussed above confirmed that this sugar is rhamnose. The small coupling constant (J = 1.4 Hz) associated with Rha H-1 indicated an α-configuration for this linkage.
The anomeric proton at δH 4.53 (δC 100.7) showed an HMBC correlation to C-13 and was assigned to Glc II H-1. Extensive COSY, 1D-TOCSY, HSQC-DEPT, and HMBC analyses established that the C-13 glycoside was a Glcβ(1-2)[Glcβ(1-3)]Glcβ1 unit. This unit is found at the C-13 position of a wide range of steviol glycosides, including rebaudioside A and M. This structure was further supported by mass spectrometry data. Accurate mass measurement of 1 using HRMS provided an exact mass at m/z 1111.4830 in the negative ESI-TOF mass spectrum. This corresponded to a molecular formula of C50H80O27 (calcd for C50H79O27: 1111.4809). The MS/MS spectrum of 1, selecting the [M-H]− ion at m/z 1111.4 for fragmentation, indicated loss of 1 glucose unit at m/z 947.4371 followed by loss of 1 rhamnose unit at m/z 803.3752 and sequential loss of 3 sugar units at m/z 641.3210, 479.2636, and 317.1950, confirming the presence of 4 glucose and 1 rhamnose units in the structure (see Supporting Information).
The key HMBC and COSY correlations used to assign the C-19 and C-13 glycoside region of 1 are provided in Figure 4. Compound 1 was therefore identified as 13-[(2-O-β-d-glucopyranosyl-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)oxy]ent-atis-16-en-19-oic acid-[(2-O-α-l-rhamnopyranosyl-β-dester]. The glycosylation pattern recapitulates that for rebaudioside J. We have termed this compound stevatisene J.
Key HMBC and COSY correlations used to assign the C-13 and C-19 glycoside regions of 1.
The structure of compound 2 was characterized in a similar manner. The 1H, 13C, and HSQC-DEPT data indicated an additional glucose unit in 2 when compared with 1. This additional glucose unit was supported by the presence of 6 anomeric proton and carbon resonances in the NMR data compared to 5 in 1 (see Supporting Information and Tables 1 and 2).
Analysis of the 1D and 2D NMR data further confirmed the presence of 5 glucose units and 1 rhamnose unit in 2. The rhamnose unit was attached to Glc I as in 1. The relatively higher frequency chemical shift of Glc I C-3 (δC 87.0) implied additional substitution at this position in Glc I. This was confirmed by an HMBC correlation from the anomeric proton at δH 4.54 (Glc VI H-1) to Glc I C-3 (δC 87.0) as well as a reciprocal correlation from Glc I H-3 (δH 3.79) to the anomeric carbon of Glc VI (δC 104.4 or 104.5). The coupling constant of 7.9 Hz for the Glc VI anomeric proton indicted a β-configuration for this linkage. Further 2D NMR analysis confirmed the structure of the C-13 region and also established that the C-19 glycoside unit was Glcβ(1-2)[Glcβ(1-3)]Glcβ1 as in 1. Structural determination of 2 was further supported by mass spectrometry data. Accurate mass measurement of 2 provided an exact mass of m/z 1273.5353 in the negative ESI-TOF mass spectrum. This corresponded to a molecular formula of C56H90O32 (calcd for C56H89O32: 1273.5337). The MS/MS spectrum of 2, selecting the [M-H]− ion at m/z 1273.5 for fragmentation, indicated loss of 2 glucose units at m/z 1111.4684 and 949.4500 followed by loss of 1 rhamnose unit at m/z 803.3704 and sequential loss of 3 sugar units at m/z 641.3172, 479.2629, and 317.1921, confirming the presence of 5 glucose and 1 rhamnose units in the structure.
Compound 2 was identified as 13-[(2-O-β-d-glucopyranosyl-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)oxy]ent-atis-16-en-19-oic acid-[(2-O-α-l-rhamnopyranosyl-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)ester]. The glycosylation pattern recapitulates that for rebaudioside N. We have termed this compound stevatisene N.
Structure elucidation of compound 3 was based on a similar analysis of 1D and 2D NMR data and was supported by mass spectrometry data. The 1H, 13C, and HSQC-DEPT data indicated 7 anomeric protons and carbons and thus an additional glucose unit in 3 when compared with 2 (see Supporting Information and Tables 1 and 2).
Extensive 1D and 2D NMR analyses placed this additional glucosyl unit in the C-19 glycoside region. Key observations in establishing this were the relatively higher frequency chemical shift of Rha C-3 (δC 83.0) implying substitution at this position as well as an HMBC correlation from the anomeric proton (at δH 4.58, Glc V H-1) to a carbon at δC 83.0 (Rha C-3) and a reciprocal HMBC correlation from δH 3.73 (Rha H-3) to the anomeric carbon at δC 105.1 (Glc V C-1). The coupling constant of 7.8 Hz for the Glc V anomeric proton signal observed in the 1H NMR spectrum indicated a β-configuration. As before, the C-19 glycoside region was shown to be Glcβ(1-2)[Glcβ(1-3)]Glcβ1. Furthermore, accurate mass measurement of 3 using HRMS provided an exact mass at m/z 1435.5844 in the negative ESI-TOF mass spectrum. This corresponded to a molecular formula of C62H100O37 (calcd for C62H99O37: 1435.5865). The MS/MS spectrum of 3, selecting the [M-H]- ion at m/z 1435.5 for fragmentation, indicated loss of 3 glucose units at m/z 1273.5428, 1111.4835, and 949.4512 followed by loss of 1 rhamnose unit at m/z 803.3798 and sequential loss of 3 sugar units at m/z 641.3209, 479.2545, and 317.1973. These data thus supported the presence of 6 glucose and 1 rhamnose unit.
Compound 3 was identified as 13-[(2-O-β-d-glucopyranosyl-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)oxy]ent-atis-16-en-19-oic acid-[(2-O-α-l-rhamnopyranosyl-(3-O-β-d-glucopyranosyl)-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)ester]. The glycosylation pattern recapitulates that for rebaudioside O. We have termed this compound stevatisene O.
The complete NMR spectral data analysis of compound 4 indicated that it is an isomer of compound 1. NMR data comparisons of 4 with 1 (Tables 1 and 2) as well as extensive 1D and 2D NMR analysis placed 2 glucose and rhamnose units within the C-13 glycosyl unit and 2 glucose units within the C-19 glycosyl unit in 4. The relatively higher frequency chemical shifts of Glc II C-2 (δC 77.6) and C-3 (δC 88.7) indicated substitution at those positions. An HMBC correlation from Rha anomeric proton resonance observed at δH 5.30 to Glc II C-2 (δC 77.6) and a reciprocal HMBC correlation from δH 3.50 (Glc II H-2) to the anomeric carbon at δC 102.1 (Rha C-1) placed the rhamnose unit as the Glc II C-2 substituent. Similarly, an HMBC correlation from Glc IV anomeric proton at δH 4.46 to Glc II C-3 (δC 88.7) and a reciprocal HMBC correlation from δH 3.69 (Glc II H-3) to the anomeric carbon at δC 104.5 (Glc IV C-1) placed a glucose unit at Glc II C-3. The above described structure was supported by mass spectrometry data. Accurate mass measurement of 4 provided an exact mass at m/z 1111.4907 in the negative ESI-TOF mass spectrum. This corresponded to a molecular formula of C50H80O27 (calcd for C50H79O27: 1111.4809) supporting that 4 was an isomer of 1.
Compound 4 was therefore identified as 13-[(2-O-α-l-rhamnopyranosyl-3-O-β-d-glucopyranosyl-β-d-glucopyranosyl)oxy]ent-atis-16-en-19-oic acid-[(2-O-β-d-glucopyranosyl-β-d-glucopyranosyl)ester]. The glycosylation pattern recapitulates that for rebaudioside K. We have termed this compound stevatisene K.
Compound 5 was characterized in a similar manner on the basis of extensive analysis of the NMR data which indicated the presence of 5 glucose and 1 deoxypentose units attached to the ent-atisene central core of the structure.
NMR data established the deoxypentose unit in 5 was xylose. Xylose is relatively rare in Stevia glycosides, and its structural assignment as well as its position at Glc I C-2 was based on 1D and 2D NMR data (see Supporting Information). The anomeric proton resonance observed at δH 4.94 showed an HMBC correlation to Glc I C-2 (δC 77.8) and was assigned as the anomeric proton of Xyl. The reciprocal HMBC correlation from Glc I H-2 (δH 3.80) to the anomeric carbon of Xyl (δC 104.1) was also observed confirming this linkage.
The COSY and 1D-TOCSY data allowed the assignments of Xyl H-1 (δH 4.94), H-2 (δH 3.11), H-3 (δH 3.32), H-4 (δH 3.50), and H-5 (δH 3.21 and 3.82). The HSQC-DEPT data then allowed the assignments of the carbons as Xyl C-1 (δC 104.1), C-2 (δC 75.7), C-3 (δC 77.9-78.4), C-4 (δC 71.3), and C-5 (δC 67.0). These assignments were further confirmed by HMBC correlations of H-1/C-3, C-5; H-2/C-1, C-3; H-3/C-4; H-4/C-5; and H-5/C-3, C-4. In the 1H NMR spectrum, a coupling value of 7.9 Hz for the Xyl anomeric proton indicated a β-configuration. The assigned structure was further supported by the mass spectrometry data. Accurate mass measurement of 5 provided an exact mass at m/z 1259.4926 in the negative ESI-TOF mass spectrum. This corresponded to a molecular formula of C55H88O32 (calcd for C55H87O32: 1259.5180). The MS/MS spectrum of 5, selecting the [M-H]− ion at m/z 1259.0 for fragmentation, indicated loss of 2 glucose units at m/z 1097.4545 and 935.4002 followed by loss of 1 xylose unit at m/z 803.3441 and sequential loss of 3 sugar units at m/z 641.2988, 479.2617, and 317.1986.
Compound 5 was therefore identified as 13-[((2-O-β-d-glucopyranosyl-3-O-β-d-glucopyranosyl)-β-d-glucopyranosyl)oxy]ent-atis-16-en-19-oic acid-[((2-O-β-d-xylopyranosyl-3-O-β-d-glucopyranosyl)-β-d-glucopyranosyl)ester]. The glycosylation pattern is similar to that for rebaudioside T.6 We have termed this compound stevatisene T.
Diterpene glycosides from S. rebaudiana are generally characterized by the presence of an ent-kaurene aglycone core. There is at least one report of labdane diterpenes from S. rebaudiana7; labdanes have also been found in S. seleriana.8 Clerodanes have been found in S. polycephala9 and grindelanes in S. subpubescens.10
Here we report on the isolation and structure elucidation of 5 compounds. These ent-atisenes (1-5) had glycosylation patterns identical to rebaudiosides J, N, O, K, and T, respectively. The ent-atisene core has been found in a limited set of plants including, for example, Aconitum heterophyllum (Ranunculaceae),11Isodon albopilosus (Lamiaceae)12 and Viguiera insignis (Asteraceae).13 Its presence in a commercially relevant plant such as S. rebaudiana is therefore significant.
Experimental
Isolation of Compounds 1 to 5 by Preparative HPLC
The fractionation of ~300 g of S. rebaudiana leaf extract was performed in successive chromatographic steps. The first step used a Waters XBridge RP18 column (50 × 250 mm, 7 µm) or Phenomenex Luna C18 (2) column (50 × 250 mm, 10 µm) with the following elution method: mobile phase A: 35% MeOH (v/v) in 0.05% HOAc; mobile phase B: 60% MeOH (v/v) in 0.05% HOAc; mobile phase C: MeOH: elution: 0 to 35 minutes (100A:0B:0C), 50 to 63 min (0A:100B:0C), 63.1 to 68 min (0A:0B:100C); injection load: 4 to 10 g of extract dissolved in 80 L of mobile phase A; flow rate was 100 mL/min; detection at 220 nm. Fractions containing compounds of interest (based on chromatographic and MS profiles, see below for methods) were concentrated from the eluent by rotary evaporation. These concentrated aqueous solutions were used “as is” in the second chromatographic step. This step used a Waters XBridge Phenyl column (19 × 250 mm, 5 µm) and isocratic elution with 18% MeCN in H2O as mobile phase: flow rate was 20 mL/min; detection at 210 nm. The mobile phase used to isolate compound 5 in this step was slightly modified: 20% MeCN in H2O. All other parameters remained the same. Compounds of interest were collected for further fractionation in a third chromatographic step. This step used a Waters XBridge Amide column (19 × 250 mm, 5 µm) with the following elution method for fractions containing 1, 2, and 4; mobile phase A: 85% MeCN (v/v) in H2O; mobile phase B: 70% MeCN (v/v) in H2O: elution: 0 to 30 minutes linear ramp from 100% mobile phase A to 100% mobile phase B and hold at 100% mobile phase B for 15 minutes; flow rate was 20 mL/min; detection at 210 nm. Compound 3 was isolated using a slight modification of the elution procedure as follows: 0 to 30 minutes linear ramp from 100% mobile phase A to 100% mobile phase B with 5 minutes isocratic holds at 87%, 47%, and 30% mobile phase A composition. Compound 5 was also isolated using a slight modification in the elution procedure. Here mobile phase A was 80% MeCN (v/v) in H2O and the gradient was a 0 to 60 minutes linear ramp from 100% mobile phase A to 100% mobile phase B.
Amounts of sample available for spectroscopic and spectrometric analyses were 1 (4.5 mg), 2 (7.9 mg), 3 (3.8 mg), 4 (0.8 mg), and 5 (0.3 mg).
Fraction Analyses
Analysis of preparative purification fractions from the first step was performed using the following method: Phenomenex Synergi Hydro-RP (4.6 × 250 mm, 4 µm); column temp: 55°C; mobile phase A: 0.00284% NH4OAc and 0.0116% AcOH in water; mobile phase B: MeCN; flow Rate: 1.0 mL/min; injection volume: 10 µL.
Primary fractions were analyzed using the following method: gradient: 0 to 8.5 minutes (75A:25B), 10 minutes (71A:29B), 16.5 minutes (70A:30B), 18.5 to 23 minutes (66A:34B), 23.5 to 27.0 minutes (30A:70B), 27.5 minutes (75A:25B). Fractions from the second processing step were analyzed using the following method: Waters XBridge Phenyl (4.6 × 150 mm, 5 µm); column temp: ambient; mobile phase A: H2O; mobile phase B: MeCN; flow rate: 1.0 mL/min; injection volume: 10 µL; gradient: 0 to 20 minutes (80A:10B), 20 to 25 minutes (50A:50B). Fractions from the third isolation step were analyzed using the following method: Waters XBridge Amide (4.6 × 150 mm, 3.5 µm); column temp: ambient; mobile phase A: H2O; mobile phase B: MeCN; flow rate: 1.0 mL/min; injection volume: 10 µL; gradient: 0 minutes (15A:85B), 20 minutes (30A:70B), 20 to 25 minutes (50A:50B). All gradient segments are linear. Detection for all methods was by UV (210 nm) and CAD.
Mass Spectrometry
The ESI-TOF mass spectra were generated with a Waters Q-Tof Micro or Premier mass spectrometer equipped with an electrospray ionization source. The sample was diluted with H2O-MeCN (1:1) or H2O-MeCN (1:1) with 0.1% formic acid and introduced via infusion using the onboard syringe pump and analyzed by negative ESI.
Nuclear Magnetic Resonance Spectroscopy
NMR data for 1 to 5 were acquired from Bruker Avance 500 MHz instrument with a 5 mm broadband probe and 2.5 mm inverse detection probe. Some of the NMR data were also acquired at 600 MHz. The 1H NMR and 13C NMR spectra were referenced to the MeOD signal at δH 3.30 and δC 49.0 ppm, respectively. NMR data were acquired either at 300 K or at 286 K.
HRMS (ESI-TOF): m/z [M-H]– (calcd for C55H87O32: 1259.5180); found: 1259.4926.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
1.
Lemus-MondacaR.Vega-GálvezA.Zura-BravoL.Ah-HenK. Stevia rebaudiana Bertoni, source of a high-potency natural sweetener: a comprehensive review on the biochemical, nutritional and functional aspects. Food Chem. 2012;132(3):1121-1132.doi:10.1016/j.foodchem.2011.11.140
2.
CeunenS.GeunsJMC. Steviol glycosides: chemical diversity, metabolism, and function. J Nat Prod. 2013;76(6):1201-1228.doi:10.1021/np400203b
3.
IbrahimMA.RodenburgDL.AlvesKet al. Rebaudiosides R and S, minor diterpene glycosides from the leaves of Stevia rebaudiana. J Nat Prod. 2016;79(5):1468-1472.doi:10.1021/acs.jnatprod.6b00048
4.
PrakashI.MaG.BundersCet al. A novel diterpene glycoside with nine glucose units from Stevia rebaudiana Bertoni. Biomolecules. 2017;7(4):10-19.doi:10.3390/biom7010010
5.
PereraW.GhivirigaI.RodenburgDet al. Tetra-glucopyranosyl diterpene ent-Kaur-16-en-19-oic acid and ent-13(S)-hydroxyatisenoic acid derivatives from a commercial extract of Stevia rebaudiana (Bertoni) Bertoni. Molecules. 2018;23(12):3328-3342.doi:10.3390/molecules23123328
6.
PereraWH.GhivirigaI.RodenburgDLet al. Rebaudiosides T and U, minor C-19 xylopyranosyl and arabinopyranosyl steviol glycoside derivatives from Stevia rebaudiana (Bertoni) Bertoni. Phytochemistry. 2017;135:106-114.doi:10.1016/j.phytochem.2016.12.001
7.
McGarveyBD.AttygalleAB.StarrattANet al. New non-glycosidic diterpenes from the leaves of Stevia rebaudiana. J Nat Prod. 2003;66(10):1395-1398.doi:10.1021/np0302091
8.
EscamillaEM.OrtegaA. Labdane diterpenoids from Stevia seleriana. Phytochemistry. 1991;30(2):599-602.doi:10.1016/0031-9422(91)83733-2
9.
AngelesE.FoltingK.GriecoPA.HuffmanJC.MirandaR.SalmónM. Isolation and structure of stephalic acid, a new clerodane diterpene from Stevia polycephala. Phytochemistry. 1982;21(7):1804-1806.doi:10.1016/S0031-9422(82)85072-3
PelletierSW.AteyaA-MM.Finer-MooreJ.ModyNV.SchrammLC. Atisenol, a new ent-atisene diterpenoid lactone from Aconitum heterophyllum. J Nat Prod. 1982;45(6):779-781.doi:10.1021/np50024a028