Two new flavonol glycosides, rhamnocitrin 3-O-α-L-rhamnopyranosyl-(1→6)-[α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside (1) and quercetin 3-O-6-Z-p-coumaroyl-β-D-glucopyranosyl-(1→2)-α-L-rhamnopyranoside (2), along with 3 flavonol glycosides, isoquercitrin (3), rutin (4), and quercetin 3-O-α-L-rhamnopyranosyl-(1→6)-β-D-galactopyranoside (5), and two known sesquiterpenes, alismol (6) and spathulenol (7), were isolated from the leaves of Phoebe poilanei Kosterm. Their chemical structures were elucidated by analyses of their high-resolution electrospray ionization mass spectral data and nuclear magnetic resonance spectral data and comparison with those reported in the literature. Two sesquiterpenes 6 and 7 were found to exhibit moderate cytotoxic activity with IC50 values ranging from 21.6 to 29.8 µM.
Phoebe is a genus of evergreen trees and shrubs belonging to the Laurel family, Lauraceae. There are approximately 100 species of the Phoebe genus, distributed in tropical and subtropical Asia and neotropical America.1 In Vietnam, genus Phoebe is composed of about 12 species scattered throughout the country and is used to treat skin injuries and headaches.2Phoebe is mainly recognized for its alkaloidal contents particularly for oxoaporphine and aporphine alkaloids of isoquinoline class.1 Previously, the presence of flavonol glycosides in the chemical constituents from P. poilanei has been confirmed by our group.3 Herein, we report the isolation, structural elucidation, and cytotoxic activity of 2 new flavonol glycosides and 10 known compounds from the leaves of P. poilanei.
Compound 1 was obtained as a yellow amorphous powder, characterizes a flavonol, and its molecular formula was determined as C34H42O19 by a quasi-molecular ion peak at m/z 777.2218 [M+Na]+ (calcd for [C34H42O19Na]+, 777.2213) in high-resolution electrospray ionization mass spectrometry (HR-ESI-MS). The 1H-NMR spectrum of 1 (in methanol-d4) showed the following signals: 4 aromatic protons at δH 6.88 (2H, d, J = 8.4 Hz) and 8.02 (2H, d, J = 8.4 Hz), assigning to the AA′BB′ coupling system of B ring; two aromatic protons of A ring at δH 6.23 (1H, d, J = 2.4 Hz) and 6.50 (1H, d, J = 2.4 Hz); one methoxy group at δH 3.82 (3H, s); 3 anomeric protons at δH 4.48 (1H, br s), 5.22 (1H, br s), and 5.61 (d, J = 7.6 Hz); and two secondary methyl groups at δH 0.98 (d, J = 6.4 Hz) and 1.06 (d, J = 6.4 Hz), suggesting the appearance of 3 sugar units. The 13C-NMR and heteronuclear single quantum coherence (HSQC) spectra revealed the signals of 34 carbon, of which 15 were assigned to a flavonol aglycone, 18 were assigned to 3 monosaccharide moieties, and 1 to a methoxy carbon (Table 1). The heteronuclear multiple bond correlation (HMBC) cross peaks from H-2′/H-6′ (δH 8.02) to C-2 (δC 159.3)/C-1′ (δC 123.0)/C-4′ (δC 161.2) confirmed that the hydroxy group was at C-4′. The HMBCs from H-6 (δH 6.23) to C-5 (δC 162.7)/C-7 (δC 166.9)/C-8 (δC 93.1)/C-10 (δC 106.7), from H-8 (δH 6.50) to C-6 (δC 98.9)/C-7 (δC 166.9)/C-9 (δC 158.2)/C-10 (δC 106.7), and from methoxy protons (δH 3.82) to C-7 (δC 166.9) confirmed the position of hydroxy and methoxy groups at C-5 and C-7, respectively. Acid hydrolysis of 1 revealed D-glucose and L-rhamnose (identified as trimethylsilyl [TMS] derivatives by a gas chromatography method). In addition, multiplicity of glc H-1″ [5.61 (d, J = 7.6 Hz)], rha H-1″′ [5.22 (br s)], and rha H-1″″ [4.48 (br s)] in the 1H-NMR spectral data of 1 showed the presence of one β-D-glucopyranosyl and two α-L-rhamnopyranosyl moieties. The HMBC (Figure 1) between rha H-1″′ (δH 5.22) and glc C-2″ (δC 79.8) and between rha H-1″″ (δH 4.48) and glc C-6″ (δC 68.3) suggested sugar linkages as α-L-rhamnopyranosyl-(1→6)-[O-α-L-rhamnopyranosyl (1→2)]-β-D-glucopyranoside.4 The position of trisaccharide at C-3 of flavonol was proved by the observation of HMBC between glc H-1″ (δH 5.61) and C-3 (δC 134.5). Consequently, the new compound 1 was determined to be rhamnocitrin 3-O-α-L-rhamnopyranosyl-(1→6)-[α-L-rhamnopyrano-syl-(1→2)]-β-D-glucopyranoside.
The 1H- and 13C-NMR Data for Compounds 1 and 2 in CD3OD.
C
1
C
2
δC
δH (mult., J, Hz)
δC
δH (mult., J, Hz)
Aglycone
Aglycone
2
159.3
-
2
159.0
-
3
134.5
-
3
137.4
-
4
179.2
-
4
179.7
-
5
162.7
-
5
163.3
-
6
98.9
6.23 (d, 2.4)
6
99.9
6.17 (d, 1.6)
7
166.9
-
7
165.9
-
8
93.1
6.50 (d, 2.4)
8
94.8
6.33 (d, 1.6)
9
158.2
-
9
158.5
-
10
106.7
-
10
106.1
-
1′
123.0
-
1′
122.9
-
2′
132.2
8.02 (d, 8.4)
2′
117.1
7.31 (d, 2.0)
3′
116.1
6.88 (d, 8.4)
3′
146.3
-
4′
161.2
-
4′
149.7
-
5′
116.1
6.88 (d, 8.4)
5′
116.3
6.88 (d, 8.4)
6′
132.2
8.02 (d, 8.4)
6′
122.9
7.23 (dd, 2.0, 8.4)
7-OMe
56.4
3.82 (s)
3-O-Glc
3-O-Rha
1″
100.4
5.61 (d, 7.6)
1″
103.4
5.53 (br s)
2″
79.8
3.60 (dd, 7.6, 8.4)
2″
83.8
4.33 (br d, 2.0)
3″
78.8
3.57 (t, 8.4)
3″
71.7
3.86 (dd, 2.0, 8.5)
4″
71.9
3.24 (t, 8.4)
4″
73.7
3.35 (t, 8.5)
5″
76.9
3.39 (m)
5″
71.8
3.73 (m)
6″
68.3
3.37 (dd, 4.8, 11.6)3.81 (dd, 2.0, 11.6)
6″
17.7
1.01 (d, 6.0)
2″-O-Rha
2″-O-Glc
1′′′
102.5
5.22 (br s)
1′′′
107.1
4.31 (d, 8.0)
2′′′
72.3
4.00 (br d, 2.0)
2′′′
75.1
3.35 (dd, 8.0, 8.5)
3′′′
72.0
3.80 (dd, 2.0, 8.4)
3′′′
77.5
3.36 (t, 8.5)
4′′′
73.9
3.34 (t, 8.4)
4′′′
71.6
3.20 (t, 8.5)
5′′′
69.9
4.06 (m)
5′′′
75.3
3.22 (m)
6′′′
17.6
0.98 (d, 6.4)
6′′′
64.3
4.18 (dd, 6.0, 12.0)4.23 (dd, 2.8, 12.0)
6″-O-Rha
6′′′-p-Cou
1′′′′
102.3
4.48 (br s)
1′′′′
127.3
-
2′′′′
72.2
3.59 (br d, 2.0)
2′′′′, 6′′′′
133.8
7.21 (d, 8.5)
3′′′′
72.3
3.46 (dd, 2.0, 8.4)
3′′′′, 5′′′′
115.8
6.69 (d, 8.5)
4′′′′
73.8
3.23 (t, 8.4)
4′′′′
159.9
-
5′′′′
69.7
3.40 (m)
7′′′′
145.1
6.39 (d, 12.8)
6′′′′
17.8
1.06 (d, 6.4)
8′′′′
115.7
5.36 (d, 12.8)
9′′′′
167.8
-
Key HMBCs of compounds 1 and 2. HMBC, heteronuclear multiple bond correlation.
Compound 2 was also isolated as a yellow amorphous powder, characterizes a flavonol, and its molecular formula was confirmed as C36H38O18 by HR-ESI-MS [M–H]– ion at m/z 755.1821 (calcd for [C36H37O18]–, 755.1829). The 1H-NMR spectrum suggested that compound 2 is a flavonol glycoside with the following signals: two meta-protons of A ring at δH 6.17 (d, J = 1.6 Hz) and 6.33 (d, J = 1.6 Hz); 3 ABX protons of B rings at δH 6.88 (d, J = 8.4 Hz), 7.23 (dd, J = 2.0, 8.4 Hz), and 7.31 (d, J = 2.0 Hz). In addition, two anomeric protons at δH 5.53 (br s) and 4.31 (d, J = 8.0 Hz) were assigned to two sugar moieties. The proton signals at δH 6.69 (2H, d, J = 8.5 Hz), 7.21 (2H, d, J = 8.5 Hz), 5.36 (d, J = 12.8 Hz), and 6.39 (d, J = 12.8 Hz) were assigned to a coumaroyl moiety. The 13C-NMR and HSQC spectra showed the signals of 36 carbons, including 2 carbonyl, 11 non-protonated carbons, 21 methines, 1 methylene, and 1 methyl carbon (Table 1). The combination of 1H-, 13C-NMR, HSQC, and HMBC experiments allowed the assignments of all protons and carbons as well as the connectivity among quercetin, coumaroyl, rhamnopyranosyl, and glucopyranosyl moieties. On comparison, the 1H- and 13C-NMR spectra of 2 with those of quercetin 3-O-6-E-p-coumaroyl-β-D-glucopyranosyl-(1→2)-α-L-rhamnopyranoside5 were found to be similar. The only difference between them is the configuration of p-coumaroyl moiety. The HMBCs between H-2″″/H-6″″ (δH 7.21) and C-4″″ (δC 159.9)/C-7″″ (δC 145.1) and between H-7″″ (δH 6.39) and C-2″″ (C-6″″) (δC 133.8)/C-9″″ (δC 167.8) as well as the coupling constant between H-7″″ and H-8″″ (J = 12.8 Hz) confirmed the presence of (Z)-p-coumaroyl moiety. Acid hydrolysis of 2 revealed D-glucose and L-rhamnose (identified as TMS derivatives by a gas chromatography method).
In addition, the multiplicity of rha H-1″ [5.53 (br s)] and glc H-1″′ [4.31 (d, J = 8.0 Hz)] proved the configurations of sugar moieties as α-L-rhamnopyranosyl and β-D-glucopyranosyl. The HMBCs from glc H-6″′ (δH 4.18 and 4.23) cou C-9″″ (δC 167.8); from glc H-1″′ (δH 4.31) to rha C-2″ (δC 83.8); and from rha H-1″ (δH 5.53) to C-3 (δC 137.4) suggested the acyl saccharide linkages as 6-Z-p-coumaroyl-β-D-glucopyranosyl-(1→2)-α-L-rhamnopyranosyl and the position of saccharide at C-3 of the quercetin aglycone. Based on the above evidence, the new structure of 2 was elucidated as quercetin 3-O-6-Z-p-coumaroyl-β-D-glucopyranosyl-(1→2)-α-L-rhamnopyranoside.
The known compounds were elucidated as kaempferol 3-O-β-D-glucopyranoside (2),6 isoquercitrin (3),4 rutin (4),7 quercetin 3-O-α-L-rhamnopyranosyl-(1→6)-β-D-galactopyranoside (5),8 alismol (6),9 and spathulenol (7)10 (Figure 2) by analyses of HR-ESI-MS, 1D-NMR, and 2D-NMR spectral data and comparison with those reported in the literature. The sesquiterpenes were also reported from Phoebe genus.11
Chemical structures of compounds 1 to 12.
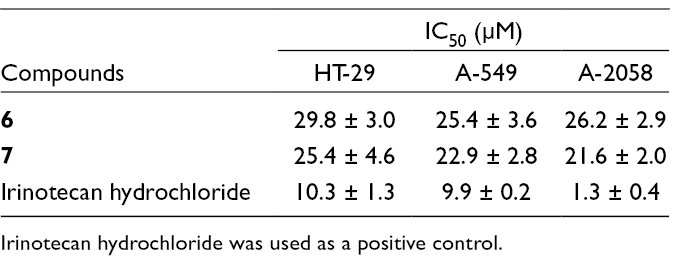
To investigate the cytotoxic effects, all the isolated compounds were evaluated against 3 human cancer cell lines, HT-29, A-549, and A-2058 using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Irinotecan hydrochloride, an anticancer agent,12 was used as a positive control with IC50 values ranging from 1.3 to 10.3 µM for all the tested human cancer cell lines (Table 2). Among the isolated compounds, two sesquiterpenes 6 and 7 exhibited moderate activity with IC50 values ranging from 21.6 to 29.8 µM. All flavonol glycosides did not show any cytotoxic activity on tested human cancer cell lines (IC50 > 30 µM).
Effects of 6 and 7 on the Growth of Human Cancer Cells.
Compounds
IC50 (µM)
HT-29
A-549
A-2058
6
29.8 ± 3.0
25.4 ± 3.6
26.2 ± 2.9
7
25.4 ± 4.6
22.9 ± 2.8
21.6 ± 2.0
Irinotecan hydrochloride
10.3 ± 1.3
9.9 ± 0.2
1.3 ± 0.4
Irinotecan hydrochloride was used as a positive control.
Experimental
General
Optical rotations were determined on a Jasco DIP-370 automatic polarimeter. The nuclear magnetic resonance (NMR) spectra were recorded using a Agilent 400-MR spectrometer (400 MHz for 1H and 100 MHz for 13C). The HR-ESI-MS were obtained using Agilent 6550 iFunnel QTOF LC/MS system. Column chromatography was performed using silica gel (Kieselgel 60, 70-230 mesh and 230-400 mesh, Merck, Darmstadt, Germany) or RP-18 resins (30-50 μm, Fujisilisa Chemical Ltd., Kasugai Aichi, Japan), and thin layer chromatography was performed using precoated silica gel 60 F254 (0.25 mm, Merck) and RP-18 F254S plates (0.25 mm, Merck).
Plant Materials
The plant samples were collected at Tam Duong, Lao Cai Province, Vietnam, in April 2016. The scientific name was identified as Phoebe poilanei Kosterm by Dr Vu Van Thanh, Institute of Ecology and Biological Resources, Vietnam Academy of Science and Technology (VAST). A voucher specimen (NCCT-P28) was deposited at the Institute of Marine Biochemistry, VAST.
Extraction and Isolation
The dried leaves of Phoebe poilanei (3.2 kg) were extracted 3 times with MeOH using a sonicator to yield 280 g of a dark solid residue, which was then suspended in water and successively partitioned with dichloromethane and ethyl acetate (EtOAc) to obtain the corresponding dichloromethane (PP1, 105.0 g), EtOAc (PP2, 52.0 g), and water layers (PP3) after removal of solvent in vacuo. PP1 was subjected to a silica gel column chromatography (CC), eluting with gradient solvent system of acetone and n-hexane (0% to 100% volume of acetone) to give five fractions, PP1A to PP1E. PP1C (3.7 g) was chromatographed on a silica gel column eluting with n-hexane/EtOAc (5/1, v/v) to give two smaller fractions (PP1C1 and PP1C2). Compounds 6 (10.0 mg) and 7 (11.0 mg) were obtained from PP1C1 eluting with MeOH/water (8/1, v/v). PP1D was chromatographed on a RP-18 column eluting with MeOH/water (12/1, v/v) to give two smaller fractions (PP1D1 and PP1D2). PP1D1 was chromatographed on a RP-18 column eluting with acetone/water (5/1, v/v) to yield compound 3 (20.0 mg). PP3 was chromatographed on a Diaion HP-20 column first eluting with water to remove sugar components, then increasing concentration of MeOH in water (25%, 50%, 75%, and 100%) to obtain 4 fractions, PP3A to PP3D, respectively. PP3B was subjected to a silica gel CC, eluting with gradient solvent system of chloroform/MeOH (20/1, 10/1, 5/1, 2.5/1, v/v) to give 4 fractions, PP3B1 to PP3B4. PP3B2 was chromatographed on a RP-18 column eluting with MeOH/water (1/1.3, v/v) to give 3 smaller subfractions, PP3B2A to PP3B2C. Compound 1 (48.0 mg) was obtained from PP3B2B by chromatography on a RP-18 column eluting with methanol/water (5/1, v/v). PP3C was subjected to a silica gel CC, eluting with gradient solvent system of chloroform/MeOH (20/1, 10/1, 5/1, 2.5/1, v/v) to give 4 fractions, PP3C1 to PP3C4. PP3C3 was chromatographed on a RP-18 column eluting with acetone/water (1/1, v/v) to give 4 smaller subfractions, PP3C3A to PP3C3D. PP3C3A was chromatographed on a RP-18 column eluting with acetone/water (2/1, v/v) to obtain compounds 4 (9.0 mg) and 5 (8.0 mg). PP3C3C was chromatographed on a RP-18 column eluting with acetone/water (2/1, v/v) to yield compound 2 (15.0 mg).
HR-ESI-MS: found m/z 755.1821 [M - H]- (calcd for [C36H37O18]- 755.1829).
Acid Hydrolysis
Each compound (1 and 2, 2.0 mg) was dissolved in 1.0 N HCl (dioxane–H2O, 1:1, v/v, 1.0 mL) and then heated to 80°C in a water bath for 3 hours. The acidic solution was neutralized with silver carbonate and the solvent thoroughly driven out under N2 gas overnight. After extraction with CHCl3, the aqueous layer was concentrated to dryness using N2 gas. The residue was dissolved in 0.1 mL of dry pyridine, and then L-cysteine methyl ester hydrochloride in pyridine (0.06 M, 0.1 mL) was added to the solution. The reaction mixture was heated at 60°C for 2 hours, and 0.1 mL of trimethylsilylimidazole solution was added, followed by heating at 60°C for 1.5 hours. The dried product was partitioned with n-hexane and H2O (0.1 mL, each), and the organic layer was analyzed by gas chromatography: column of SPB-1 (0.25 mm × 30 m); flame ionization detector, column temperature 210°C, injector temperature 270°C, detector temperature 300°C, carrier gas He (2.0 mL/min). The retention times of persilylated glucose and rhamnose were found to be 14.11 and 4.50 minutes, respectively, when compared with the standard solutions prepared by the same reaction from the standard monosaccharides. The retention times of persilylated D-glucose, L-glucose, and L-rhamnose were 14.11, 14.26, and 4.50 minutes, respectively.
Cytotoxic Assays
The effects of compounds 1 to 7 on the growth of human cancer cells were determined by MTT assay. The cancer cell lines, HT-29 (human colon cancer), A-549 (human lung cancer), and A-2058 (human melanoma cancer cell) were grown in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/mL and 100 g/mL, respectively) at 37°C in a humidified 5% CO2 atmosphere. The MTT assays were performed as follows: human cancer cells (1.5-2.5 × 105 cells/mL) were treated for 48 hours with the samples in dimethylsulfoxide (DMSO) and positive control, irinotecan hydrochloride (1.0, 3, 10, 30 µM). After incubation, 0.1 mg (50 µL of a 2 mg/mL solution) MTT (Sigma, St Louis, MO, USA) was added to each well and the cells were then incubated at 37°C for 4 hours. The plates were centrifuged at 1000 rpm for 5 minutes at room temperature and the supernatant was then carefully aspirated. DMSO (150 µL) was then added to each well to dissolve the formazan crystals. The plates were read immediately at 540 nm on a microplate reader (Amersham Pharmacia Biotech., NewYork, USA). All the assays were performed in triplicate and the mean absorbance values were calculated. The results are expressed as the relative cell viability percentage presented by a reduction in the absorbance after the treatment of the tested compounds compared to the untreated controls. A dose-response curve was generated and the IC50 value was determined for each compound as well as each cell line.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2015.22.
Supplemental Material
References
1.
Kumar SemwalD.Badoni SemwalR.SemwalDK.SemwalRB. Ethnobotany, pharmacology and phytochemistry of the genus Phoebe (Lauraceae). Mini Rev Org Chem. 2013;10(1):12-26.doi:10.2174/1570193X11310010002
2.
ChiVV. The dictionary of Vietnamese medicinal plants. Hanoi Medicine: Hanoi; 1999.
KazumaK.NodaN.SuzukiM. Malonylated flavonol glycosides from the petals of Clitoria ternatea. Phytochemistry. 2003;62(2):229-237.doi:10.1016/S0031-9422(02)00486-7
5.
OhH.KangD-G.KwonJ-Wet al. Isolation of angiotensin converting enzyme (ACE) inhibitory flavonoids from Sedum sarmentosum. Biol Pharm Bull. 2004;27(12):2035-2037.doi:10.1248/bpb.27.2035
6.
ParkSY.KimJS.LeeSY.BaeKH.KangSS. Chemical Constituents of Lathyrus davidii. Nat Prod Sci. 2008;14:281-288.
7.
BeckMA.HäberleinH. Flavonol glycosides from Eschscholtzia californica. Phytochemistry. 1999;50(2):329-332.doi:10.1016/S0031-9422(98)00503-2
8.
JaramilloK.DawidC.HofmannT.FujimotoY.OsorioC. Identification of antioxidative flavonols and anthocyanins in Sicana odorifera fruit peel. J Agric Food Chem. 2011;59(3):975-983.doi:10.1021/jf103151n
9.
YoshikawaM.HatakeyamaS.TanakaN.FukudaY.MurakamiN.YamaharaJ. Orientalols a, B, and C, sesquiterpene constituents from Chinese Alismatis Rhizoma, and revised structures of alismol and alismoxide. Chem Pharm Bull. 1992;40(9):2582-2584.doi:10.1248/cpb.40.2582
10.
NhiemNX.TuongNT.KyPTet al. Chemical Components from Phaeanthus vietnamensis and Their Inhibitory NO Production in BV2 Cells. Chem Biodivers. 2017;14(8):e1700013.doi:10.1002/cbdv.201700013
11.
DingW.NingL.WangY.LiF.ZhangL.XiongY. Chemical constituents of the ethanol extract from the xylem of Phoebe hui. Chem Nat Compd. 2017;53(5):966-967.doi:10.1007/s10600-017-2171-4
12.
FujiiH.YamadaY.WatanabeDet al. Dose adjustment of irinotecan based on UGT1A1 polymorphisms in patients with colorectal cancer. Cancer Chemother Pharmacol. 2019;83(1):123-129.doi:10.1007/s00280-018-3711-8
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.